PD-L1 Targeting Immune-Microbubble Complex Enhances Therapeutic Index in Murine Colon Cancer Models



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
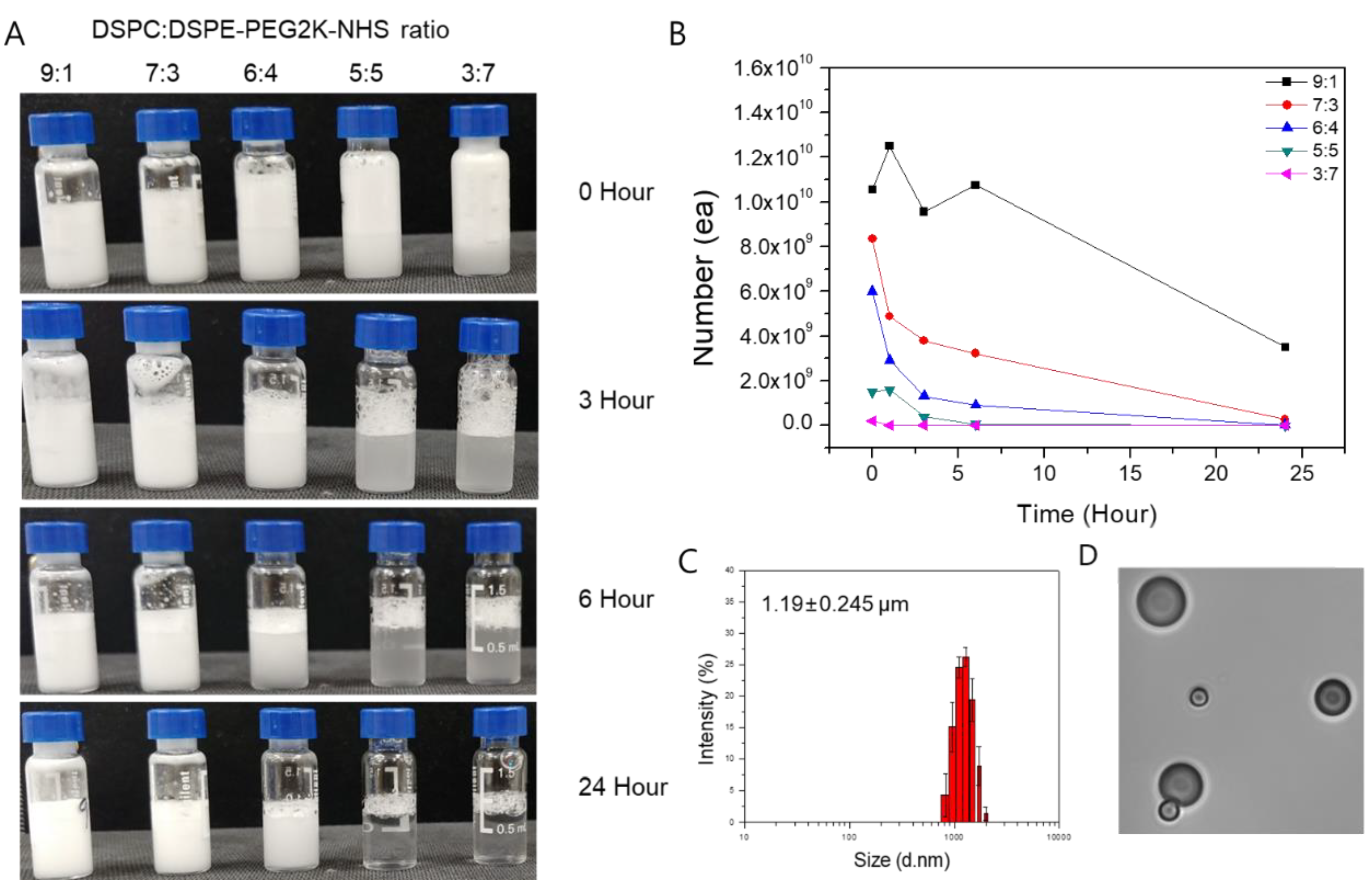
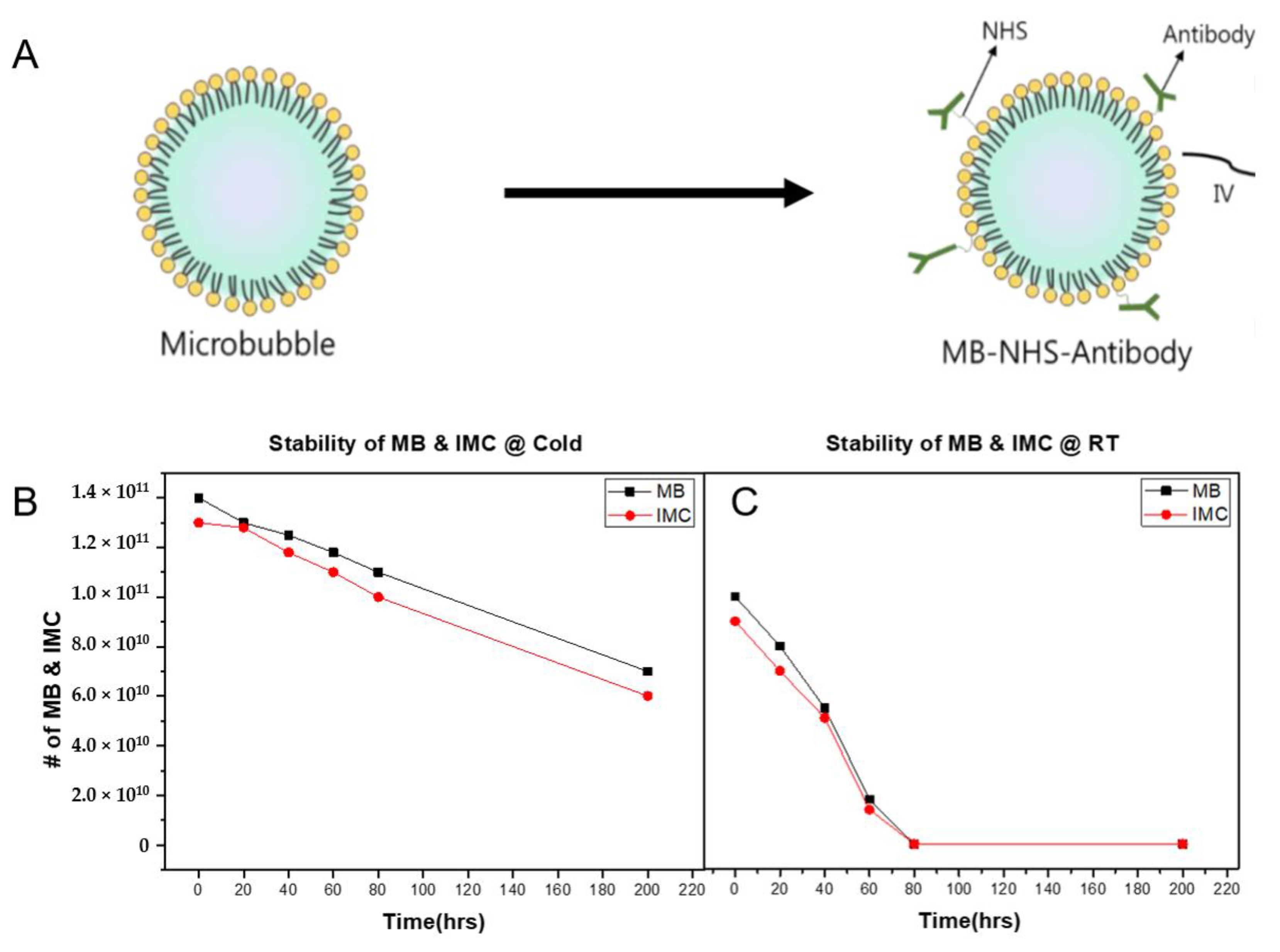
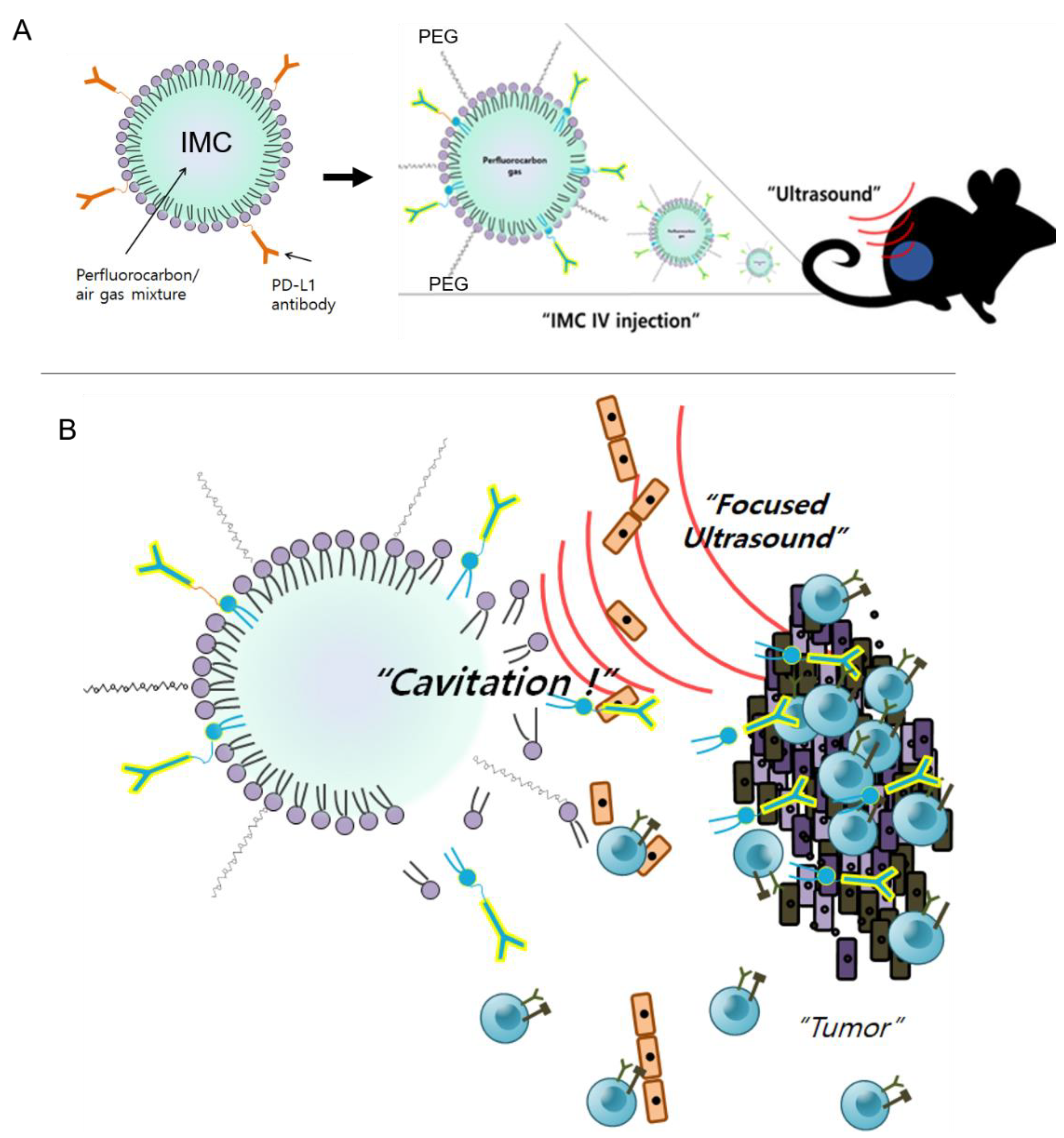
2.1. Characterization of MBs and IMC
2.2. Confocal Image of IMC
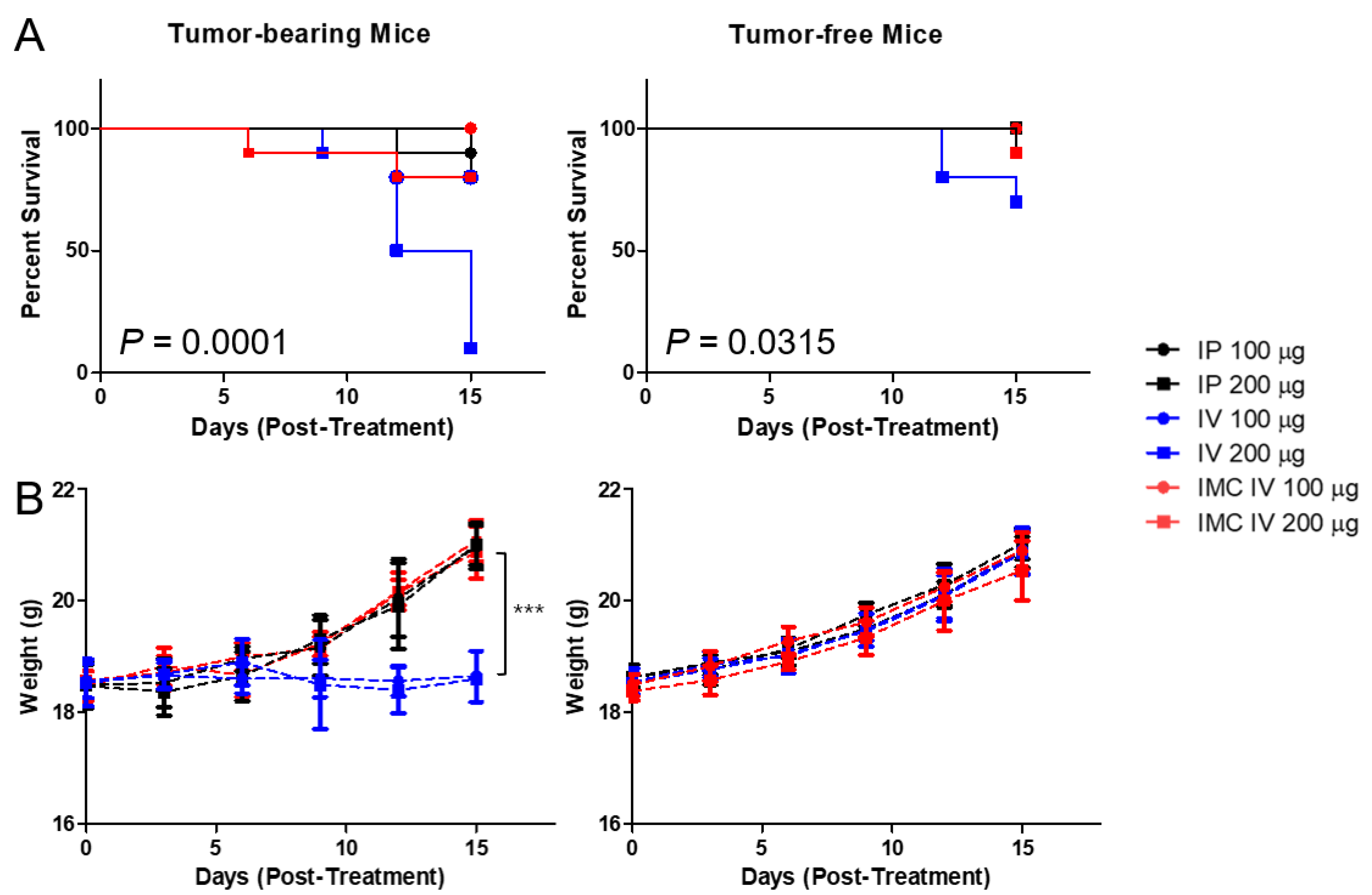
2.3. Improved Toxicological Profiles of IMC over the PD-L1 Antibodies In Vivo
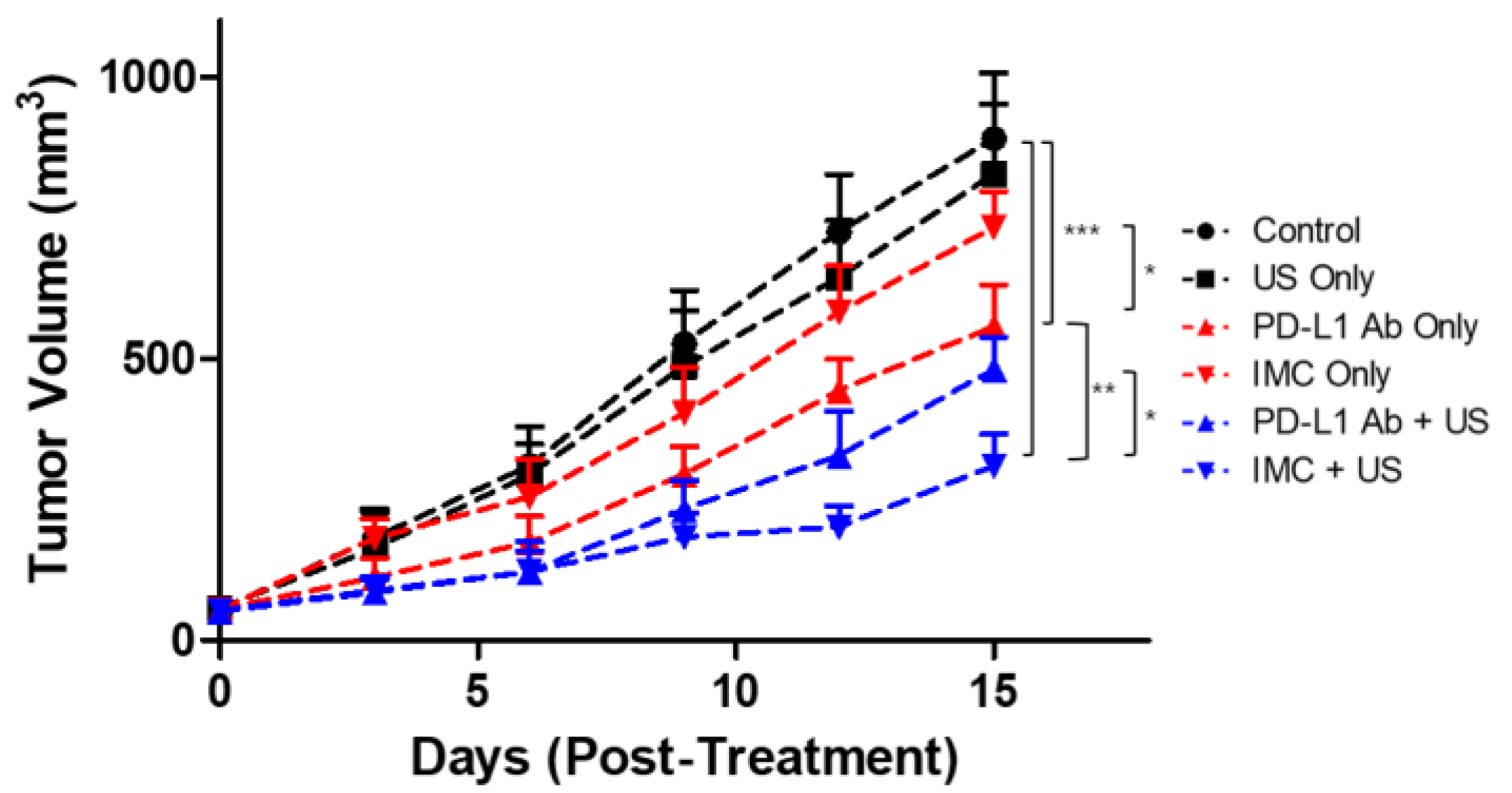
2.4. Inhibition of Tumor Growth by the IMC-FUS Combination Therapy
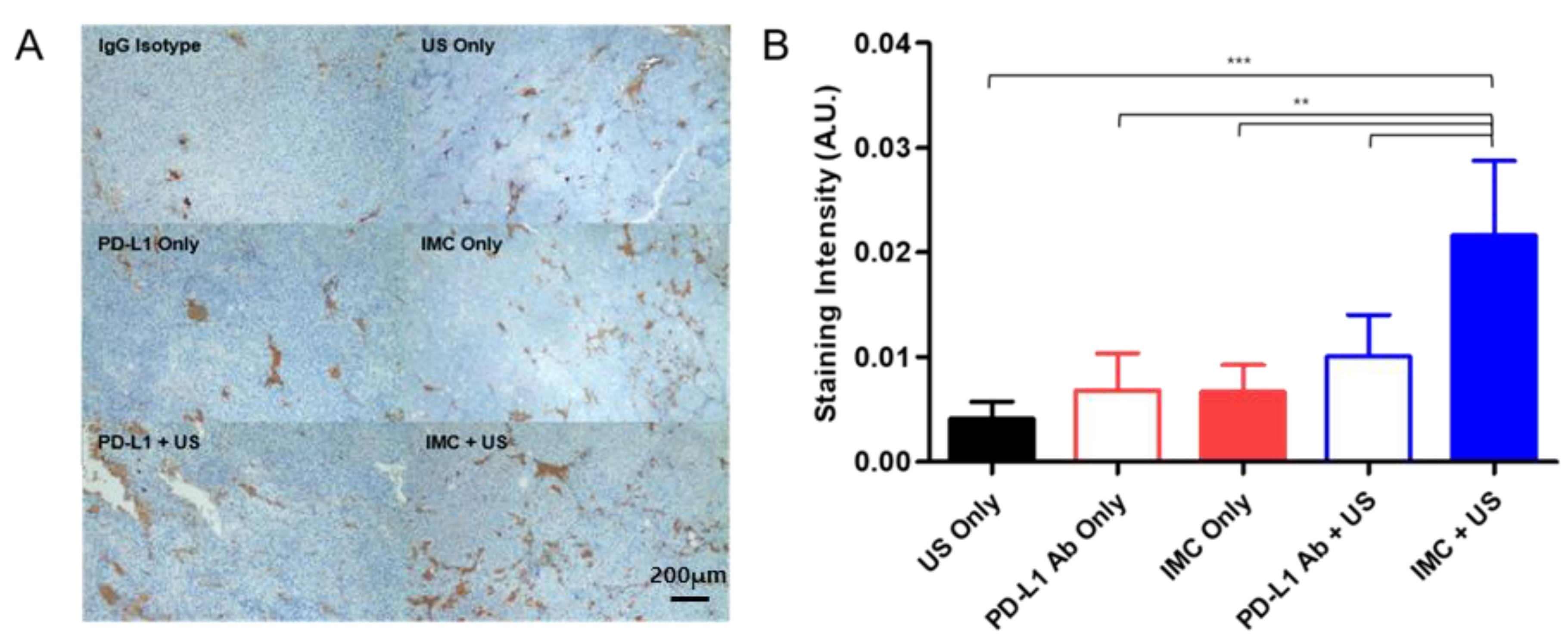
2.5. Immunohistochemical Staining of the Tumor Confirms Enhanced Localization of PD-L1 Antibodies
3. Discussion
4. Materials and Methods
4.1. Preparation of the Lipid Microbubbles
4.2. Preparation and Characterization of the IMC
4.3. Stability Test of MB and IMC
4.4. Confocal Imaging
4.5. Animal Studies
4.6. Preparation of Immunohistochemistry
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Disis, M.L. Mechanism of action of immunotherapy. Semin Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Hoefsmit, E.P.; Smyth, M.J.; Blank, C.U.; Teng, M.W. The promise of neoadjuvant immunotherapy and surgery for cancer treatment. Clin. Cancer Res. 2019, 25, 5743–5751. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, P.; Chamoto, K.; Honjo, T. Combination therapy strategies for improving PD-1 blockade efficacy: a new era in cancer immunotherapy. J. Intern. Med. 2018, 283, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, G.L.; Hansen, H.J. Targeted combination immunotherapy of cancer. U.S. Patent No. 6,077,499, 20 January 2000. [Google Scholar]
- Devaud, C.; John, L.B.; Westwood, J.A.; Darcy, P.K.; Kershaw, M.H. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology 2013, 2, e25961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, J.; Son, S.; Park, K.S.; Zou, W.; Shea, L.D.; Moon, J.J. Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 2019, 4, 398–414. [Google Scholar] [CrossRef]
- Peach, R.J.; Bajorath, J.; Naemura, J.; Leytze, G.; Greene, J.; Aruffo, A.; Linsley, P.S. Both extracellular immunoglobin-like domains of CD80 contain residues critical for binding T cell surface receptors CTLA-4 and CD28. J. Biol. Chem. 1995, 270, 21181–21187. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhang, Y.; Rolfe, P.A.; Hernández, V.M.; Guzman, W.; Kradjian, G.; Marelli, B.; Qin, G.; Qi, J.; Wang, H. Combination therapy with NHS-muIL12 and avelumab (anti-PD-L1) enhances antitumor efficacy in preclinical cancer models. Clin. Cancer Res. 2017, 23, 5869–5880. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A. Releasing the brakes on cancer immunotherapy. N. Engl. J. Med. 2015, 373, 1490–1492. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Horn, L.A.; Haile, S.T. The programmed death-1 immune-suppressive pathway: Barrier to antitumor immunity. J. Immunol. 2014, 193, 3835–3841. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Song, X.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017, 407, 57–65. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.A.; Minn, A.J. Combination cancer therapy with immune checkpoint blockade: Mechanisms and strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yu, J.X.; Hubbard-Lucey, V.M.; Neftelinov, S.T.; Hodge, J.P.; Lin, Y. The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Publ. Group 2018, 17, 854–855. [Google Scholar] [CrossRef]
- Lindley, C.; McCune, J.S.; Thomason, T.E.; Lauder, D.; Sauls, A.; Adkins, S.; Sawyer, W.T. Perception of chemotherapy side effects cancer versus noncancer patients. Cancer Pract. 1999, 7, 59–65. [Google Scholar] [CrossRef]
- Scotté, F.; Ratta, R.; Beuzeboc, P. Side effects of immunotherapy: A constant challenge for oncologists. Curr. Opin. Oncol. 2019, 31, 280–285. [Google Scholar] [CrossRef]
- Macy, E. Immune-Related Adverse Drug Reactions and Immunologically Mediated Drug Hypersensitivity. Immunol. Allergy Clin. N. Am. 2020, 40, 635–647. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1714. [Google Scholar] [CrossRef]
- Baldo, B. Adverse events to monoclonal antibodies used for cancer therapy: Focus on hypersensitivity responses. Oncoimmunology 2013, 2, e26333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mall, C.; Sckisel, G.D.; Proia, D.A.; Mirsoian, A.; Grossenbacher, S.K.; Pai, C.-C.S.; Chen, M.; Monjazeb, A.M.; Kelly, K.; Blazar, B.R. Repeated PD-1/PD-L1 monoclonal antibody administration induces fatal xenogeneic hypersensitivity reactions in a murine model of breast cancer. Oncoimmunology 2016, 5, e1075114. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, Z.-B.; Lu, P.; Xu, Z.-L.; Chen, W.-Z.; Zhu, H.; Jin, C.-B. Activated anti-tumor immunity in cancer patients after high intensity focused ultrasound ablation. Ultrasound Med. Biol. 2004, 30, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Wu, F. High intensity focused ultrasound ablation and antitumor immune response. J. Acoust. Soc. Am. 2013, 134, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Curley, C.T.; Stevens, A.D.; Mathew, A.S.; Stasiak, K.; Garrison, W.J.; Miller, G.W.; Sheybani, N.D.; Engelhard, V.H.; Bullock, T.N.; Price, R.J. Immunomodulation of intracranial melanoma in response to blood-tumor barrier opening with focused ultrasound. Theranostics 2020, 10, 8821. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hu, Z.; Qiu, L.; Hui, C.; Li, C.; Zhong, P.; Zhang, J. Boosting high-intensity focused ultrasound-induced anti-tumor immunity using a sparse-scan strategy that can more effectively promote dendritic cell maturation. J. Transl. Med. 2010, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-Y.; Liu, T.-M.; Chen, C.-Y.; Huang, Y.-L.; Huang, W.-K.; Sun, C.-K.; Chang, F.-H.; Lin, W.-L. Quantitative and qualitative investigation into the impact of focused ultrasound with microbubbles on the triggered release of nanoparticles from vasculature in mouse tumors. J. Control. Release 2010, 146, 291–298. [Google Scholar] [CrossRef]
- Gao, Y.; Gao, S.; Zhao, B.; Zhao, Y.; Hua, X.; Tan, K.; Liu, Z. Vascular effects of microbubble-enhanced, pulsed, focused ultrasound on liver blood perfusion. Ultrasound Med. Biol. 2012, 38, 91–98. [Google Scholar] [CrossRef]
- Stern, M.; Herrmann, R. Overview of monoclonal antibodies in cancer therapy: Present and promise. Crit. Rev. Oncol. Hematol. 2005, 54, 11–29. [Google Scholar] [CrossRef]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Muller, P.Y.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2010, 2, 233–255. [Google Scholar] [CrossRef] [Green Version]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milling, L.; Zhang, Y.; Irvine, D.J. Delivering safer immunotherapies for cancer. Adv. Drug. Deliv. Rev. 2017, 114, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Lee, J.J.; Siu, L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 2009, 101, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Shurin, M.; Shvedova, A.A. Current understanding of interactions between nanoparticles and the immune system. Toxicol. Appl. Pharm. 2016, 299, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barenholz, Y. Doxil(R)--the first FDA-approved nano-drug: Lessons learned. J. Control Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Mumper, R.J. Paclitaxel Nano-Delivery Systems: A Comprehensive Review. J. Nanomed. Nanotechnol. 2013, 4, 1000164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, W.G.; Husseini, G.A.; Staples, B.J. Ultrasonic drug delivery--a general review. Expert Opin. Drug Deliv. 2004, 1, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Mullick Chowdhury, S.; Lee, T.; Willmann, J.K. Ultrasound-guided drug delivery in cancer. Ultrasonography 2017, 36, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Konofagou, E.E.; Tung, Y.S.; Choi, J.; Deffieux, T.; Baseri, B.; Vlachos, F. Ultrasound-induced blood-brain barrier opening. Curr. Pharm. Biotechnol. 2012, 13, 1332–1345. [Google Scholar] [CrossRef]
- Kim, D.; Lee, S.S.; Yoo, W.Y.; Moon, H.; Cho, A.; Park, S.Y.; Kim, Y.S.; Kim, H.R.; Lee, H.J. Combination Therapy with Doxorubicin-Loaded Reduced Albumin Nanoparticles and Focused Ultrasound in Mouse Breast Cancer Xenografts. Pharmaceuticals 2020, 13, 235. [Google Scholar] [CrossRef]
- Li, T.; Wang, Y.N.; Khokhlova, T.D.; D’Andrea, S.; Starr, F.; Chen, H.; McCune, J.S.; Risler, L.J.; Mashadi-Hossein, A.; Hingorani, S.R.; et al. Pulsed High-Intensity Focused Ultrasound Enhances Delivery of Doxorubicin in a Preclinical Model of Pancreatic Cancer. Cancer Res. 2015, 75, 3738–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eranki, A.; Farr, N.; Partanen, A.; Sharma, K.V.; Rossi, C.T.; Rosenberg, A.Z.; Kim, A.; Oetgen, M.; Celik, H.; Woods, D.; et al. Mechanical fractionation of tissues using microsecond-long HIFU pulses on a clinical MR-HIFU system. Int. J. Hyperth. 2018, 34, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Zhong, M.; Ye, F.; Zhang, X. Low-frequency HIFU induced cancer immunotherapy: Tempting challenges and potential opportunities. Cancer Biol. Med. 2019, 16, 714–728. [Google Scholar] [CrossRef] [PubMed]
- Eranki, A.; Srinivasan, P.; Ries, M.; Kim, A.; Lazarski, C.A.; Rossi, C.T.; Khokhlova, T.D.; Wilson, E.; Knoblach, S.M.; Sharma, K.V.; et al. High-Intensity Focused Ultrasound (HIFU) Triggers Immune Sensitization of Refractory Murine Neuroblastoma to Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2020, 26, 1152–1161. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Lee, S.S.; Moon, H.; Park, S.Y.; Lee, H.J. PD-L1 Targeting Immune-Microbubble Complex Enhances Therapeutic Index in Murine Colon Cancer Models. Pharmaceuticals 2021, 14, 6. https://doi.org/10.3390/ph14010006
Kim D, Lee SS, Moon H, Park SY, Lee HJ. PD-L1 Targeting Immune-Microbubble Complex Enhances Therapeutic Index in Murine Colon Cancer Models. Pharmaceuticals. 2021; 14(1):6. https://doi.org/10.3390/ph14010006
Chicago/Turabian StyleKim, Daehyun, Seung Soo Lee, Hyungwon Moon, So Yeon Park, and Hak Jong Lee. 2021. "PD-L1 Targeting Immune-Microbubble Complex Enhances Therapeutic Index in Murine Colon Cancer Models" Pharmaceuticals 14, no. 1: 6. https://doi.org/10.3390/ph14010006
APA StyleKim, D., Lee, S. S., Moon, H., Park, S. Y., & Lee, H. J. (2021). PD-L1 Targeting Immune-Microbubble Complex Enhances Therapeutic Index in Murine Colon Cancer Models. Pharmaceuticals, 14(1), 6. https://doi.org/10.3390/ph14010006