1. Introduction
G protein-coupled receptors (GPCRs) are one of the most widely studied families of membrane proteins in the human genome due to their extensive involvement in a plethora of cell signaling pathways [
1]. These seven-transmembrane eukaryotic receptors contribute to the normal function of a cell but can also be responsible for a widespread variety of disease states. While approximately 40% of all drugs target these receptors [
2], this must be contrasted with a relatively low coverage of the GPCR family overall (around 19%), leaving many conditions with little to no prophylactic options [
3]. Advances in drug design and discovery are therefore needed to expand this coverage; further understanding of the structure–function relationship of GPCRs will aid with the development of orthosteric and allosteric ligands against these receptors, widening the pool of potentially druggable GPCRs [
4]. This paper therefore presents a novel application of a mathematical technique, geometric morphometrics with principal component analysis, to further enhance the field’s knowledge and understanding of GPCR structures.
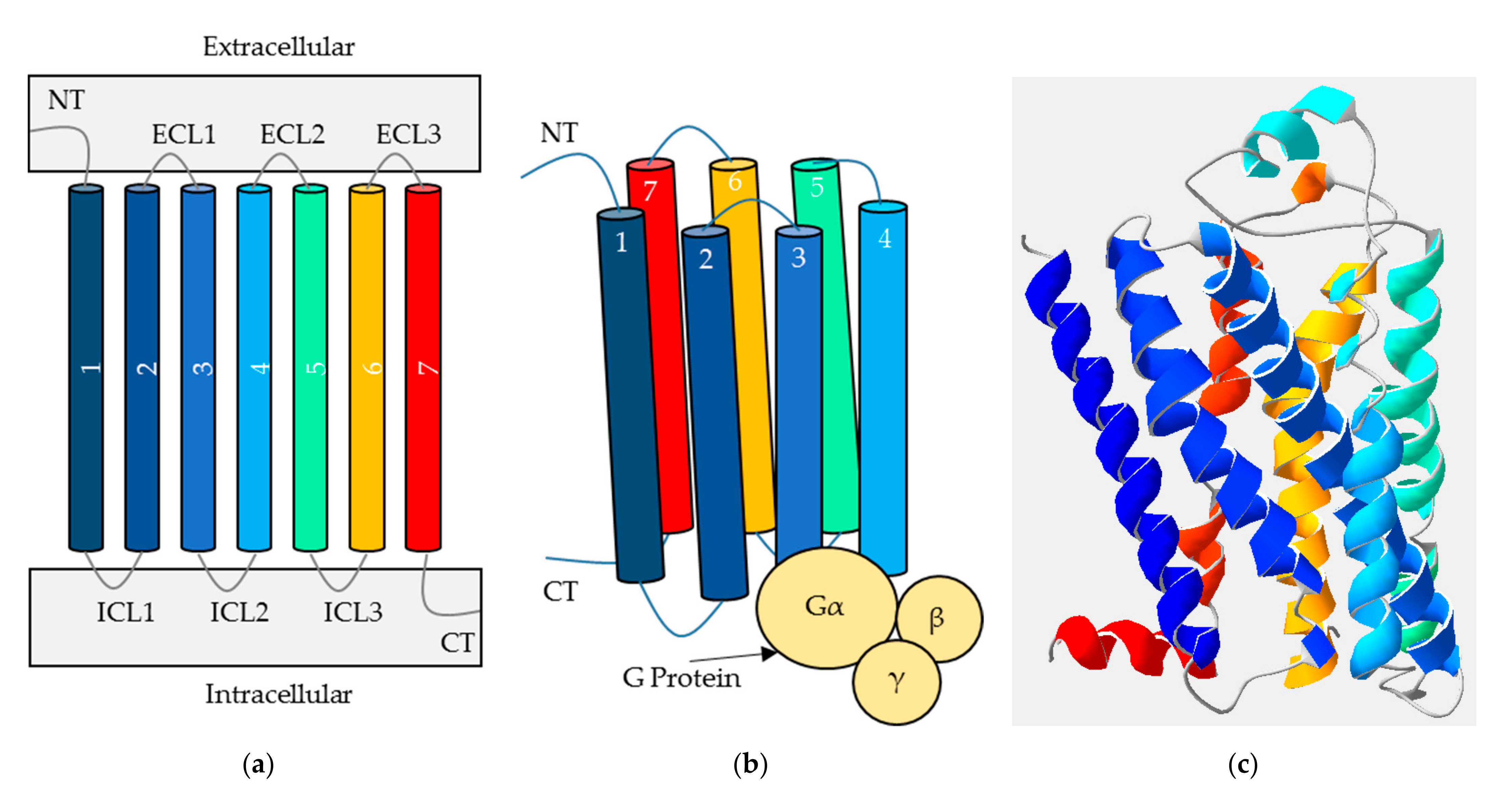
GPCRs are typically composed of three domains as shown in
Figure 1; the N terminus which is extracellular, the transmembrane (TM) domain and the C terminus which is intracellular. These are connected by three extra- and three intracellular loops [
5]. While the TM domain always contains seven alpha-helices and the C terminus is relatively short, the N terminus varies in size depending on the classification of the receptor. As the N terminus can form a part of the location of orthosteric ligand binding, the ligands also vary in size and characteristic, ranging between ions and proteins [
6]. Referring to the original system of classification, there are six main GPCR families (named A–F), as summarised in
Table 1. Additionally, the alternative GRAFS system describes the main five vertebrate GPCR families: (G)lutamate, (R)hodopsin, (A)dhesion, (F)rizzled/Taste2 and (S)ecretin [
7]. This paper will continue to use the classical system, alongside naming the appropriate receptor sub-groups (for example, the neurotensin receptors within family A).
Following binding of the extracellular ligand, the receptor undergoes a characteristic conformational change, promoting a signalling cascade within the cell [
9]. This movement is proposed to include a series of molecular switching and repacking of key contacts within the intra- and interhelical arrangement, followed by the outward movement of TMs 5 and 6 away from the TM bundle [
10]. TM6 is then thought to undergo a mechanical rotation at the intracellular face, allowing exposure of a binding cleft which enables recruitment of the G protein, and subsequent propagation of the signalling cascade. Movement is therefore thought to be focussed around the intracellular end of TM6 and more recently, TM7 has also been observed to move towards TM3 [
11]; overall, the molecular activation of GPCRs remains complicated to elucidate exactly. As shown in
Figure 1b, heterotrimeric G proteins are composed of three subunits (α, β and γ); the α subunit facilitates an exchange of GDP for GTP, while the βγ complex splits away to signal independently from Gα [
9]. As humans encode 18, 5 and 12 different α, β and γ subunits respectively, these combine into a variety of stimulatory or inhibitory effects on the subsequent signalling pathways. These intracellular signals often result in changes to protein activity or transcription factors, affecting cell behaviour (second messenger signaling, for example) depending on the combination of ligand and receptor in question [
9]. Nevertheless, the exact molecular determinants underlying GPCR-G protein coupling also remain to be fully elucidated.
Overall, the relationship between structure and function of GPCRs is of utmost importance as the tertiary structure of the receptor contributes to determining the orthosteric and allosteric ligand binding domains, the efficiency of the conformational change and the receptors’ interaction with other proteins and lipids as well [
12]. It would therefore seem logical to conclude that intended or unintended modifications of a GPCRs wildtype sequence/structure may impact on its efficacy as a functional membrane protein. Despite this, researchers frequently perform such modifications including thermostabilising mutations and chimeric fusions because of the need for enhanced stability during expression and purification, for example [
13,
14,
15,
16].
We therefore hypothesised that resolved structures would not only show differences between conformational states, but that any modifications may also show a difference when compared to the unmodified structure. Any significant differences may therefore correlate with constraining the receptor to a certain conformation, of which the researcher should be aware. However, equally, no differences observed may indicate that the particular modification is ‘safe’ to use, and the GPCR structure is not significantly impacted upon. To study these potential differences between resolved GPCR structures, the design rationale highlighted the importance of an objective and robust system of collecting data, with unbiased and mathematical analyses.
Geometric morphometrics was selected for this task as it can quantify variation between shapes and fulfils the criteria of the design rationale; when the data is transformed, a morphospace is produced where objects are positioned based upon differences or similarities in shape [
17,
18,
19]. Geometric morphometrics can therefore quantifiably capture and display these shape variations between resolved GPCR structures, in a three-dimensional format. The data required for this method are Cartesian landmark coordinates and should be common reference points between the shapes [
18]. As such, the seven transmembrane helices of GPCRs can be used as they are the defining structural characteristic of these receptors and remain relatively conserved between receptors. In order to assess any shape variation at the extra- and intracellular face of the receptor, both ends of the TM helices were selected as reference points; each receptor, by definition, contains these landmarks. More specifically, the XYZ coordinates of the alpha-carbon atom (Cα) of the amino acid residue at each end of the helix (meaning the first and last residue of each TM helix) were selected as the Cartesian landmarks to minimize variation due to amino acid identity at these positions.
A comprehensive explanation of geometric morphometrics can be found elsewhere [
17,
18,
20]; however, the main elements are as follows. The landmark coordinates are first size- and rotation-adjusted by a Procrustese superimposition; this is an orthogonal transformation to standardise and scale the data for comparison [
21]. Subsequently, a covariance matrix is generated from which principal component analysis is then performed. Principal components are a series of vectors which show variation in the data and are composed of the eigenvectors of the data’s covariance matrix. An eigenvector is a direction with variation, and its eigenvalue is the variance of that direction [
22]. As such, the principal components of the data produce a morphospace where the first principal component is responsible for the greatest variation between the coordinate data [
23]. As principal component analysis uses covariance, its analysis is two-dimensional and therefore principal component scores can be compared; a comparison between principle components 1 and 2 (PC1 and PC2) will show the greatest variation in the morphospace. Overall, geometric morphometrics with principal component analysis breaks down three-dimensional landmark coordinates to find patterns in the data, because of their shape variations, and can be further analysed statistically by analysis of similarity (ANOSIM) and permutational multivariate analysis of variance (PERMANOVA) [
17,
23].
We therefore present an application of geometric morphometric analysis to resolved GPCR structures which is, to our knowledge, the first demonstration of its suitability for membrane protein analysis. Overall, we have been able to classify, discriminate and mathematically determine significant differences between GPCR structures based on their characteristics such as activation state, bound ligands and fusion proteins. While this is a method more often used in disciplines such as anthropology and palaeontology, we believe that this technique has wide applicability to the structural biology field. By increasing understanding of GPCR structures resolved under varied conditions, it is hoped that intelligent, structure-based ligand design will continue to improve and expand the current range of druggable receptors.
3. Discussion
GPCRs are widely studied membrane protein receptors due to their involvement in disease states, and potential as drug targets. Despite their importance, development of novel, specific and effective prophylaxis can often be hindered by a lack of structural knowledge and understanding of these dynamic receptors. Thus, the application of innovative techniques to this field will undoubtedly further enhance understanding of GPCR structures and modifications, to ultimately improve their druggability. We hypothesised that differences between GPCR structures, under varying conditions, could be detected, quantified and analysed statistically using geometric morphometrics. This mathematical technique was therefore used with principal component analysis to assess receptor shape variation in a three-dimensional, unbiased manner. The results obtained demonstrated that this technique was successfully applied to GPCR structures and gave insight into the modifications researchers frequently perform with them.
As geometric morphometrics is mainly utilised in disciplines such as anthropology and palaeontology, it was first necessary to understand if it could be applied to protein structures as well. One unpublished example exists on the bioRxiv database which examines the soluble α-amylase enzymes with geometric morphometrics [
32]. However, after an initial pilot study and further expansion of our methods, we believe this is the first application of this technique to GPCRs, and membrane proteins in general, especially on this scale.
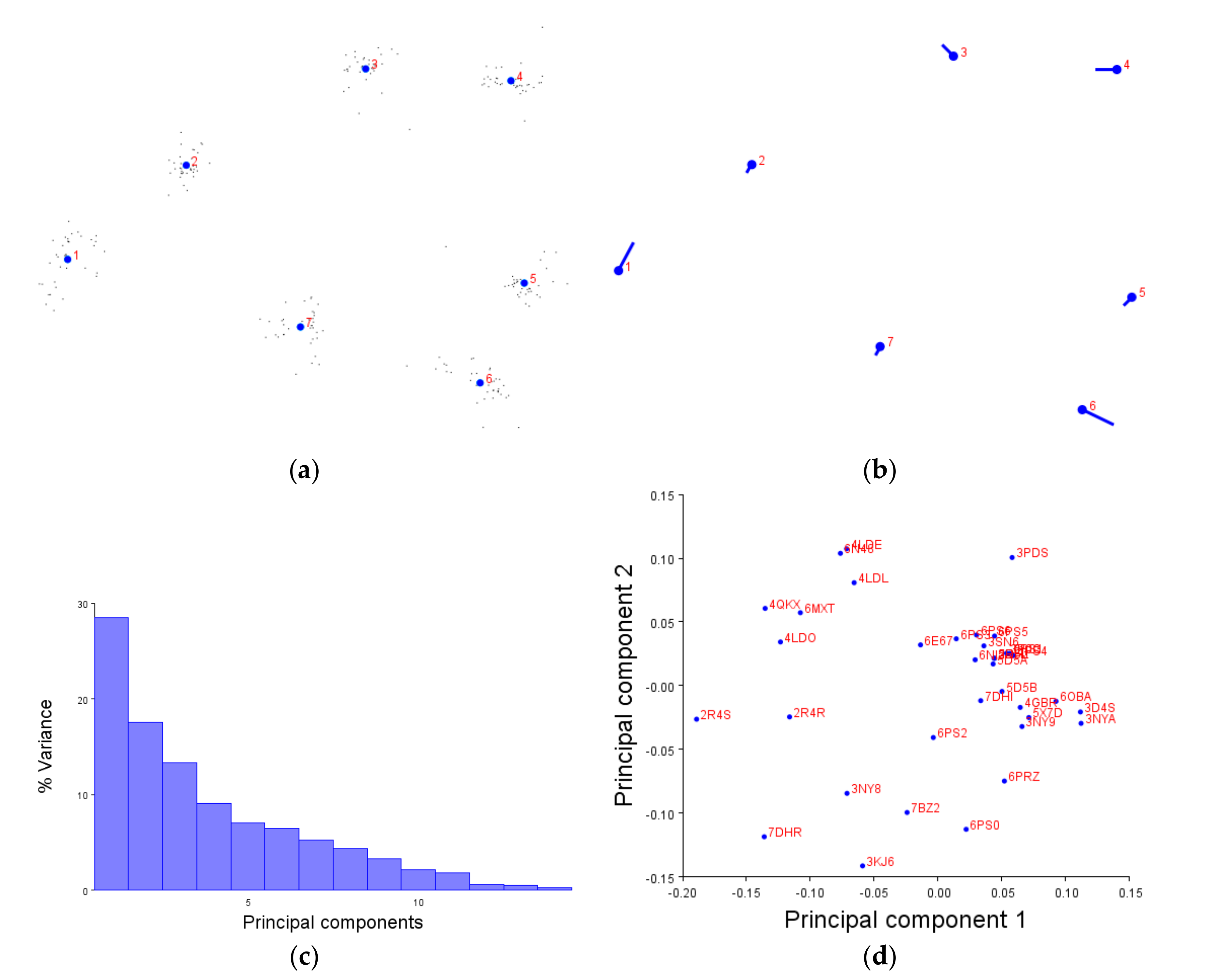
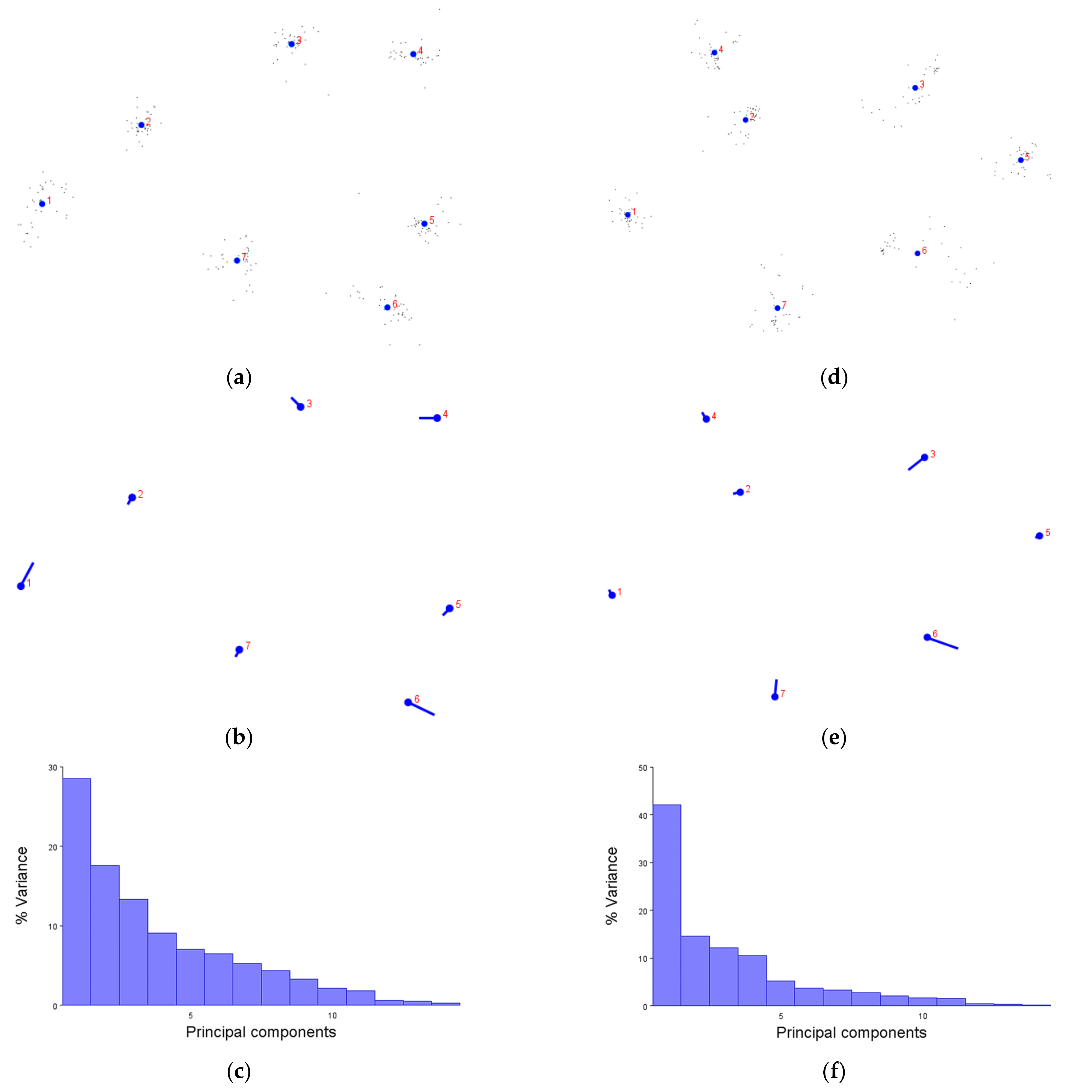
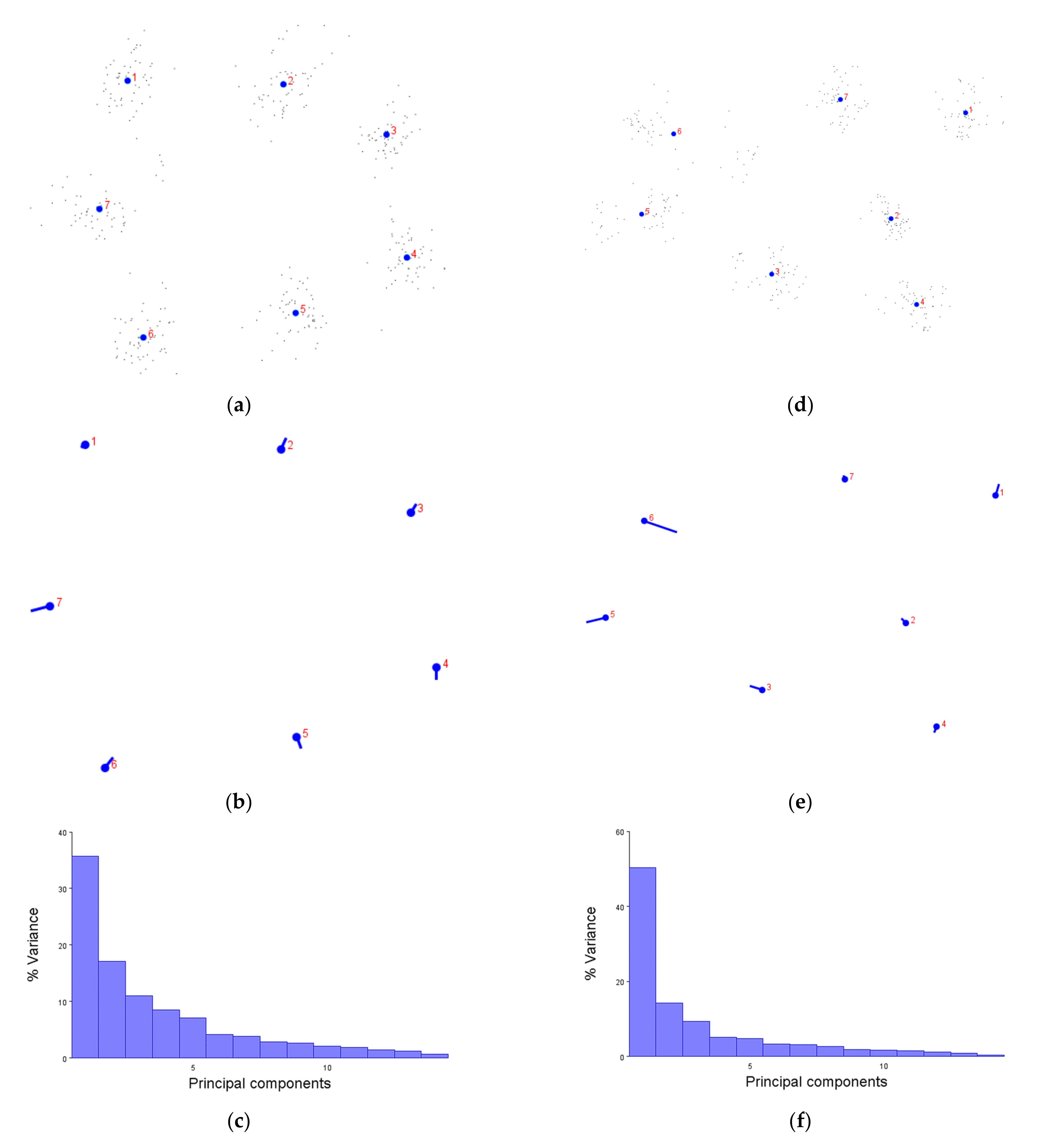
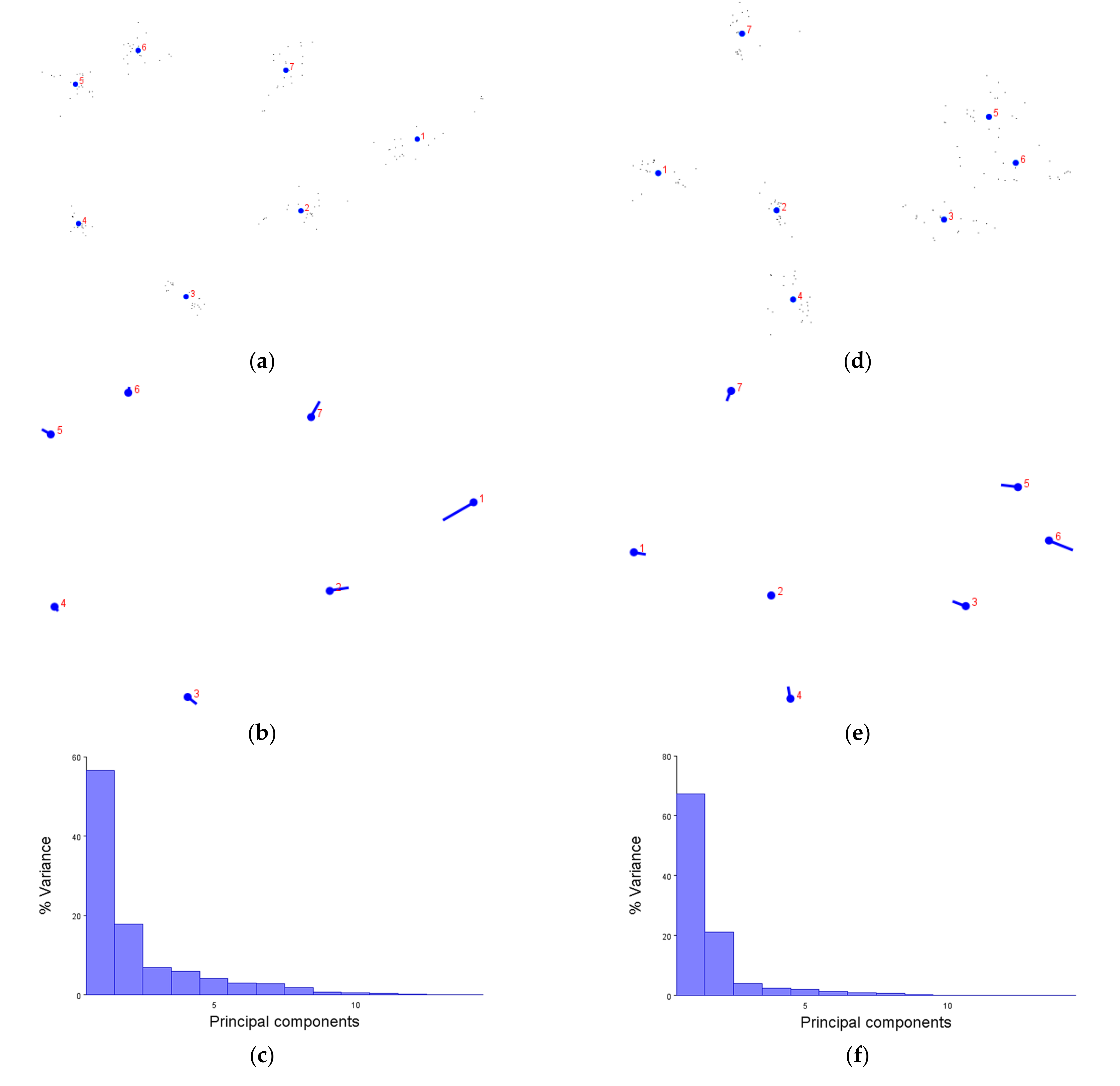
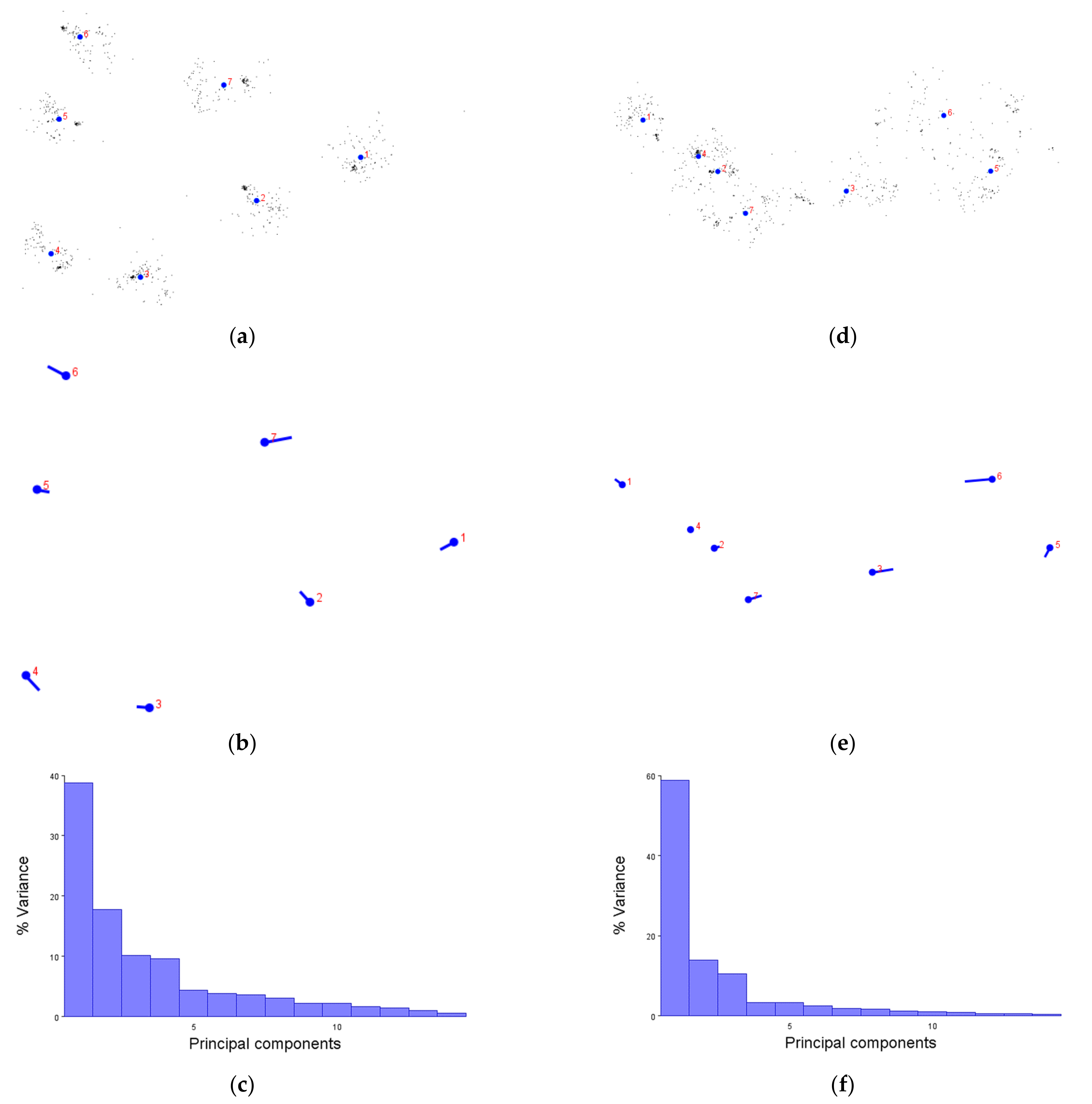
We first demonstrated that the XYZ coordinates of amino acids are not only compatible with the MorphoJ software, but produce figures which make sense. The generalised Procrustese transformations presented (e.g.,
Figure 2a) show the average helical positions which, when compared back to GPCR structures, accurately represent a view of the transmembrane helix bundle at both the extra- and intracellular faces. This meant that the coordinate data was transformed into a standardised and reliable dataset from which the subsequent analyses could be performed with confidence. Similarly, the overall direction and magnitude of the dataset’s variation was able to be portrayed by the lollipop graphs (e.g.,
Figure 2b), indicating which helices vary the most under different conditions. Finally, the principal component score comparisons (e.g.,
Figure 2d) provided an unbiased platform to then infer meaning by grouping receptors together and statistically analysing the dissimilarity between those groups.
Initially, receptor activation was selected as the characteristic to provide proof of concept as it is the main source of natural variation between structures. It is a movement native to all wild-type GPCRs, and is reasonably well understood [
33]. Based on the generally accepted model of activation, it was hypothesised that GPCRs resolved in different activation states should vary from one another. Helical movement would therefore be expected, with a focus on the intracellular face of the transmembrane bundle. Reassuringly, these predictions were proved to be correct as shown in
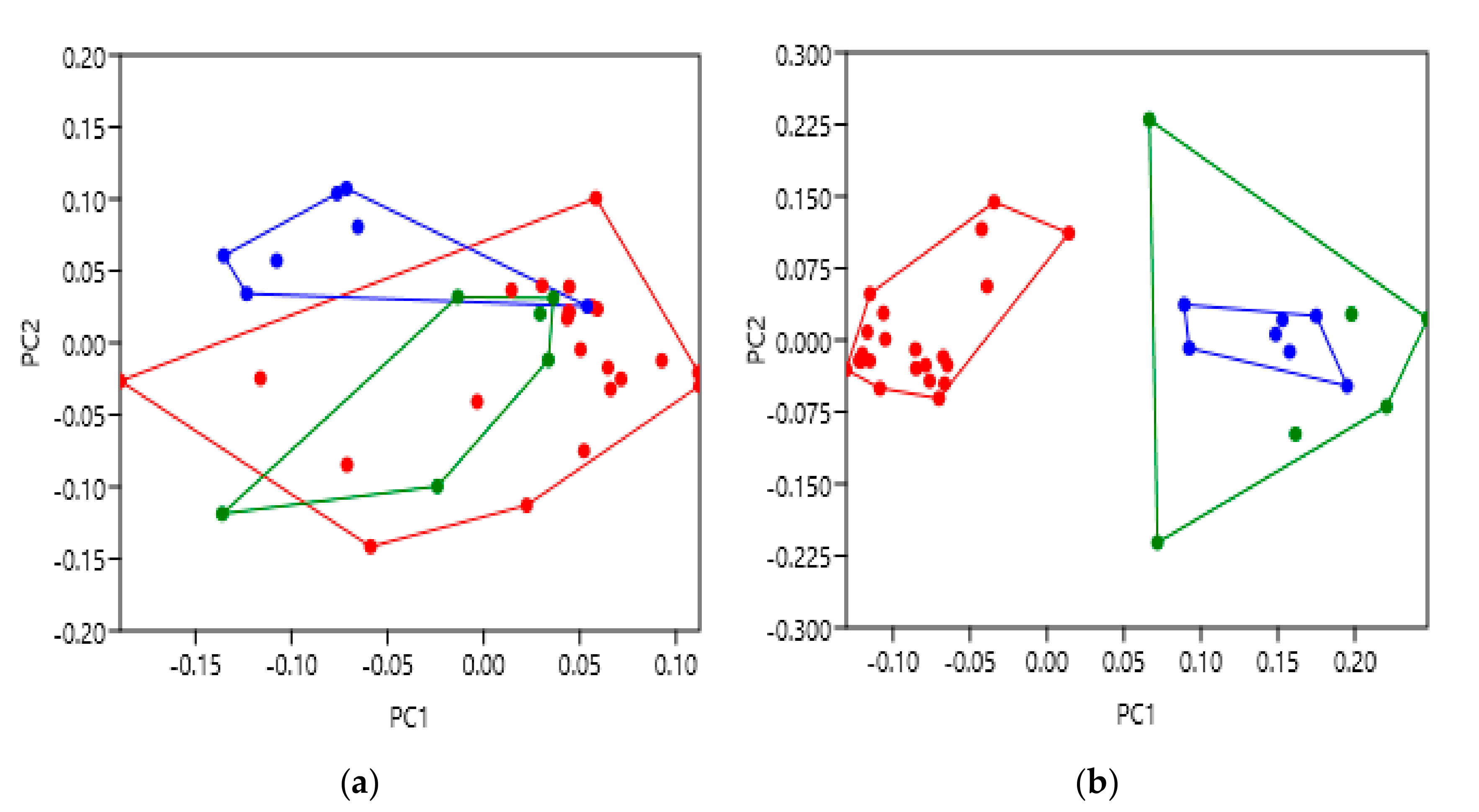
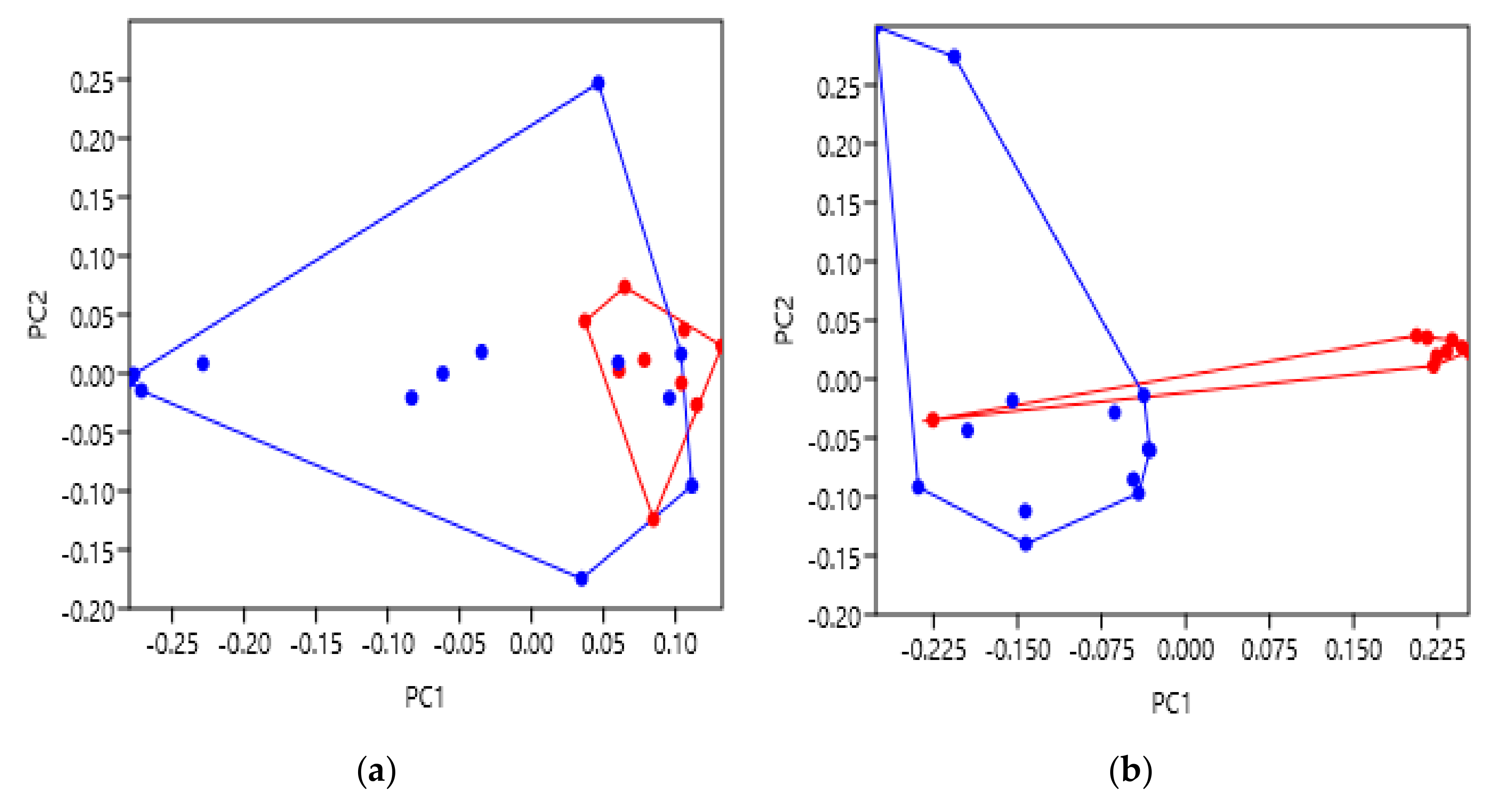
Appendix B Figure A1b,e. TM6 was highlighted as the main location of variation between activation states, moving away from the TM bundle as expected. Subsequently, comparison of the intracellular principal component scores (
Figure 3b) showed very clear separation between activation states. The group of inactive β2AR receptors were observed to significantly differ from the active or active with G
s groups, and thus further confirm our hypothetical predictions. These results are therefore likely indicative of the conformational movement to allow post-activation binding of the intracellular G protein and propagation of the appropriate signalling pathway, as predicted. Additionally, the significant separation of the active and active coupled to G
s groups at the extracellular face should also be noted (
Figure 3a). This unexpected result suggests that the binding of a stimulatory G protein possibly causes bias to the extracellular face of the β2AR TM bundle to adopt a different conformation than those with only agonist bound to them. Interestingly, this was not observed at the intracellular face despite being the location of G protein binding, providing a thought-provoking avenue for further research. Indeed, it has been observed that binding of a G protein to a GPCR can increase the affinity for ligands, which may be as a result of changes to the extracellular face [
34]. Nevertheless, these convincing results provide strong evidence to support the use of geometric morphometrics with GPCRs and that it can be further applied to highlight structural variation under additional conditions.
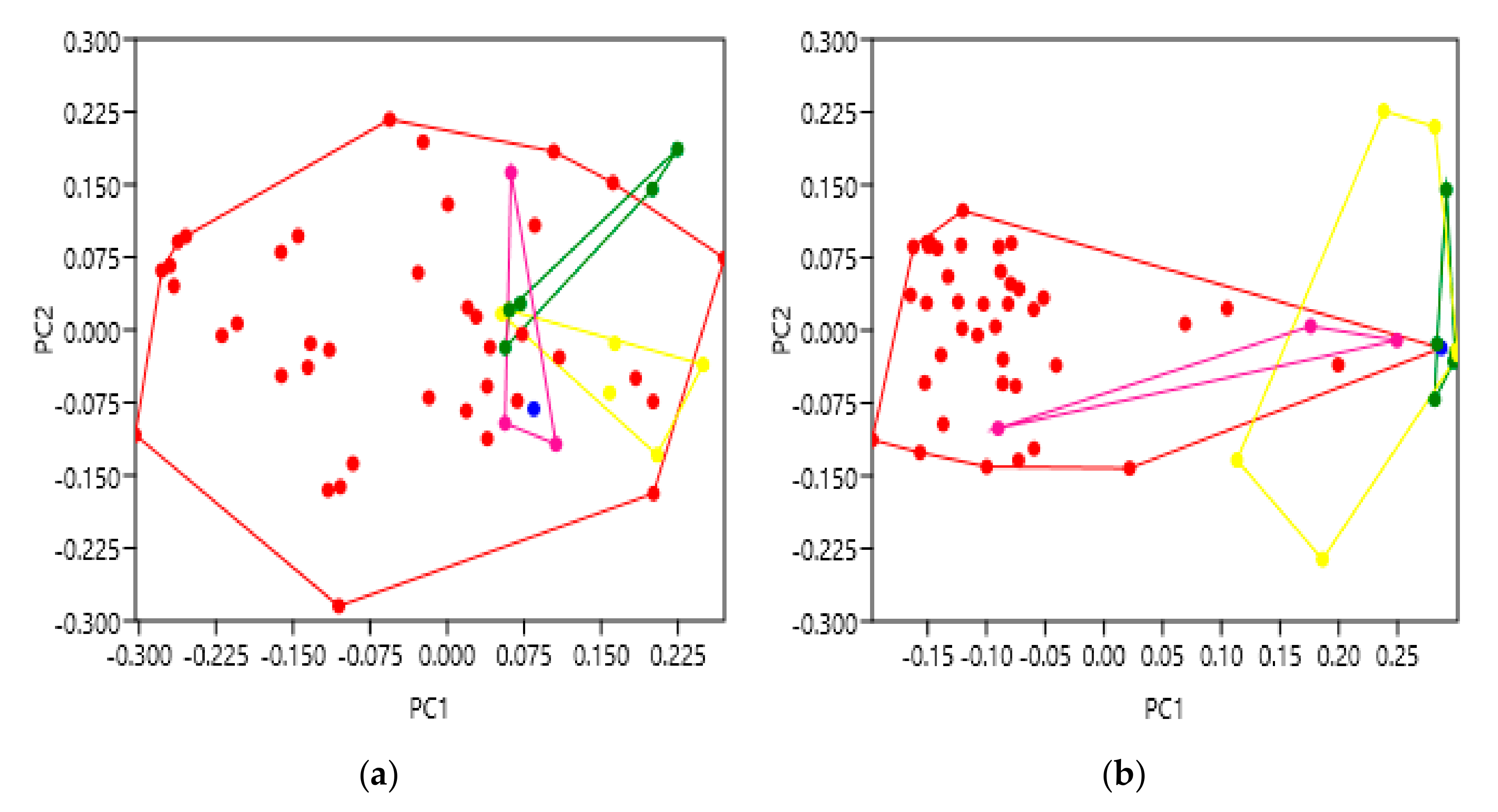
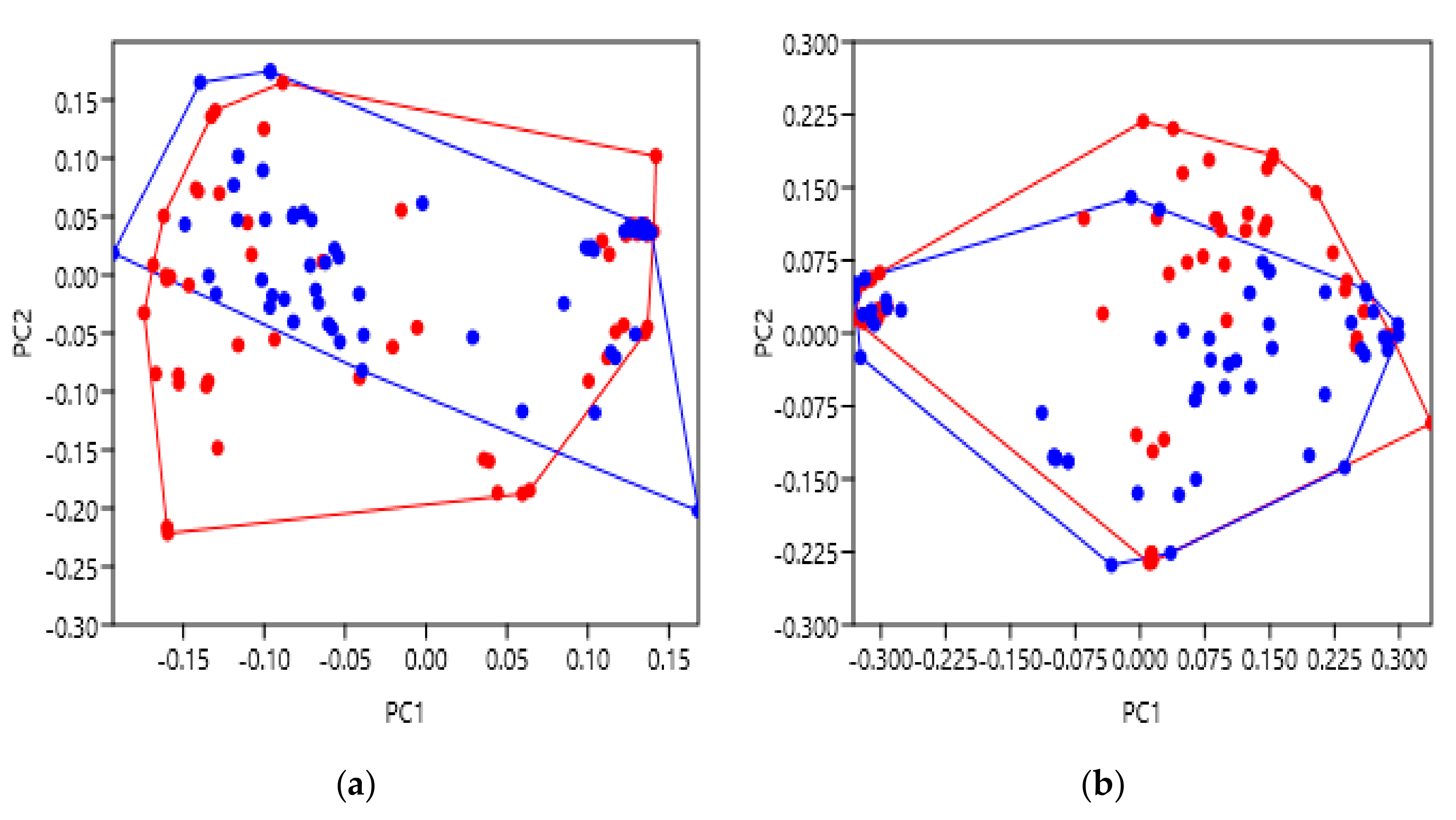
As receptor activation largely relies upon ligand binding, this was the next characteristic studied. This is especially interesting as GPCRs can interact with a variety of different ligand types, and with bias towards certain ligands or signalling pathways. One explanation may involve a specific helical movement which opens an intracellular binding cleft biased towards certain G proteins, either stimulatory or inhibitory, etc. We therefore hypothesised that GPCR structures with different ligand classes bound to them may vary from each other, again, possibly focussed at the intracellular face. The extracellular data of the family B receptors showed a general similarity with no significantly different groupings (
Figure 4a). This possibly suggests that as the N terminus of their peptide hormone ligands bind to a residue close to the TM bundle and ECL2, the extracellular face of the TM bundle arrangement remains more similar across the family B receptors, perhaps due to their shared ligand affinity [
35]. However, the intracellular face exhibited significant variation (
Figure 4b), with agonist bound structures shown to be most dissimilar to the structures resolved with antagonist or NAM bound to them, and unbound structures falling somewhere between the two. One possible explanation for this may include a greater involvement of the intracellular helix arrangement in the bias towards certain G protein pathways, as a result of ligand binding. It is also interesting to observe that the structural variation of the partial agonist-bound receptor (blue) was more similar to antagonist-bound rather than agonist-bound. Looking in more depth at this PDB file (5YQZ), it is nonetheless described as inactive which could explain this observation [
36]. Furthermore, several of these family B receptors can require co-expression with an accessory protein called a receptor activity modifying protein (RAMP) [
27]. Biologically, these RAMPs can influence trafficking to the cell membrane, glycosylation and receptor pharmacology. Crystallisation without a RAMP, in some cases, could therefore hinder a family B GPCR’s full dynamic range, and should be considered when studying their resolved structures. Nonetheless, these results demonstrate the ability of geometric morphometrics to significantly discriminate structures based upon their bound ligands, with a meaningful gradient of activation across the principal component score axis.
While the analysis of activity and bound ligands supported our hypotheses based on existing GPCR understanding, we also focussed our attention to modifications frequently performed during the experimentation of proteins. These included the insertion of an intracellular fusion protein and the introduction of receptor thermostabilising mutations, highlighting some important results to share.
Firstly, insertion of a glycogen synthase fusion protein into the ICL3 of the orexin receptors caused significant variation at the intracellular face (
Figure 5b). Despite seemingly having no effect on their pharmacology, the presence of the fusion protein appears to largely bias these receptors to a significantly different intracellular conformation. This is important for the field to be aware of during experimental design as intracellular fusion proteins could sterically hinder the receptor’s full range of motion or bias structures to more stable conformations that do not necessarily fully represent the in vivo state of the protein. One anomalous result was found to have the opposite effect where the insertion of a fusion protein caused no difference from those without the fusion. It is interesting that this structure (6V9S) was bound with a novel antagonist (JH112) made by enantiospecific synthesis [
37], whereas the other structures had their typical non-selective antagonist Suvorexant bound to them. JH112 is a sub-nanomolar antagonist to activate G
q mediated recruitment of β-arrestin to the orexin 1 receptors [
37], and further study is likely required to confirm whether JH112 can bias the orexin receptor to a different intracellular conformation than the other antagonist-bound structures. In any case, we believe this is the first demonstration of using geometric morphometrics to indicate that an intracellular fusion modification can cause significant variation at the intracellular face of GPCRs.
Finally, thermostabilising mutations remain to be a more controversial area of GPCR research as conflicting opinions on its use still persist. Some groups advocate for the advantages thermostability provides experimentally, especially when resolving difficult structures [
38]. However, others promote a degree of caution when using non-native proteins and emphasise the need to ensure proteins are active. Either way, thermostabilising mutations remain a prevalent component of the holistic approach to studying protein structures. Ultimately, our analyses did not detect any significant differences between thermostabilised and non-mutated GPCRs, from a variety of receptor sub-groups (
Figure 6); this perhaps suggests that, on a geometric level, thermostabilising mutations are “safe” to use. However, given that some receptors are no longer able to signal after this modification, it would be pragmatic to comprehensively analyse one’s GPCR of interest, nonetheless.
One downside to this method is its manual nature; the variety of annotated PDB files makes automation difficult to attempt though this may become possible in the future. Despite this, geometric morphometrics is an overwhelmingly advantageous technique. Landmark coordinates are not restricted to the ends of the transmembrane helices, other locations such as the middle of helices could be analysed, and they must simply be common to each structure. Similarly, this technique is not limited to GPCRs and could be applied to other proteins of interest such as ABC transporters and aquaporins [
39,
40]. While interesting results and structural differences were observed with regards to the characteristics presented in this paper, we have also been able to classify receptors based on further factors including organism species, highlighting possible future analyses. With regards to GPCRs, an area requiring further study is the explanation of G protein coupling specificity [
41], especially concerning intermediate states and the role of the intracellular binding cleft. Another exciting possibility involves checking the validity of the recent AlphaFold project predictions—at the time of writing, these predicted structures seem reasonable although limited to inactive states for now [
42,
43]. To conclude, the results presented in this paper provide a proof of concept for the use of geometric morphometrics in the study of GPCR structures, especially when variation may be undetectable by pharmacological assays or other structural techniques such as root-mean-square deviation (RMSD). We have therefore adapted a method to apply this mathematical technique and demonstrate meaningful and statistically significant analyses. It is ultimately intended to be a tool to aid sense-checking newly resolved structures and planning early experimental design, even beyond GPCRs.
4. Materials and Methods
A database of all currently known GPCR structures was first obtained from the
GPCRdb website (
https://gpcrdb.org/structure/ (accessed on 21 August 2021)) and cross-referenced to the
mpstruc database (
https://blanco.biomol.uci.edu/mpstruc/ (accessed on 21 August 2021)) to ensure the working model was based upon a comprehensive list of resolved structures (last sampled 30 June 2021) [
26,
44]. From here, the PDB code of each structure was used to download the .pdb file from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (
https://www.rcsb.org/ (accessed on 21 August 2021)). Next, the Swiss-PdbViewer (DeepView) software version 4.10 (
http://www.expasy.org/spdbv/ (accessed on 21 August 2021)) was used to visualise and manipulate the structures before data collection [
8]. Finally, data processing and statistical analysis was performed with the MorphoJ software (
https://morphometrics.uk/MorphoJ_page.html (accessed on 21 August 2021)) and the PAST data analyser software (
https://past.en.lo4d.com/windows (accessed on 21 August 2021)) [
24,
45]. All of these are freely available.
Each receptor structure was analysed using the same process as follows. Firstly, the amino acid residues located at the very end of each transmembrane helix were identified manually and the name and sequence number recorded. As GPCRs have seven transmembrane helices, this results in fourteen landmarks for each structure: Seven at the extracellular face, and seven at the intracellular face. Next, the .pdb file was opened as text and the x, y and z coordinates located and recorded for each of the fourteen identified residues. Specifically, these were the coordinates for the alpha-carbon atom (Cα) for each residue. These were then divided into two groups (extra- and intracellular) and then exported as .txt files. This manual process was repeated exactly for every structure analysed. We envisage that this process could be automated, although the disparity between presentation and annotation of the .pdb files could well hinder attempts.
A new project was then started in MorphoJ, with the relevant .txt file imported as three-dimensional data (the extracellular data was processed separately to the intracellular). Two preliminary processes were then performed. Firstly, a Procrustese fit was performed with the data aligned by principal axes, and then a covariance matrix was generated. Finally, principal component analysis was performed which gives the principal component shape changes, eigenvalues and scores [
24]. The PC score comparisons were then used to identify groups, clusters and outliers based upon the characteristics of the GPCR structure. Any overlap or differences in data have been highlighted by the addition of coloured convex hulls to the PC comparisons which demarcate the smallest area containing each group.
These PC scores were then tested statistically using PAST [
45]; one-way permutational multivariate analysis of variance (PERMANOVA) tested the distance between the centroids of each group, while one-way analysis of similarity (ANOSIM) tested group dissimilarity. Both of these multivariate tests involved 9999 permutations and pairwise comparisons were performed using Euclidean distances. PERMANOVA gives P and F values, testing for differences between groups by distance; a larger F value indicates a more pronounced group separation. ANOSIM gives P and R values, using the mean rank of distances between and within groups; an R value of 1 indicates complete dissimilarity. Significance for both tests is calculated by 10,000 permutations of group membership [
17].





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









