Repurposing the McoTI-II Rigid Molecular Scaffold in to Inhibitor of ‘Papain Superfamily’ Cysteine Proteases

 ,
,  , and
, and
Abstract
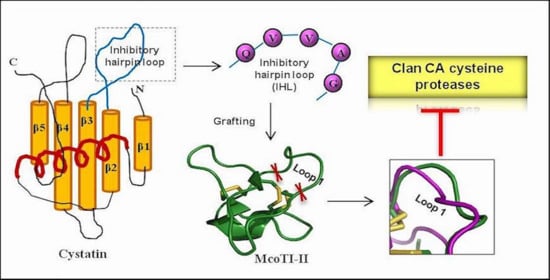
:
1. Introduction
2. Results
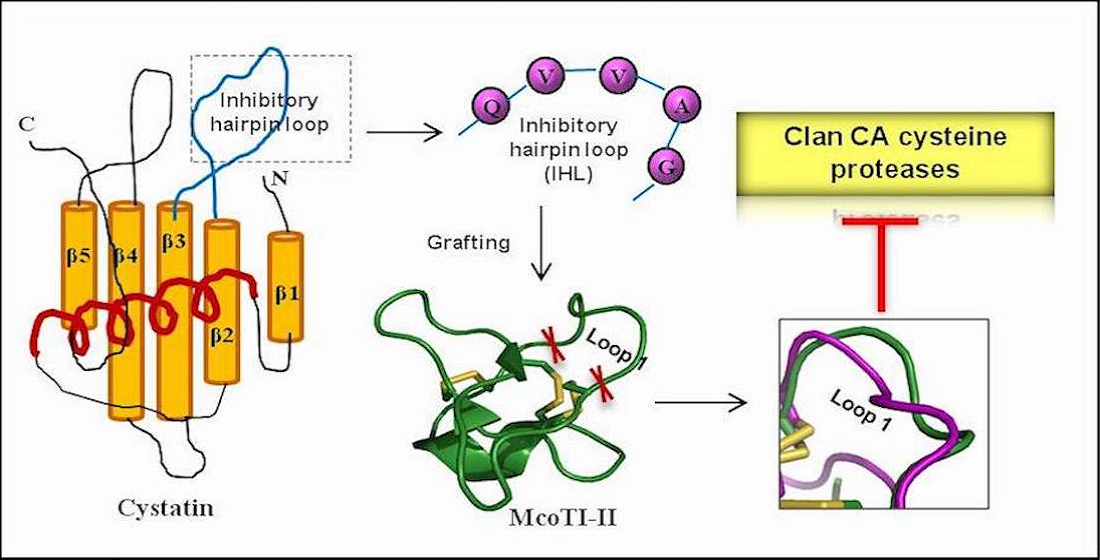
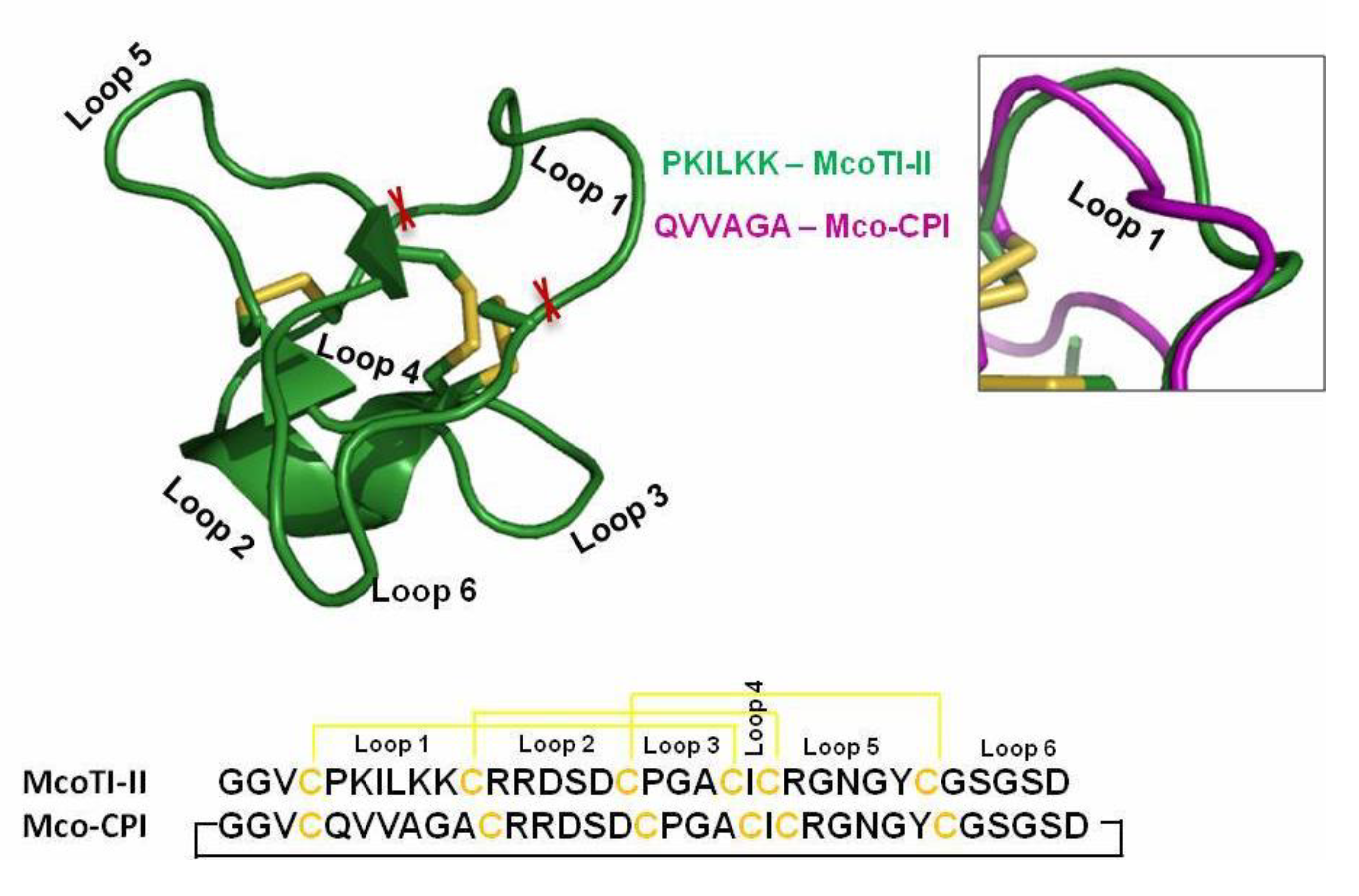
2.1. Modulation of McoTI-II Scaffold for the Design of C1A Cysteine Protease Inhibitor
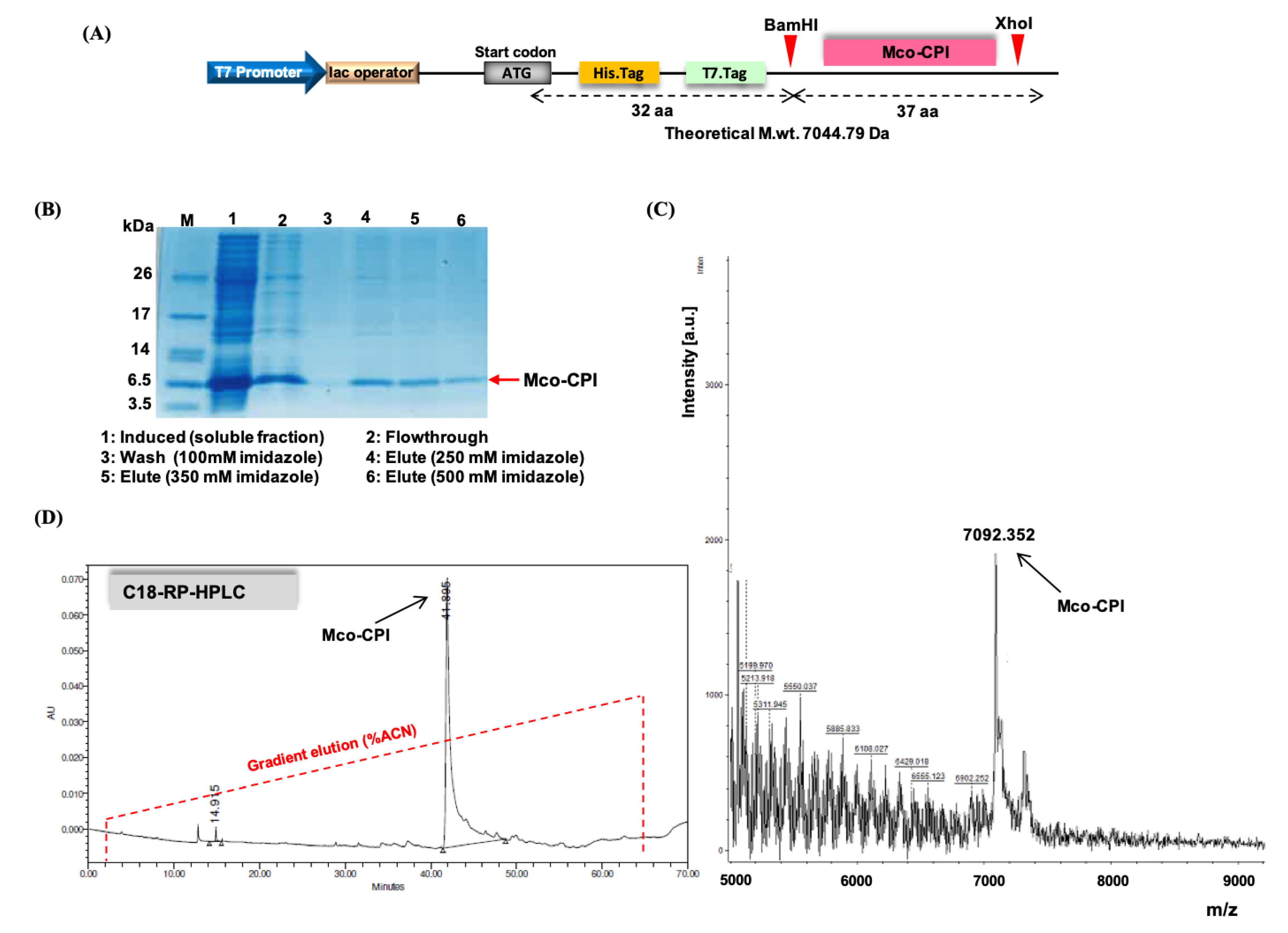
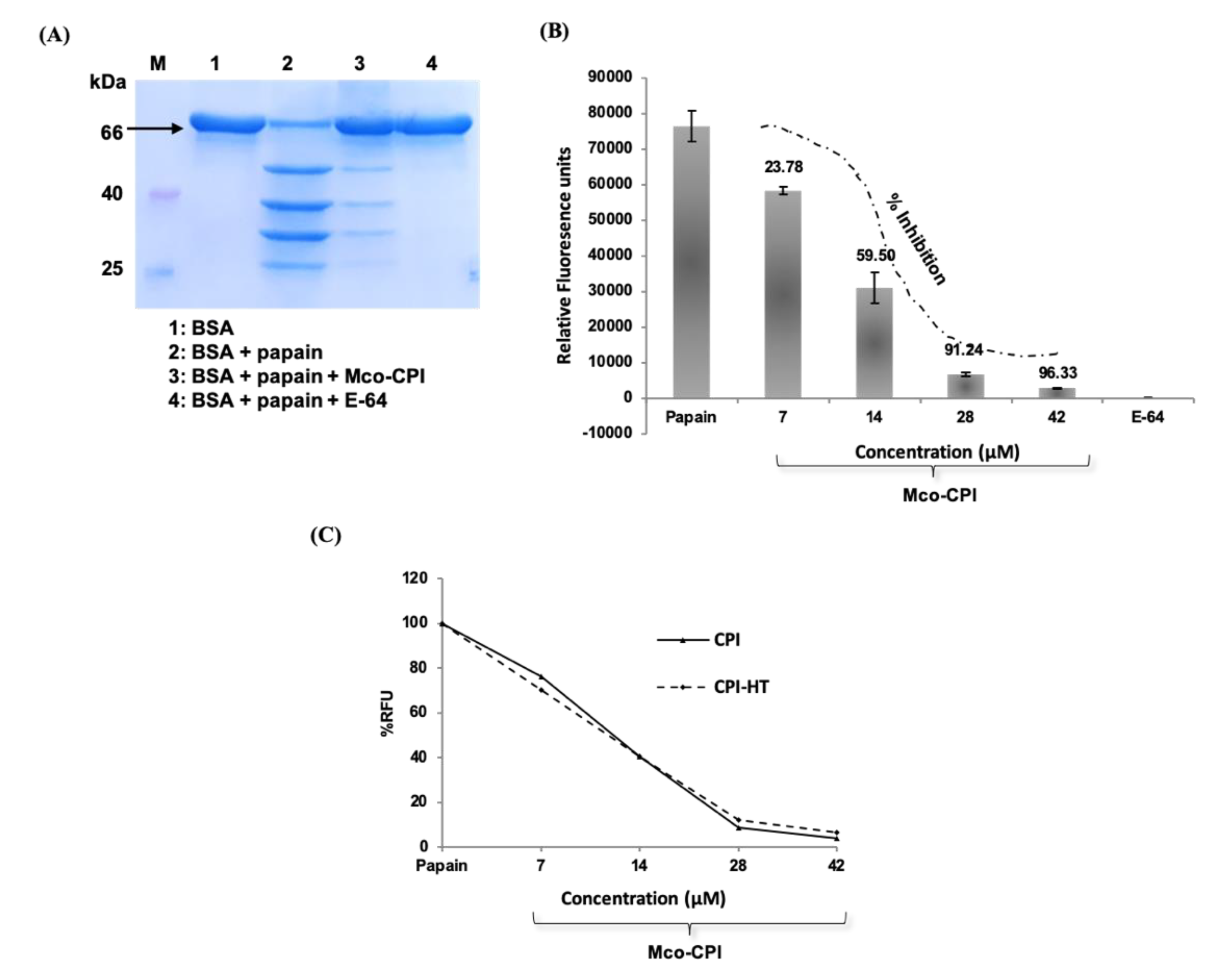
2.2. Recombinant Production and Biochemical Characterization of Mco-CPI
2.3. Inhibitory Activity and Kinetic Analysis of Mco-CPI
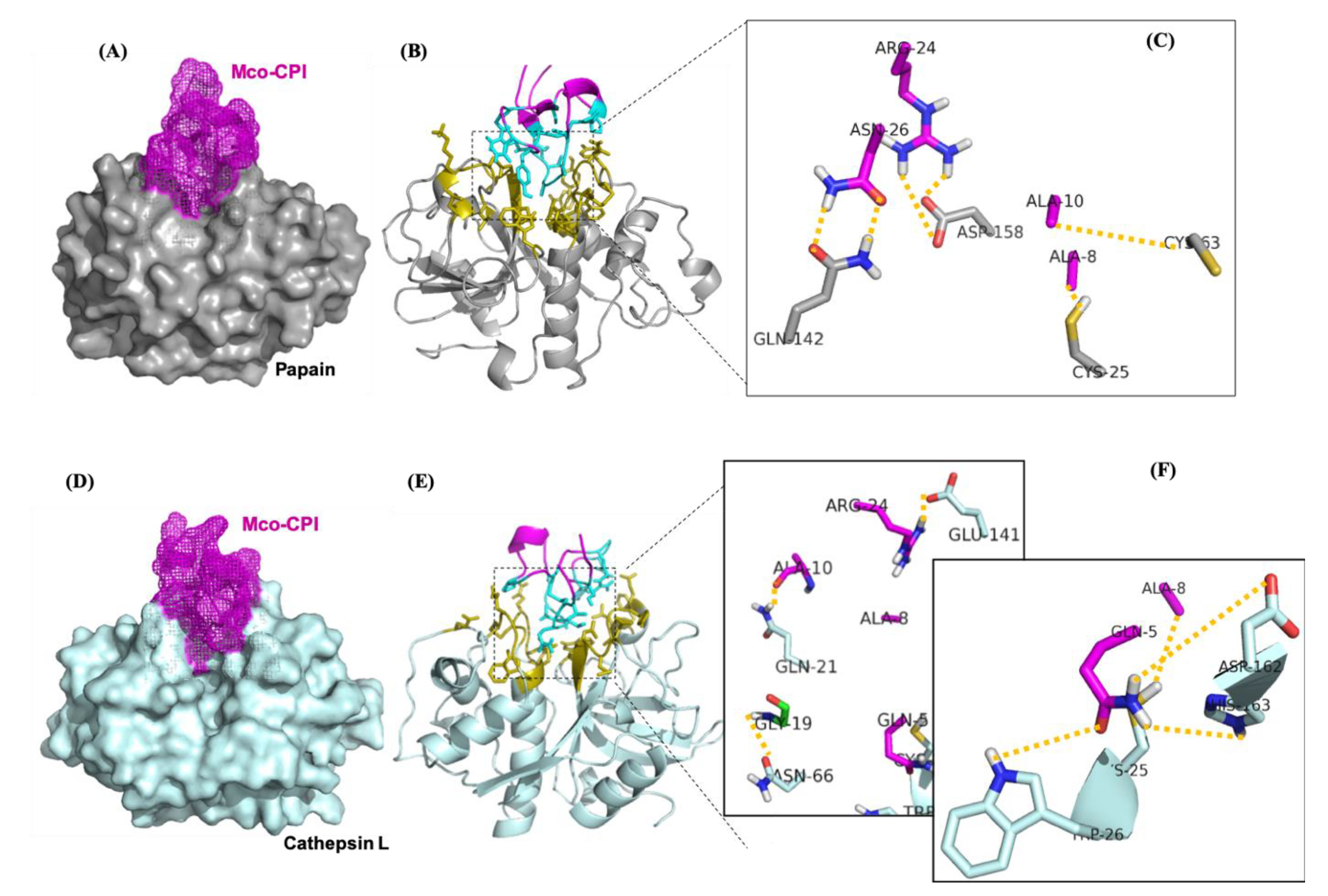
2.4. Mco-CPI Interaction with Model C1A Cysteine Proteases: Papain and Cathepsin L
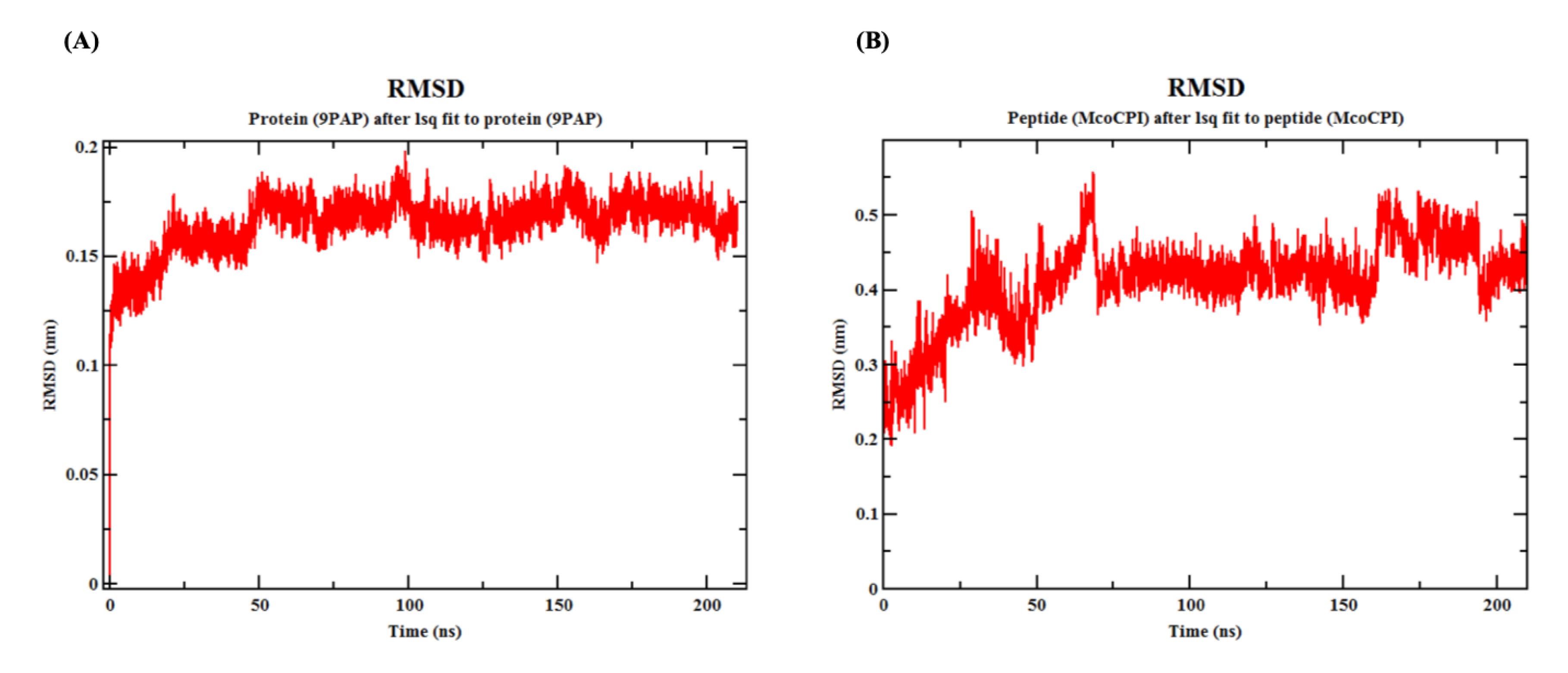
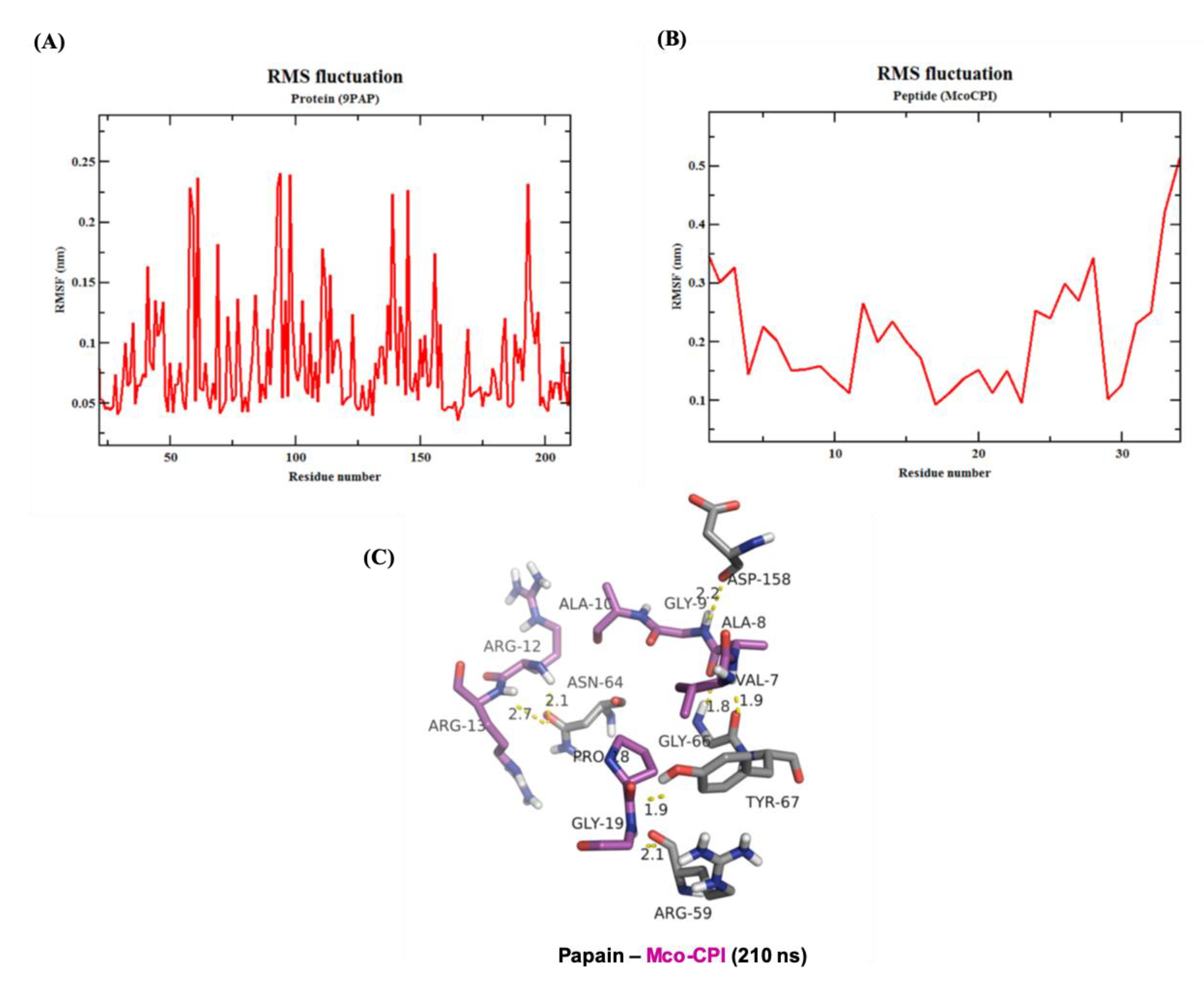
2.5. Molecular Dynamics Simulation of Mco-CPI–Papain Complex
3. Discussion
4. Materials and Methods
4.1. Production of Gene Construct
4.2. Recombinant Expression and Purification of Inhibitor
4.3. Biochemical Characterization of Mco-CPI Protein
4.3.1. HPLC
4.3.2. MALDI-TOF–MS
4.3.3. Free Thiol Content by Ellman’s Assay
4.4. Inhibitory Activity of Mco-CPI
4.4.1. Proteolytic Activity Assay
4.4.2. Inhibitory Activity Assays
4.4.3. Thermostability and Residual Inhibitory Activity
4.5. Molecular Modeling and Docking with Target C1A Cysteine Proteases: Papain, Cathepsin L
4.6. MD Simulations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PLCPs | Papain-like cysteine proteases |
| IHL | Inhibitory hairpin loop |
| CCK | Cyclic cystine knot |
| McoTI-II | Momordica cochinchinensis trypsin inhibitor-II |
| ZFR-AMC | Z-Phe-Arg-7-amino-4-methyl coumarin |
| MALDI-TOF-MS | Matrix-assisted laser desorption–ionization/time of flight–mass spectrometry |
| RP-HPLC | Reverse phase-high performance liquid chromatography |
| SDS-PAGE | Sodium dodecyl sulphate–polyacrylamide gel electrophoresis |
References
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, J.S. Proteases: History, discovery and roles in health and disease. J. Biol. Chem. 2019, 294, 1643–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttle, D.J.; Mort, J.S. Cysteine proteases. In Encyclopedia of Biological Chemistry, 2nd ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 589–592. ISBN 9780123786319. [Google Scholar]
- Grzonka, Z.; Jankowska, E.; Kasprzykowski, F.; Kasprzykowska, R.; Lankiewicz, L.; Wiczk, W.; Wieczerzak, E.; Ciarkowski, J.; Drabik, P.; Janowski, R.; et al. Structural studies of cysteine proteases and their inhibitors. Acta Biochim. Pol. 2001, 48, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. Families of cysteine peptidases. Methods Enzymol. 1994, 244, 461–484. [Google Scholar]
- McGrath, M.E. The lysosomal cysteine proteases. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 181–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Townsend, P.A.; Zamyatnin, A.A., Jr. Cysteine cathepsin protease inhibition: An update on its diagnostic, prognostic and therapeutic potential in cancer. Pharmaceuticals 2019, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Lecaille, F.; Kaleta, J.; Bromme, D. Human and parasitic papain-like cysteine proteases: Their role in physiology and pathology and recent developments in inhibitor design. Chem. Rev. 2002, 102, 4459–4488. [Google Scholar] [CrossRef]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajid, M.; McKerrow, J.H. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002, 120, 1–21. [Google Scholar] [CrossRef]
- Rosenthal, P.J. Falcipains and other cysteine proteases of malaria parasites. Adv. Exp. Med. Biol. 2011, 712, 30–48. [Google Scholar] [PubMed]
- McKerrow, J.H. The diverse roles of cysteine proteases in parasites and their suitability as drug targets. PLOS Negl. Trop. Dis. 2018, 12, e0005639. [Google Scholar] [CrossRef]
- Siqueira-Neto, J.L.; Debnath, A.; McCall, L.I.; Bernatchez, J.A.; Ndao, M.; Reed, S.L.; Rosenthal, P.J. Cysteine proteases in protozoan parasites. PLoS Negl. Trop. Dis. 2018, 12, e0006512. [Google Scholar] [CrossRef]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine proteases: Modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef] [Green Version]
- Mishra, M.; Singh, V.; Singh, S. Structural insights into key Plasmodium proteases as therapeutic drug targets. Front. Microbiol. 2019, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Craik, D.J.; Čemažar, M.; Wang, C.K.L.; Daly, N.L. The cyclotide family of circular miniproteins: Nature’s combinatorial peptide template. Biopolymers 2006, 84, 250–266. [Google Scholar] [CrossRef]
- Getz, J.A.; Rice, J.J.; Daugherty, P.S. Protease-resistant peptide ligands from a knotting scaffold library. ACS Chem. Biol. 2011, 6, 837–844. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Qiu, Y.; Taichi, M.; Wei, N.; Yang, H.; Luo, K.Q.; Tam, J.P. An Orally Active Bradykinin B1 Receptor Antagonist Engineered as a Bifunctional Chimera of Sunflower Trypsin Inhibitor. J. Med. Chem. 2016, 60, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Du, J. Cyclotides as drug design scaffolds. Curr. Opin. Chem. Biol. 2017, 38, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Hao, J.; Luo, X.; Chen, Z. Engineering varied serine protease inhibitors by converting P1 site of BF9, a weakly active Kunitz-type animal toxin. Int. J. Biol. Macromol. 2018, 120 Pt A, 1190–1197. [Google Scholar] [CrossRef]
- Simeon, R.; Chen, Z. Invitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craik, D.J.; Mylne, J.S.; Daly, N.L. Cyclotides: Macrocyclic peptides with applications in drug design and agriculture. Cell Mol. Life Sci. 2010, 67, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, K.; Camarero, J.A. Cyclotides, a promising molecular scaffold for peptide-based therapeutics. Biopolym. Pept Sci. 2010, 94, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thongyoo, P.; Roque-Rosell, N.; Leatherbarrow, R.J.; Tate, E.W. Chemical and biomimetic total syntheses of natural and engineered MCoTI cyclotides. Org. Biomol. Chem. 2008, 6, 1462–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, K.P.; Daly, N.L.; Brown, D.L.; Stow, J.L.; Craik, D.J. The cyclic cystine knot miniprotein McoTI-II is internalized into cells by macropinocytosis. Int. J. Biochem. Cell Biol. 2007, 39, 2252–2264. [Google Scholar] [CrossRef]
- Cascales, L.; Henriques, S.T.; Kerr, M.C.; Huang, Y.H.; Sweet, M.J.; Daly, N.L.; Craik, D.J. Identification and characterization of a new family of cell-penetrating peptides: Cyclic cell-penetrating peptides. J. Biol. Chem. 2011, 286, 36932–36943. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.T.T.; Rowlands, D.K.; Wong, C.-H.; Lo, T.W.C.; Nguyen, G.K.T.; Li, H.-Y.; Tam, J.P. Orally active peptidic bradykinin B-1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. Engl. 2012, 51, 5620–5624. [Google Scholar] [CrossRef]
- Gao, Y.; Cui, T.; Lam, Y. Synthesis and disulfide bond connectivity- activity studies of a kalata B1-inspired cyclopeptide against dengue NS2B-NS3 protease. Bioorg. Med. Chem. 2010, 18, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.E.; Camarero, J.A. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr. Mol. Pharmacol. 2010, 3, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Majumder, S.; Millard, M.; Borra, R.; Bi, T.; Elnagar, A.Y.; Neamati, N.; Shekhtman, A.; Camarero, J.A. In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J. Am. Chem. Soc. 2013, 135, 11623–11633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.K.; Gruber, C.W.; Cemazar, M.; Siatskas, C.; Tagore, P.; Payne, N.; Sun, G.; Wang, S.; Bernard, C.C.; Craik, D.J. Molecular grafting onto a stable framework yields novel cyclic peptides for the treatment of multiple sclerosis. ACS Chem. Biol. 2014, 9, 156–163. [Google Scholar] [CrossRef]
- D’Souza, C.; Henriques, S.T.; Wang, C.K.; Cheneval, O.; Chan, L.Y.; Bokil, N.J.; Sweet, M.J.; Craik, D.J. Using the McoTI-II cyclotide scaffold to design a stable cyclic peptide antagonist of SET, a protein overexpressed in human cancer. Biochemistry 2016, 55, 396–405. [Google Scholar] [CrossRef]
- Hernandez, J.-F.; Gagnon, J.; Chiche, L.; Nguyen, T.M.; Andrieu, J.-P.; Heitz, A.; Hong, T.T.; Pham, T.T.C.; Le Nguyen, D. Squash trypsin inhibitors from Momordica cochinchinensis exhibit an atypical macrocyclic structure. Biochemistry 2000, 39, 5722–5730. [Google Scholar] [CrossRef]
- Felizmenio-Quimio, M.E.; Daly, N.L.; Craik, D.J. Circular proteins in plants: Solution structure of a novel macrocyclic trypsin inhibitor from Momordica cochinchinensis. J. Biol. Chem. 2001, 276, 22875–22882. [Google Scholar] [CrossRef] [Green Version]
- Austin, J.; Wang, W.; Puttamadappa, S.; Shekhtman, A.; Camarero, J.A. Biosynthesis and biological screening of a genetically encoded library based on the cyclotide Mcoti-I. Chem. Biochem. 2009, 10, 2663–2670. [Google Scholar] [CrossRef] [Green Version]
- Thongyoo, P.; Bonomelli, C.; Leatherbarrow, R.J. Tate EW. Potent inhibitors of b-tryptase and human leukocyte elastase based on the MCoTI-II scaffold. J. Med. Chem. 2009, 52, 6197–6200. [Google Scholar] [CrossRef]
- Turk, V.; Bode, W. The cystatins: Protein inhibitors of cysteine proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordiš, D.; Turk, V. Phylogenomic analysis of the cystatin superfamily in eukaryotes and prokaryotes. BMC Evol. Biol. 2009, 9, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, W.; Engh, R.; Musil, D.; Thiele, U.; Huber, R.; Karshikov, A.; Brzin, J.; Kos, J.; Turk, V. A 2.0 Å x-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J. 1988, 7, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on TM-score. Nucleic Acid Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. Shuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Factories 2012, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Aitken, A.; Learmonth, M. Estimation of Disulfide Bonds Using Ellman’s Reagent. In The Protein Protocols Handbook; Springer Protocols Handbooks, Walker, J.M., Eds.; Humana Press: Totowa, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Matsumoto, K.; Mizoue, K.; Kitamura, K.; Tse, W.-C.; Huber, C.P.; Ishida, T. Structural basis of inhibition of cysteine proteases by E-64 and its derivatives. Biopolymers 1999, 51, 99–107. [Google Scholar] [CrossRef]
- Serveau, C.; Juliano, L.; Bernard, P.; Moreau, T.; Mayer, R.; Gauthier, F. New substrates of papain, based on the conserved sequence of natural inhibitors of the cystatin family. Biochimie 1994, 76, 153–158. [Google Scholar] [CrossRef]
- Lalmanach, G.; Hoebeke, J.; Moreau, T.; Brillard-Bourdet, M.; Martino, M.F.-D.; Borrás-Cuesta, F.; Gauthier, F. Interaction between cystatin-derived peptides and papain. J. Protein Chem. 1993, 12, 23–31. [Google Scholar] [CrossRef]
- Lalmanach, G.; Serveau, C.; Brillard-Bourdet, M.; Chagas, J.R.; Mayer, R.; Juliano, L.; Gauthier, F. Conserved cystatin segments as models for designing specific substrates and inhibitors of cysteine proteinases. J. Protein Chem. 1995, 14, 645–653. [Google Scholar] [CrossRef]
- Aboye, T.L.; Camarero, J.A. Biological synthesis of circular polypeptides. J. Biol. Chem. 2012, 287, 27026–27032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.K.T.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Durek, T.; Kaas, Q.; Poth, A.G.; Gilding, E.K.; Conlan, B.F.; Saska, I.; Daly, N.L.; Van Der Weerden, N.L.; Craik, D.J.; et al. Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase. Nat. Commun. 2015, 6, 10199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadish, K.; Camarero, J.A. Recombinant expression of cyclotides using split inteins. Methods Mol. Biol. 2017, 1495, 41–55. [Google Scholar]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Pu, Y.; Chen, Y.; Nguyen, T.; Xu, C.-F.; Zang, L.; Sosic, Z.; Carlage, T. Application of a label free and domain specific free thiol method in monoclonal antibody characterization. J. Chromatogr. B 2019, 1114–1115, 93–99. [Google Scholar] [CrossRef]
- Zucker, S.; Buttle, D.J.; Nicklin, M.J.H.; Barrett, A.J. The proteolytic activities of chymopapain, papain, and papaya proteinase III. Biochim. Biophys. Acta 1985, 828, 196–204. [Google Scholar] [CrossRef]
- Copeland, R.A.; Lombardo, D.; Giannaras, J.; Decicco, C.P. Estimating KI values for tight binding inhibitors from dose-response plots. Bioorg. Med. Chem. Lett. 1995, 5, 1947–1952. [Google Scholar] [CrossRef]
- Uciechowska, U.; Schemies, J.; Scharfe, M.; Lawson, M.; Wichapong, K.; Jung, M.; Sippl, W. Binding free energy calculations and biological testing of novel thiobarbiturates as inhibitors of the human NAD+ dependent histone deacetylase Sirt2. Med. Chem. Commun. 2012, 3, 167–173. [Google Scholar] [CrossRef]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the Raptor-X web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acid Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK 2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2015, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangone, A.; Bonvin, A.M.J.J. Contact-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Xue, L.; Rodrigues, J.; Kastritis, P.; Bonvin, A.M.J.J.; Vangone, A. PRODIGY: A web-server for predicting the binding affinity in protein-protein complexes. Bioinformatics 2016, 32, 514–3678. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, S.R.B.L.D.G.H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Interface Residues | Interface Area (Å2), (%) | No. of Hydrogen Bonds | No. of Salt Bridges | Free Energy of Binding (ΔG) in Kcal mol−1 | |
|---|---|---|---|---|---|
| Papain | 25 | 558.9, 6 | 6 | 2 | −10.7 |
| Mco-CPI | 15 | 646.5, 25.3 | |||
| Cathepsin L | 25 | 642.2, 6.5 | 7 | 2 | −10.4 |
| Mco-CPI | 17 | 716.2, 28.1 |
| Papain | Dist (Å) H-Bonds | Mco-CPI |
|---|---|---|
| CYS, 25 (HG) | 1.70 | ALA, 8 (O) |
| GLN, 142 (HE22) | 1.87 | ASN, 26 (OD1) |
| CYS, 63 (O) | 3.10 | ALA, 10 (N) |
| ASP, 158 (OD2) | 1.89 | ARG, 24 (HH12) |
| ASP, 158 (OD2) | 1.78 | ARG, 24 (HH22) |
| GLN, 142 (OE1) | 2.02 | ASN, 26 (HD22) |
| Cathepsin L | Dist (Å) H-Bonds | Mco-CPI |
|---|---|---|
| CYS, 25 (HG) | 1.74 | ALA, 8 (O) |
| TRP, 26 (N) | 3.49 | GLN, 5 (OE1) |
| HIS, 163 (ND1) | 3.04 | GLN, 5 (O) |
| GLN, 21 (HE21) | 2.00 | ALA, 10 (O) |
| ASP, 162 (O) | 2.49 | GLN, 5 (HE22) |
| ASN, 66 (OD1) | 3.12 | GLY, 19 (N) |
| GLU, 141 (OE1) | 1.54 | ARG 24, (HH11) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, M.; Singh, V.; Tellis, M.B.; Joshi, R.S.; Singh, S. Repurposing the McoTI-II Rigid Molecular Scaffold in to Inhibitor of ‘Papain Superfamily’ Cysteine Proteases. Pharmaceuticals 2021, 14, 7. https://doi.org/10.3390/ph14010007
Mishra M, Singh V, Tellis MB, Joshi RS, Singh S. Repurposing the McoTI-II Rigid Molecular Scaffold in to Inhibitor of ‘Papain Superfamily’ Cysteine Proteases. Pharmaceuticals. 2021; 14(1):7. https://doi.org/10.3390/ph14010007
Chicago/Turabian StyleMishra, Manasi, Vigyasa Singh, Meenakshi B. Tellis, Rakesh S. Joshi, and Shailja Singh. 2021. "Repurposing the McoTI-II Rigid Molecular Scaffold in to Inhibitor of ‘Papain Superfamily’ Cysteine Proteases" Pharmaceuticals 14, no. 1: 7. https://doi.org/10.3390/ph14010007
APA StyleMishra, M., Singh, V., Tellis, M. B., Joshi, R. S., & Singh, S. (2021). Repurposing the McoTI-II Rigid Molecular Scaffold in to Inhibitor of ‘Papain Superfamily’ Cysteine Proteases. Pharmaceuticals, 14(1), 7. https://doi.org/10.3390/ph14010007