
The Specificity and Broad Multitarget Properties of Ligands for the Free Fatty Acid Receptors FFA3/GPR41 and FFA2/GPR43 and the Related Hydroxycarboxylic Acid Receptor HCA2/GPR109A

 , ,
, ,  , , and
, , and
Abstract
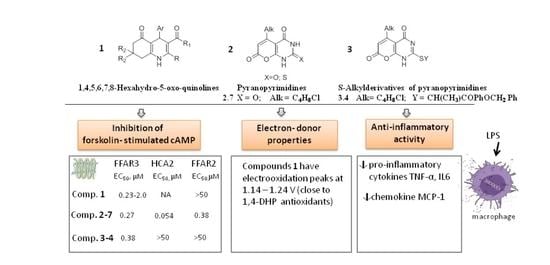
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Compounds—Putative Ligands
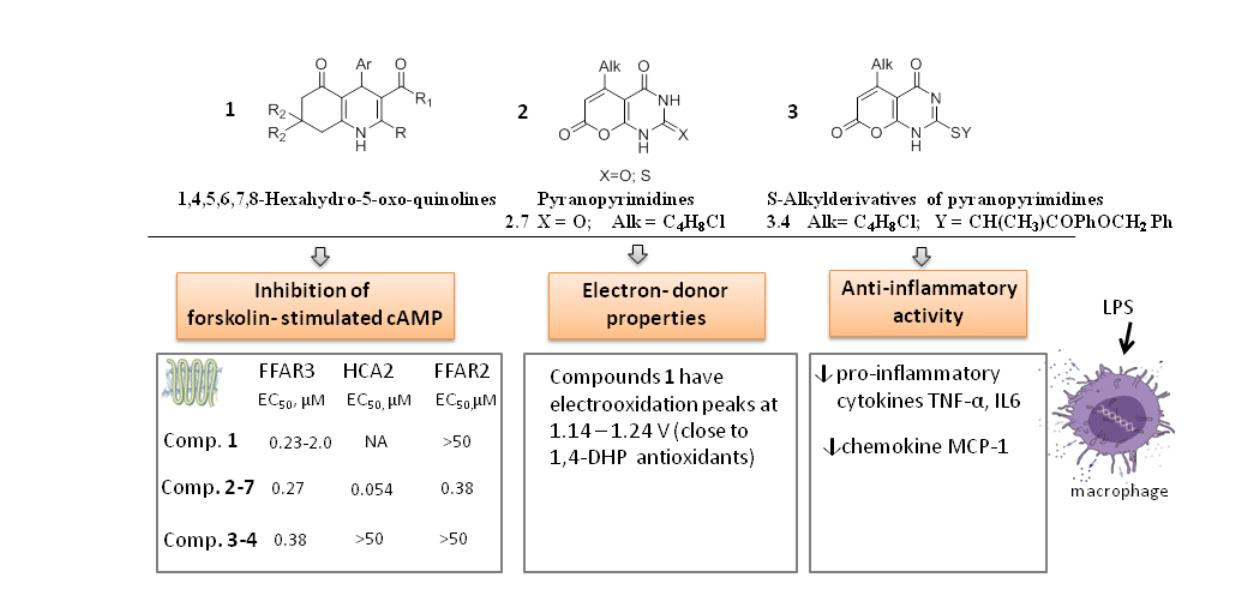
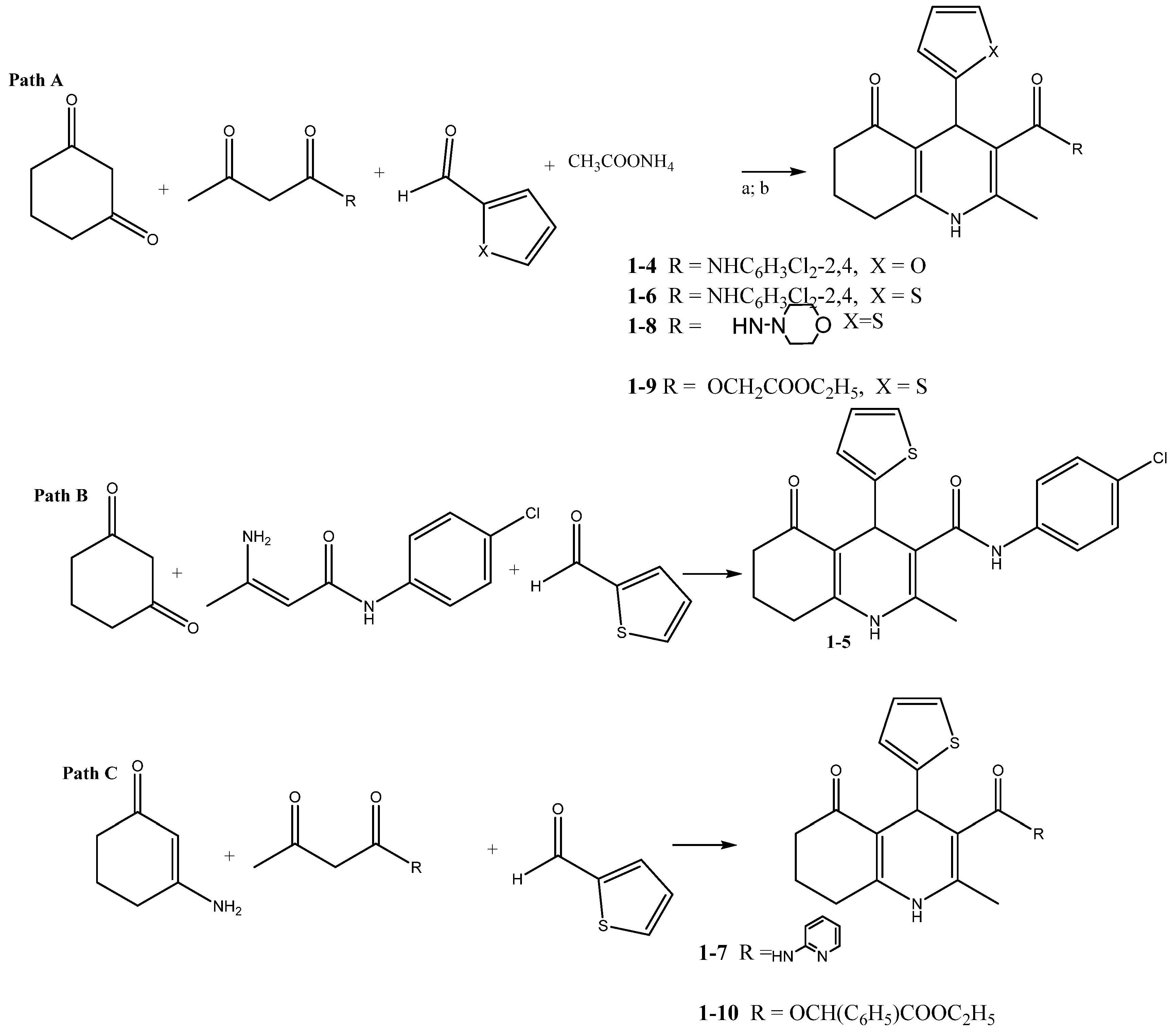
2.1.1. The Synthesis of 2-Methyl-5-oxo-4-(Aryl, Heteryl)-1,4,5,6,7,8-Hexahydroquinoline (HHQ) Derivatives 1-1–1-14
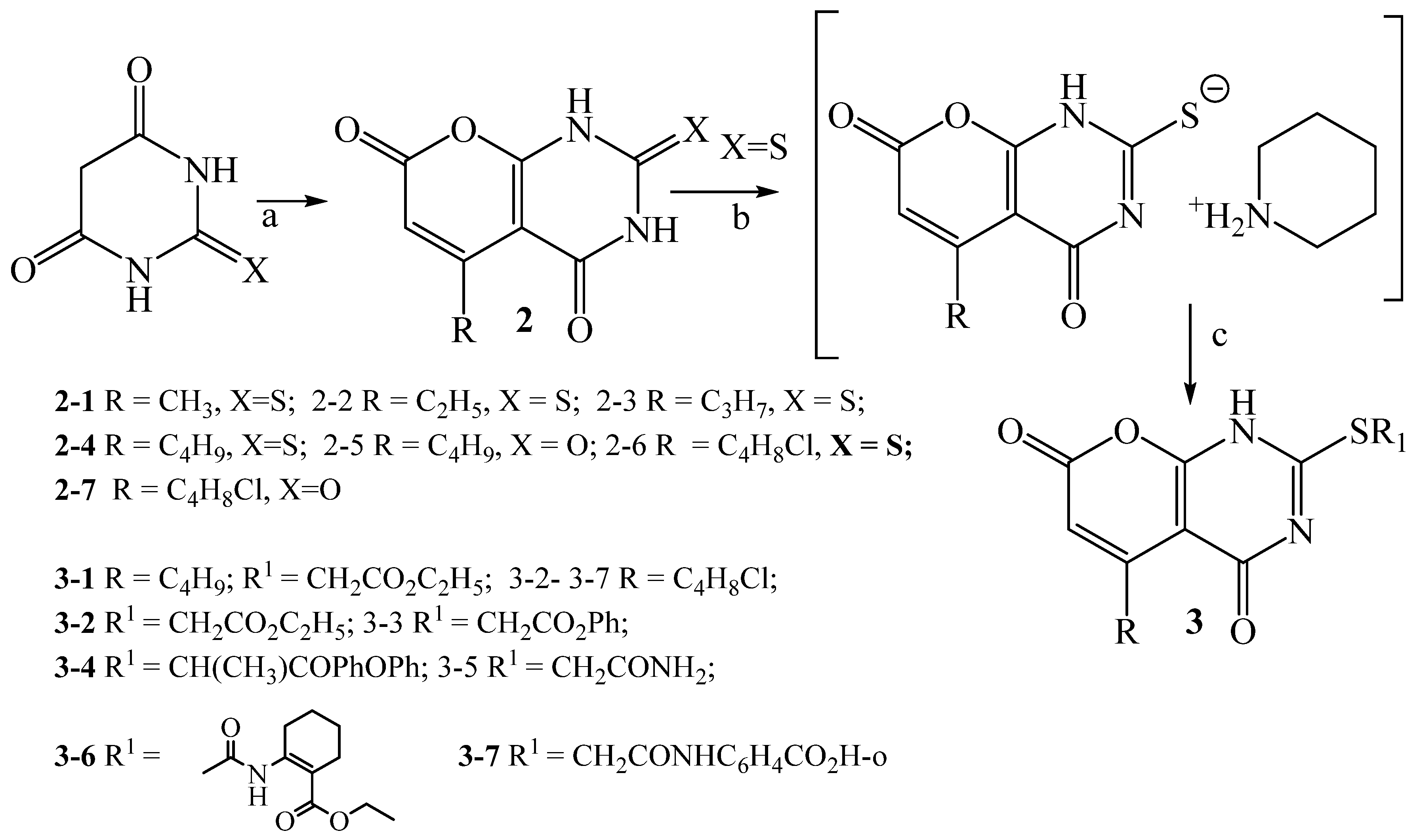
2.1.2. The Synthesis of 5-(4-Chlorobutyl)-1H-Pyrano[2,3-d]Pyrimidine-2,4,7-Triones (Pyranopyrimidinetriones) 2–5 and 2–7, 5-Alkyl-2-Thioxo-2,3-Dihydro-4H-Pyrano[2,3-d]Pyrimidine-4,7(1H)Diones (Thioxopyranopyrimidinediones) 2-3, 2-4, 2-6 and 2(Substituted thio)-5-Alkyl-4H-Pyrano[2,3-d]Pyrimidine(1H)-Diones (2-Alkylthiopyranopyrimidinediones) 3-1–3-7
2.2. The Impact of Oxo-Hexahydroquinolines on HCA2/GPR109A, FFA3/GPR41, and FFA2/GPR43 Receptors
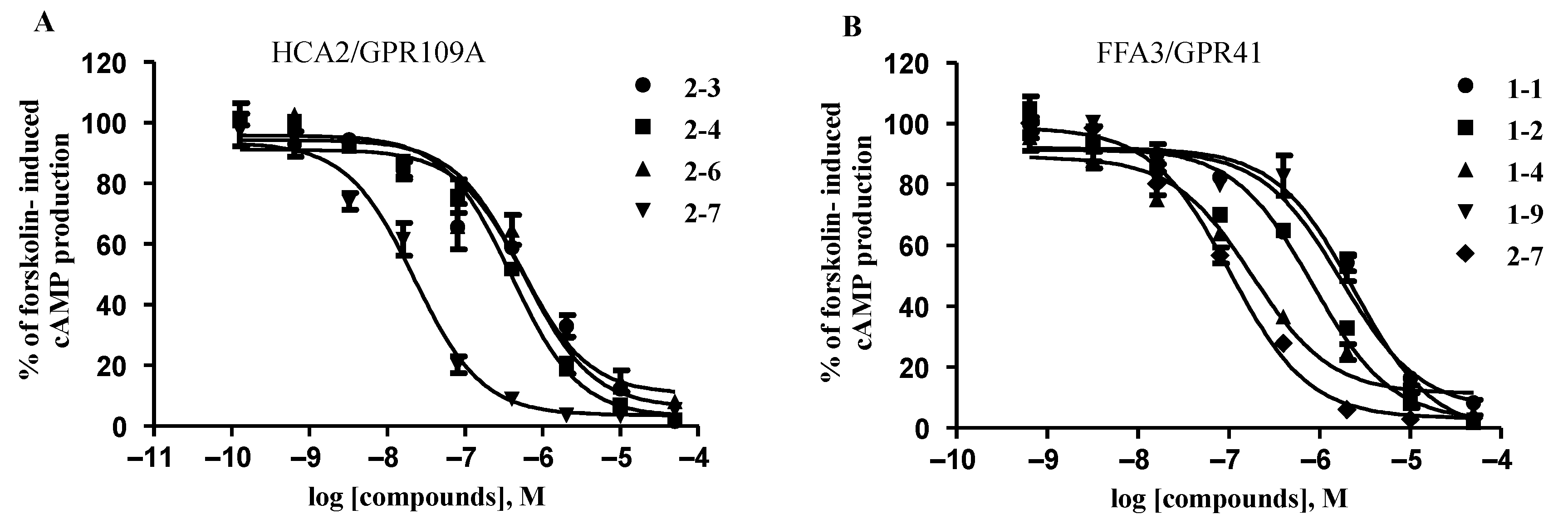
2.3. The Effects of Pyranopyrimidine Derivatives on HCA2/GPR109A, FFA2/GPR43, and FFA3/GPR41 Receptors
- High potency for HCA2/GPR109A, low potencies for FFA3/GPR41 and FFA2/GPR43: compounds 2-3 and 2-5 (5-n-propyl-2-thioxo and 5-n-butyl-2-oxo derivatives of pyranopyrimidines).
- High and comparable potencies towards all three studied receptors: compounds 2-1, 2-2, 2-4, 2-6, and 2-7, representing 2-thioxo- and 2-oxopyranopyrimidines.
2.4. The Impact of S–Alkylpyranopyrimidine Derivatives on HCA2/GPR109A, FFA3/GPR41, and FFA2/GPR43 Receptors
2.5. Electron-Donating Properties of Hexahydroquinolines and Pyranopyrimidines
2.6. Lipophilicity and Polar Surface Area of Studied Compounds
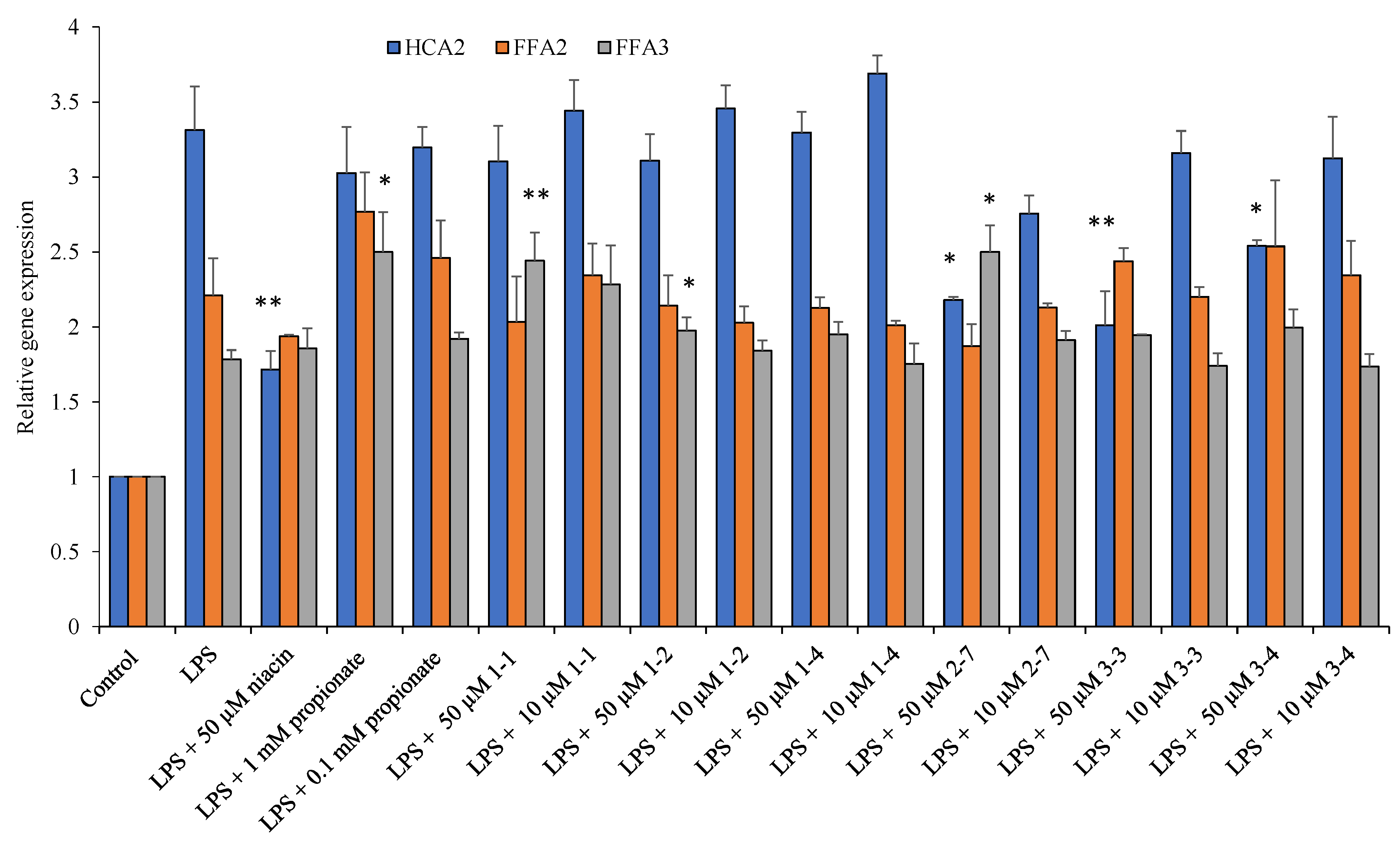
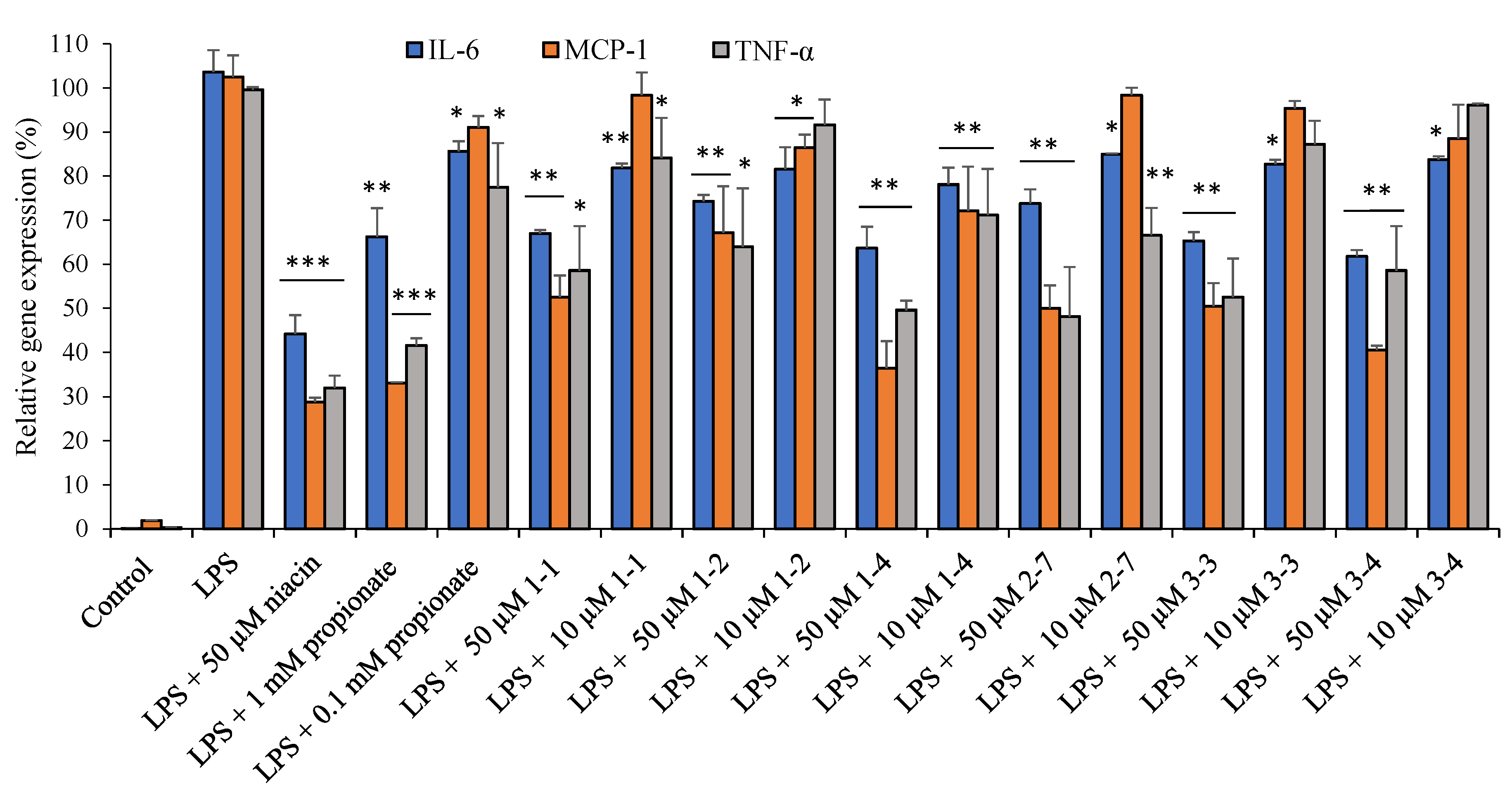
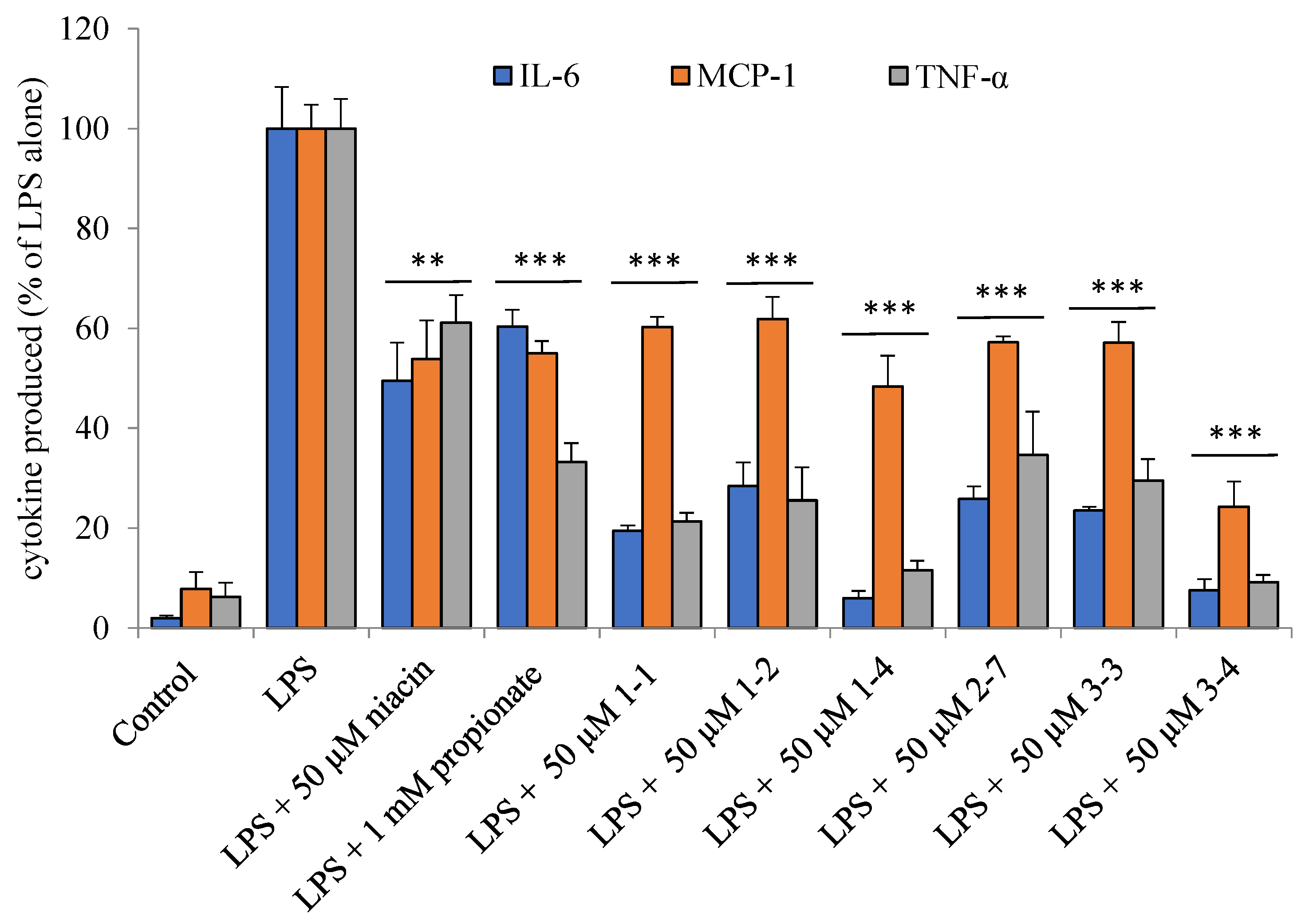
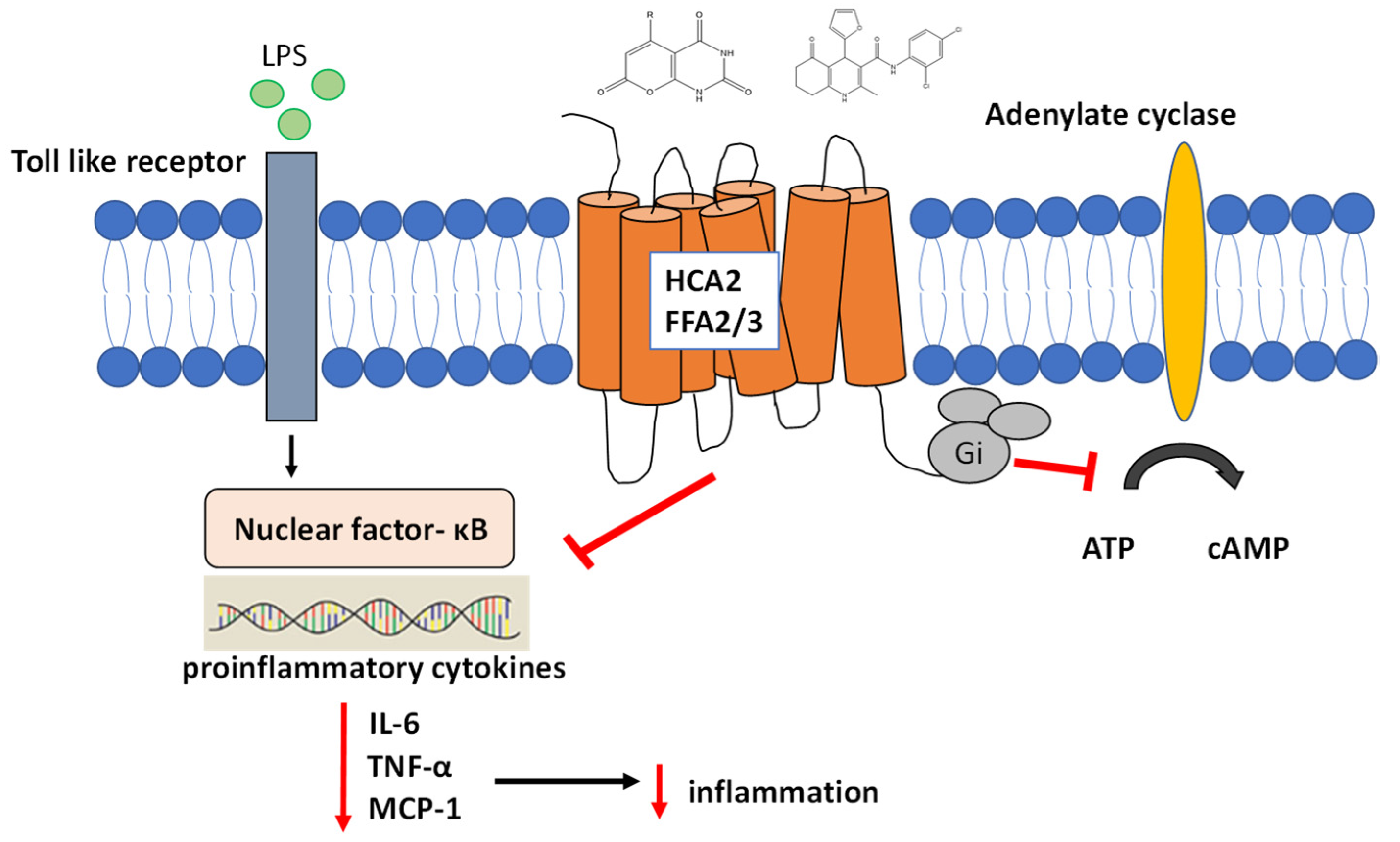
2.7. Ligands of HCA2/GPR109A, FFA3/GPR41, and FFA2/GPR43 Receptors Suppress Gene Expression and Secretion of Inflammatory Cytokines in THP-1 Macrophages
3. Materials and Methods
3.1. Materials
3.2. Synthesis
3.2.1. Hexahydroquinolines
General Procedure for the Synthesis of 5-Oxo-hexahydroquinolines 1-4, 1-6, 1-8, and 1-9
N-(2,4-Dichlorophenyl)-4-(furan-2-yl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide 1-4
N-(2,4-Dichlorophenyl)-2-methyl-5-oxo-4-(thiophen-2-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide 1-6
2-Methyl-3-(morpholine-4-carbonyl)-4-(thiophen-2-yl)-4,6,7,8-tetrahydro-1H-quinolin-5-one 1-8
Ethoxycarbonylmethyl 2-Methyl-3-(morpholine-4-carbonyl)-4-(thiophen-2-yl)-4,6,7,8-tetrahydr 1-9
N-(4-Chlorophenyl)-2-methyl-5-oxo-4-(thiophen-2-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide 1-5
General Procedure for the Synthesis of 5-Oxo-hexahydroquinolines 1-7, 1-10
2-Methyl-5-oxo-N-(pyridin-2-yl)-4-(thiophen-2-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide 1-7
(2-Ethoxy-2-oxo-1-phenylethyl)2-methyl-5-oxo-4-(2-thienyl)-4,6,7,8-tetrahydro-1H-quinoline-3-carboxylate 1-10
3.2.2. Pyranopyrimidines and Thioxopyranopyrimidines
5-(4-Chlorobutyl)-1H-pyrano[2,3-d]pyrimidine-2,4,7-trione 2-7
General Procedure for the Synthesis of 5-Alkyl-2-thioxo-2,3-dihydro-4H-pyrano[2,3-d]pyrimidine-4,7(1H)-diones 2-3, 2-4, 2-6
5-Propyl-2-thioxo-2,3-dihydro-1H-pyrano[2,3-d]pyrimidine-4,7-dionee 2-3
5-Butyl-2-thioxo-2,3-dihydro-1H-pyrano[2,3-d]pyrimidine-4,7-dione 2-4
5-(4-Chlorobutyl)-2-thioxo-2,3-dihydro-1H-pyrano[2,3-d]pyrimidine-4,7-dione 2-6
General Procedure for the Synthesis of 2-(Substituted-thio)-5-alkyl-4H-pyrano]2,3-d]pyrimidine(1H)-diones 3-1–3-7
Ethyl 5-butyl-4,7-dioxo-1,7-dihydro-4H-(pyrano[2,3-d]pyrimidin-2-ylsulfanyl)acetate 3-1
[5-(4-Chlorobutyl)-4,7-dioxo-1,7-dihydro-4H-(pyrano[2,3-d]pyrimidin-2-ylsulfanyl)aceticacid ethyl ester 3-2
2-[5-(4-Chlorobutyl)-4,7-dioxo-1,7-dihydro-4H-]pyrano[2,3-d]pyrimidin-2-ylsulfanyl]-acetic acid benzyl ester 3-3
2-[2-(4-Benzyloxy-phenyl)-1-methyl-2-oxo-ethylsulfanyl]-5-(4-chlorobutyl)-1H-pyrano[2,3-d]pyrimidine-4,7-dione 3-4
2-[5-(4-Chlorobutyl)-4,7-dioxo-1,7-dihydro-4H-pyrano[2,3-d]pyrimidin-2-ylsulfanyl]-acetamide 3-5
2-2-[5-(4-Chlorobutyl)-4,7-dioxo-1,7-dihydro-4H-pyrano[2,3-d]pyrimidin-2-ylsulfanyl]-acetamino-cyclohex-1-enecarboxylic acid ethyl ester 3-6
2-2-[5-(4-Chlorobutyl)-4,7-dioxo-1,7-dihydro-4H-pyrano[2,3-d]pyrimidin-2-ylsulfanyl]-acetamino-benzoic acid 3-7
3.3. Biological Tests
3.3.1. Preparation of Cell Cultures and the Generation of Stable Cell Lines
3.3.2. THP-1 Cell Differentiation and Stimulation
3.3.3. Intracellular cAMP Inhibition Assay
3.3.4. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
3.3.5. Quantification of Cytokines and Chemokines
3.3.6. Statistical Analysis
3.3.7. Characterisation of the Synthesized Compounds
3.3.8. The Evaluation of Redox Potentials
3.3.9. The Calculation of logP and TPSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramsay, R.R.; Popovic-Nicolic, M.R.; Nicolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Trans. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; M Babu, M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.J.; M Goldsworthy, S.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K. Biological roles and therapeutic potential of hydroxy-carboxylic acid receptors. Front. Endocrinol. 2011, 2, 51. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.P.; Karunakar, P.; Taraphder, S.; Yadav, H. Free fatty acid receptors 2 and 3 as microbial metabolite sensors to shape host health: Pharmacophysiological view. Biomedicines 2020, 8, 154. [Google Scholar] [CrossRef] [PubMed]
- Gille, A.; Bodor, E.T.; Ahmed, K.; Offermanns, S. Nicotinic acid: Pharmacological effects and mechanisms of action. Ann. Rev. Pharmacol. Toxicol. 2008, 48, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, I.; Hasegawa, S.; Kasubuchi, M.; Ichimura, A.; Nakajima, A.; Kimura, I. Nutritional signalling via free fatty acid receptors. Int. J. Mol. Sci. 2016, 17, 450. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Snelling, T.; Umlai, U.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolp, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.C.A.M.; Wagenaar, G.T.M.; Garssen, J.; Folkerts, G.; Henricks, P.A.J. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef]
- Pongkorpsakol, P.; Moonwiriyakit, A.; Muanprasat, C. Fatty acid and mineral receptors as drug targets for gastrointestinal disorders. Future Med. Chem. 2017, 9, 315–334. [Google Scholar] [CrossRef]
- Carrette, M.D.; Quiroga, J.; López, R.; Hidalgo, M.A.; Burgos, R.A. Participation of short-chain fatty acids and their receptors in gut inflammation and colon cancer. Front. Physiol. 2021, 12, 662739. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free fatty acid receptors in health and disease. Phys. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Hauser, A.; Atwood, M.M.; Back-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Grundmann, M.; Bender, E.; Schamberger, J.; Eitner, F. Pharmacology of free fatty acid receptors and their allosteric modulators. Int. J. Mol. Sci. 2021, 22, 1763. [Google Scholar] [CrossRef]
- Palani, A.; Rao, A.; Chen, X.; Huang, X.; Su, J.; Tang, H.; Huang, Y.; Qin, J.; Xiao, D.; Degrado, S.; et al. Discovery of SCH 900271, a potent nicotinic acid receptor agonist for the treatment of dyslipidemia. ACS Med. Chem. Lett. 2012, 3, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Su, J.; Rao, A.U.; Tang, H.; Zhou, W.; Zhu, X.; Chen, X.; Liu, Z.; Huang, Y.; Degrado, S.; et al. SAR studies of C2 ethers of 2H-pyrano[2,3-d]pyrimidine-2,4,7(1H,3H)triones as nicotinic acid receptor (NAR) agonists. Bioorg. Med. Chem. Lett. 2012, 22, 858. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.N.; Chu, Z.L.; Bruce, M.A.; Boatman, P.D. GPR41 and modulators thereof for the treatment of insulin-related disorders. U.S. Patent 11/666,910, 18 May 2016. [Google Scholar]
- Hudson, B.D.; Christiansen, E.; Murdoch, H.; Jenkins, L.; Hansen, A.H.; Madsen, O.; Ulven, T.; Milligan, G. Complex pharmacology of novel allosteric free fatty acid 3 receptor ligands. Mol. Pharm. 2014, 86, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulven, E.R.; Quon, T.; Sergeev, E.; Barki, N.; Brvar, M.; Hudson, B.D.; Dutta, P.A.H.; Bielefeldt, L.Ø.; Tobin, A.B. Structure-activity relationship studies of tetrahydroquinolone free fatty acid receptor 3 modulators. J. Med. Chem. 2020, 63, 3577–3595. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Ma, H.; Boshi, H.; Zhang, Y. Recent advances in multitarget-directed ligands targeting G-protein-coupled receptors. Drug Discov. Today 2020, 25, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- De Silva, M.F.; Dias, K.S.T.; Gontijo, V.S.; Ortiz, C.J.C.; Viegas, C., Jr. Multi-target directed drugs as a modern approach for drug design towards Alzheimer’s disease: An update. Curr. Med. Chem. 2018, 25, 3491–3525. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Harding, J.W. The development of multi-target-directed ligands (MTDL) to treat Alzheimer’s disease. Front. Clin. Drug Res. Alzheimer Disord. 2013, 1, 86–108. [Google Scholar] [CrossRef] [Green Version]
- Perone, R.; Albertini, C.; Uliassi, E.; Di Pietri, F.; de Pinheiro, P.S.M.; Petralla, S.; Rizzardi, N.; Fato, R.; Pulkrabkova, L.; Soukup, O.; et al. Turning Donepezil into a multi-target directed ligand through a merging strategy. ChemMedChem 2021, 16, 187–198. [Google Scholar] [CrossRef]
- Ramalakshmi, N.; Remya, R.S.; Nalini, C.N. Multitarget directed ligand approaches for Alzheimer’s disease: A Comprehensive Review. Mini-Rev. Med. Chem. 2021, 20. [Google Scholar] [CrossRef]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable drug discovery of multi-target directed ligands for Alzheimer’s disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef]
- Digby, J.E.; Martinez, F.; Jefferson, A.; Ruparelia, N.; Chai, J.; Wamil, M.; Greaves, D.R.; Choudhury, R.P. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Ang, Z.; Er, J.; Tan, N.; Lu, J.; Liou, Y.-C.; Grosse, J.; Ding, J.L. Human and mouse monocytes display distinct signalling and cytokine profiles upon stimulation with FFAR2/FFAR3 short-chain fatty acid receptor agonists. Sci. Rep. 2016, 6, 34145. [Google Scholar] [CrossRef]
- Benfeito, S.; Oliveira, C.; Fernandes, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Garrido, J.; Martins, C.; Sarmento, B.; Silva, R.; et al. Fine-tuning the neuroprotective and blood-brain barrier permeability profile of multi-target agents designed to prevent progressive mitochondrial dysfunction. Eur. J. Med. Chem. 2019, 167, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Langle, D.; Marquardt, V.; Heider, E.; Vigante, B.; Duburs, G.; Luntena, I.; Flötgen, D.; Golz, C.; Strohmann, C.; Koch, O.; et al. Design, synthesis and 3D-QSAR studies of novel 1,4-dihydropyridines as TGFb/Smad inhibitors. Eur. J. Med. Chem. 2015, 95, 249–266. [Google Scholar] [CrossRef] [PubMed]
- Stankevich, E.; Vanags, G. Polynuclear heterocyclic compounds.VII. Reaction of bis-dimedonyl methanes with compounds containing the amino group. Reduction of octahydroacridinediones. Latvijas PSR Zinātņu Akadēmijas Vēstis 1961, 2, 223–227. (In Russian) [Google Scholar]
- Kin, J.; Rao, A.; Chen, X.; Zhu, X.; Liu, Z.; Huang, X.; Degrado, S.; Huang, Y.; Xiao, D.; Aslanian, R.; et al. Discovery of a potent nicotinic acid receptor agonist for the treatment of dyslipidemia. ACS Med. Chem. Lett. 2011, 10, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Ridi, M.; Feroci, G. Barbituric acid and its derivatives. VII Some reaction with ethyl acetate. Gazz. Chim. Ital. 1950, 80, 121–128. [Google Scholar]
- Vijesh, A.M.; Isloor, A.M.; Peethambar, S.K.; Shivananda, K.N.; Arulmoli, T.; Isloor, N.A. Hantzsch reaction: Synthesis and characterization of some new 1,4-dihydropyridine derivatives as potent antimicrobial and antioxidant agents. Eur. J. Med. Chem. 2011, 46, 5591–5597. [Google Scholar] [CrossRef]
- Augustyniak, A.; Bartosz, G.; Čipak, A.; Duburs, G.; Horáková, L.; Luczaj, W.; Majekova, M.; Odysseos, A.D.; Rackova, L.; Skrzydlewska, E.; et al. Natural and synthetic antioxidants: An updated overview. Free Radic. Res. 2010, 44, 1216–1262. [Google Scholar] [CrossRef]
- Velena, A.; Zarkovic, N.; Gall Troselj, K.; Bisenieks, E.; Krauze, A.; Poikans, J.; Duburs, G. 1,4-Dihydropyridine derivatives: Dihydronicotinamide analogues—model compounds targeting oxidative stress. Oxid. Med. Cell Longev. 2016, 2016, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Da Costa Cabrera, D.; Santa-Helena, E.; Leal, H.P.; de Moura, R.R.; Nery, L.E.M.; Neves Gonçalves, C.A.; Russowsky, D.; D’Oca, M.G.M. Synthesis and antioxidant activity of new lipophilic dihydropyridines. Bioorg. Chem. 2019, 84, 1–16. [Google Scholar] [CrossRef]
- Rojstaczer, N.; Triggle, D.J. Structure-function relationships of calcium antagonists. Effect on oxidative modification of lower density lipoprotein. Biochem. Pharmacol. 1996, 51, 141–150. [Google Scholar] [CrossRef]
- Rosenkranz, A.C.; Lob, H.; Breitenboch, T.; Berkels, R.; Roesen, R. Endothelial antioxidant actions of dihydropyridines and angiotensin coverting enzyme inhibitors. Eur. J. Pharmacol. 2006, 529, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Malek, R.; Maj, M.; Wnorowski, A.; Jóźwiak, K.; Martin, H.; Iriepa, I.; Moraleda, I.; Chabchoub, F.; Marco-Contelles, J.; Ismaili, L. Multi-target 1,4-dihydropyridines showing calcium channel blockade and antioxidant capacity for Alzheimer’s disease therapy. Bioorg. Chem. 2019, 91, 103205. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Vukovic, T.; Zarkovic, N.; Tatzber, F.; Bisenieks, E.; Kalme, Z.; Bruvere, I.; Ogle, Z.; Poikans, J.; Velena, A.; et al. Antioxidative 1,4-dihydropyridine derivatives modulate oxidative stress and growth of human osteoblast-like cells in vitro. Antioxidants 2018, 7, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sürücü, O.; Bolat, G.; El-Khouly, A.; Gözde Gündüz, M.; Simşek, R.; Abacı, S.; Kuralay, F.; Şafak, C. Electrochemical detection of antioxidant activity of 1,4-dihydropyridine derivatives. Hacet. J. Biol. Chem. 2016, 44, 535–548. [Google Scholar] [CrossRef]
- Macha, R.; Ravindrachary, K.; Jayasree, G.P.L.; Tigulla, P. Spectrophotometric antioxidant bioassay and molecular modelling studies of ethyl 4-substituted-1,4,5,6,7,8-hexahydro-2,7,7-trimethyl-5-oxoquinoline-3-carboxylate derivatives. IJPSR 2017, 1, 67–79. [Google Scholar] [CrossRef]
- Brinkerhoff, R.C.; Santa-Helena, E.; do Amaral, P.C.; da Cabrera, D.; Ongaratto, R.F.; de Oliveira, P.M.; da D’Oca, C.M.; Gonçalves, C.A.N.; Nery, L.E.M.; D’Oca, M.G.M. Evaluation of the antioxidant activities of fatty polyhydroquinolines synthesized by Hantzsch multicomponent reactions. RSC Adv. 2009, 9, 24688. [Google Scholar] [CrossRef] [Green Version]
- Tavakkoli, Z.; Goljani, H.; Gunduz, M.G.; Tahir, M.N.; Nematolllahi, D. Electrochemical studies of newly synthesized 1,4-dihydropyridine-based hexahydroquinoline derivatives. J. Electrochem. Soc. 2020, 167, 125502. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Zhong, H.A.; Mashinson, V.; Woolman, T.A.; Zha, M. Understanding the molecular properties and metabolism of top prescribed drugs. Curr. Top. Med. Chem. 2013, 13, 1290–1307. [Google Scholar] [CrossRef]
- Wakade, C.; Chong, R.; Bradley, E.; Thomas, B.; Morgan, J. Upregulation of GPR109A in Parkinson’s Disease. PLoS ONE 1098, e18. [Google Scholar] [CrossRef] [Green Version]
- Falomir-Lockhart, L.J.; Cavazzutti, G.F.; Gimenez, E.; Toscani, A.M. Fatty Acid Signaling Mechanism in Neural Cells: Fatty Acid Receptors. Front. Cell. Neurosci. 2019, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Neuhaus, W.; Dandekar, T.; Förster, C. Analysing molecular polar surface descriptors to predict blood-brain barrier permeation. Int. J. Comput. Biol. Drug Des. 2013, 6, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.A.; Jackson, J.; Stanton, M.; Rojas-Triana, A.; Bober, L.; Laverty, M.; Yang, X.; Zhu, F.; Liu, J.; Wang, S.; et al. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World. J. Gastroenterol. 2009, 15, 5549–5557. [Google Scholar] [CrossRef] [PubMed]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The role of inflammation in Diabetes: Current concepts and future perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, M.; Lednovich, K.; Xu, K.; Gough, S.; Wicksteed, B.; Layden, B.T. FFAR from the gut microbiome crowd: SCFA receptors in T1D pathology. Metabolites 2021, 11, 302. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Structural Formula | HCA2/ GPR109A, % of Inhibition b | FFA3/ GPR41 EC50, µM a or % of Inhibition b; Emax,%c | FFA2/ GPR43 % of Inhibition b | logP d | TPSA d | El.ox. pot., V e | Ref. (for Synthesis) |
|---|---|---|---|---|---|---|---|---|
| 1-1 |  | NA | 1.79 ±0.3 a 96 ± 3 c | NA | 2.11 2.65 [22] d1 | 58.20 | 1.19 1.64 | [22] |
| 1-2 |  | NA | 0.61± 0.09 a 93 ± 5 c | NA | 1.69 2.18 [22] d1 | 67.43 | 1.19 1.99 | [21] |
| 1-3 |  | NA | 0.32 ± 0.05 a 99 ± 4 c | NA | 2.71 2.90 [22] d1 | 67.43 | [21] | |
| 1-4 |  | NA | 0.23± 0.07 a 86 ± 7 c | NA | 2.71 | 67.43 | 1.24 1.67 | M |
| 1-5 |  | NA | 45 ± 9 b | NA | 2.51 | 58.20 | M | |
| 1-6 |  | NA | 1.4± 0.17 a 82 ± 7 c | NA | 3.13 | 58.20 | M | |
| 1-7 |  | NA | NA | 49 ± 8 | 1.35 | 70.56 | M | |
| 1-8 |  | NA | 28 ± 2 b | NA | 0.06 | 58.64 | M | |
| 1-9 |  | NA | 1.9 ± 0.4 a 91 ± 6 c | 40 ± 2 | 0.97 | 81.70 | 1.18 1.68 | M |
| 1-10 |  | 60 ± 5 | 49 ± 15 b | 31 ± 4 | 2.54 | 72.01 | 1.18 1.68 | M |
| 1-11 |  | 52 ± 5 | 51 ± 6 b | 55 ± 6 | 5.43 | 81.70 | [33] | |
| 1-12 |  | 16 ± 4 | 16 ± 8 b | 21 ± 4 | 4.35 | 49.41 | [33] | |
| 1-13 |  | 26 ± 8 | 30 ± 9 b | 32 ± 7 | 4.35 | 63.68 | [34] | |
| 1-14 |  | NA | NA | NA | 4.70 | 92.70 | [33] |

| Compound | R | X | HCA2/ GPR109A EC50, µM a; Emax,% c | FFA3/ GPR41 EC50, µM a, or % of Inhibition b; Emax,% c | FFA2/GPR43 EC50, µM a, or % of Inhibition b; Emax,% c | LogP d | TPSA d | El.ox. pot., V e | Ref. (for Synthesis) |
|---|---|---|---|---|---|---|---|---|---|
| 2-1 | CH3 | S | 7.10 ± 1.31 a 96 ± 5 c | 4.8 ± 0.36 a 89 ± 7 c | 19 ± 6.8 a 87 ± 8 c | 0.10 | 67.43 | [36] | |
| 2-2 | C2H5 | S | 0.49 ± 0.06 a 99 ± 2 c | 0.37 ± 0.08 a9 6 ± 5 c | 0.92 ± 0.04 a 91 ± 4 c | 0.57 | 67.43 | [19] | |
| 2-3 | C3H7 | S | 0.87 ± 0.11 a 86 ± 3 c | 38 ± 2 b | 29 ± 3 b | 1.12 | 67.43 | M | |
| 2-4 | C4H9 | S | 0.55 ± 0.04 a 97 ± 4 c | 0.19 ± 0.03 a 95 ± 2 c | 0.74 ± 0.08 a 93 ± 6 c | 1.67 | 67.43 | M | |
| 2-5 | C4H9 | O | 0.051 ±0.007 a 93 ± 2 c | 28 ± 4 b | NA | 1.25 | 84.50 | 1.94 | [18] |
| 2-6 | C4H8Cl | S | 0.35 ± 0.02 a 94 ± 4 c | 0.34 ± 0.05 a 89 ± 3 c | 2.5 ± 0.4 a 83 ± 9 c | 1.36 | 67.43 | M | |
| 2-7 | C4H8Cl | O | 0.054 ± 0.006 a 98 ± 3 c | 0.22 ± 0.03 a 89 ± 5 c | 0.38 ± 0.06 a 88 ± 7 c | 0.94 | 84.50 | 1.94 | M |

| Com-pound | R | R1 | HCA2/ GPR109A EC50, µM a or % of Inhibition b; Emax, % c | FFA3/ GPR41 EC50, µM a or % of Inhibition b; Emax, % c | FFA2/ GPR43v% of Inhibition b | logP d | TPSA d | El. ox.pot. V e | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 3-1 | C4H9 | CH2CO2C2H5 | 75 ± 11a 93 ± 8c | 1.6± 0.5a 95 ± 3c | 65 ± 1 | 1.81 | 97.74 | 1.97 | M |
| 3-2 | C4H8Cl | CH2CO2C2H5 | 43 ±8a 89 ± 4c | 1.7± 0.2a 91 ± 5c | 64 ± 2 | 1.50 | 97.74 | M | |
| 3-3 | C4H8Cl | CH2CO2CH2Ph | 17 ± 3a 85 ± 10c | 72 ± 2b | 66 ± 6 | 2.79 | 97.74 | M | |
| 3-4 | C4H8Cl |  | 33 ± 16b | 0.38± 0.1a 95 ± 2c | 49 ± 2 | 4.62 | 97.74 | 1.98 | M |
| 3-5 | C4H8Cl | CH2CONH2 | 62± 10a 87 ± 6c | 20 ± 4b | 42± 2 | 0.02 | 114.53 | 1.93 | M |
| 3-6 | C4H8Cl |  | 48 ± 8b | 8.2 ± 1.3a 89 ± 7c | 62 ± 3 | 2.24 | 126.84 | M | |
| 3-7 | C4H8Cl |  | 30 ± 6b | 4.6 ± 0.6 a 87 ± 9c | 52 ± 10 | 2.70 | 134.16 | M |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisenieks, E.; Vigante, B.; Petrovska, R.; Turovska, B.; Muhamadejev, R.; Soloduns, V.; Velena, A.; Pajuste, K.; Saso, L.; Klovins, J.; et al. The Specificity and Broad Multitarget Properties of Ligands for the Free Fatty Acid Receptors FFA3/GPR41 and FFA2/GPR43 and the Related Hydroxycarboxylic Acid Receptor HCA2/GPR109A. Pharmaceuticals 2021, 14, 987. https://doi.org/10.3390/ph14100987
Bisenieks E, Vigante B, Petrovska R, Turovska B, Muhamadejev R, Soloduns V, Velena A, Pajuste K, Saso L, Klovins J, et al. The Specificity and Broad Multitarget Properties of Ligands for the Free Fatty Acid Receptors FFA3/GPR41 and FFA2/GPR43 and the Related Hydroxycarboxylic Acid Receptor HCA2/GPR109A. Pharmaceuticals. 2021; 14(10):987. https://doi.org/10.3390/ph14100987
Chicago/Turabian StyleBisenieks, Egils, Brigita Vigante, Ramona Petrovska, Baiba Turovska, Ruslan Muhamadejev, Vitalijs Soloduns, Astrida Velena, Karlis Pajuste, Luciano Saso, Janis Klovins, and et al. 2021. "The Specificity and Broad Multitarget Properties of Ligands for the Free Fatty Acid Receptors FFA3/GPR41 and FFA2/GPR43 and the Related Hydroxycarboxylic Acid Receptor HCA2/GPR109A" Pharmaceuticals 14, no. 10: 987. https://doi.org/10.3390/ph14100987
APA StyleBisenieks, E., Vigante, B., Petrovska, R., Turovska, B., Muhamadejev, R., Soloduns, V., Velena, A., Pajuste, K., Saso, L., Klovins, J., Duburs, G., & Mandrika, I. (2021). The Specificity and Broad Multitarget Properties of Ligands for the Free Fatty Acid Receptors FFA3/GPR41 and FFA2/GPR43 and the Related Hydroxycarboxylic Acid Receptor HCA2/GPR109A. Pharmaceuticals, 14(10), 987. https://doi.org/10.3390/ph14100987