
Computational and In Vitro Experimental Investigations Reveal Anti-Viral Activity of Licorice and Glycyrrhizin against Severe Acute Respiratory Syndrome Coronavirus 2


 ,
,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
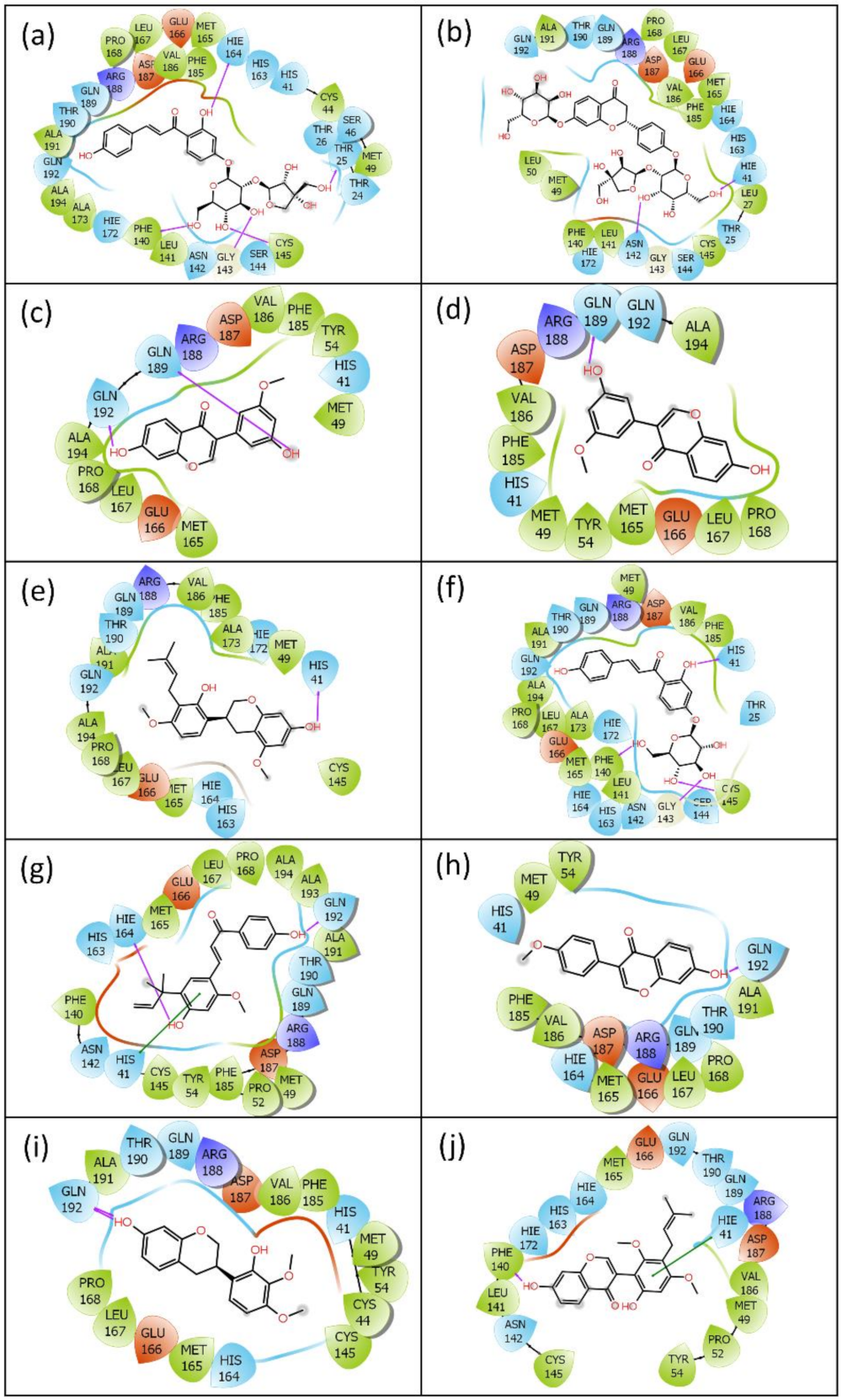
2.1. Virtual Screening and Re-Docking
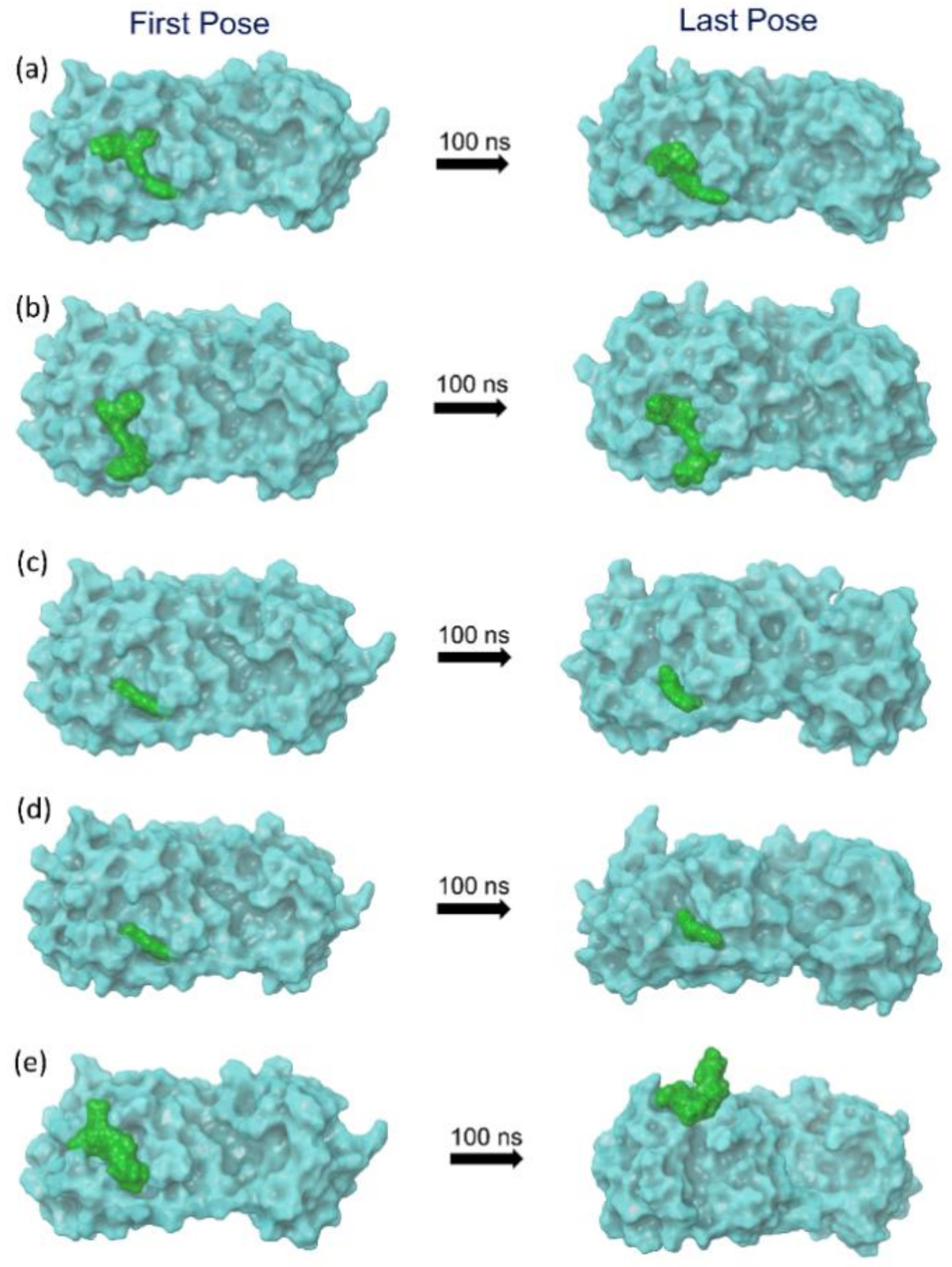
2.2. Molecular Dynamics Simulation Analysis
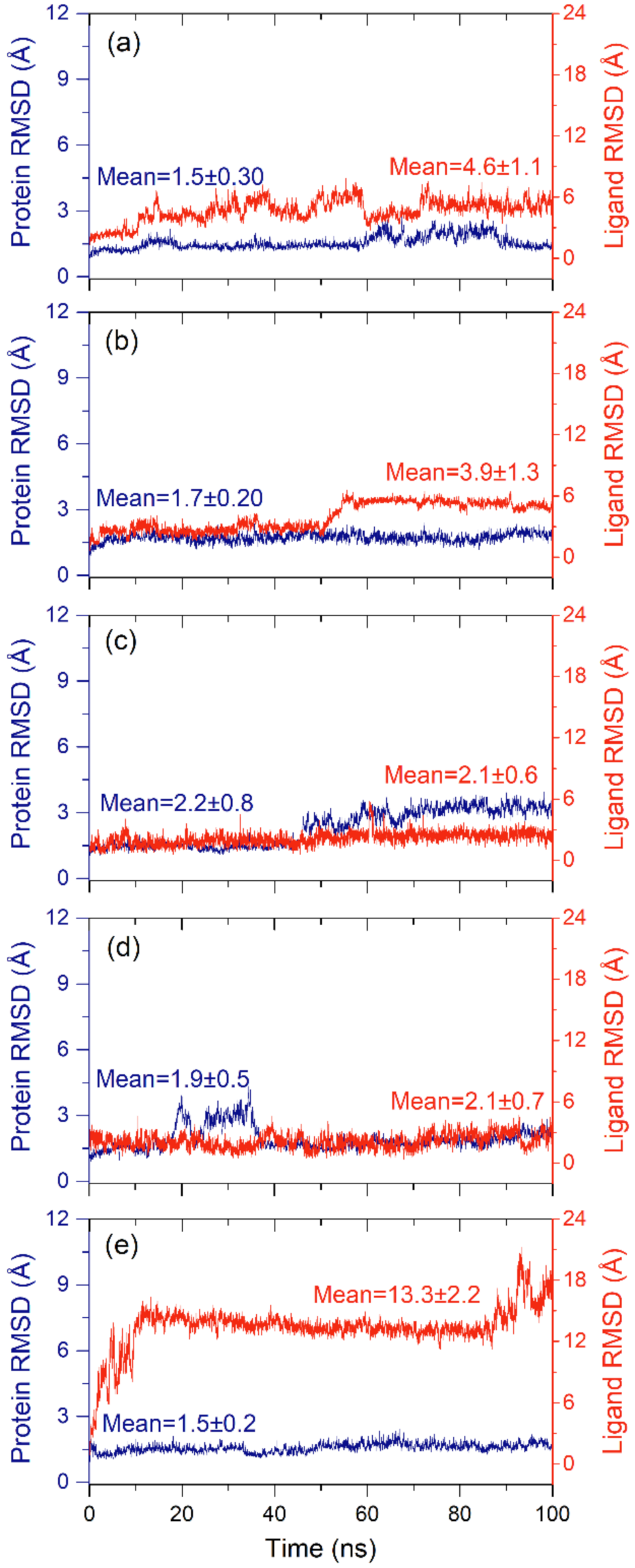
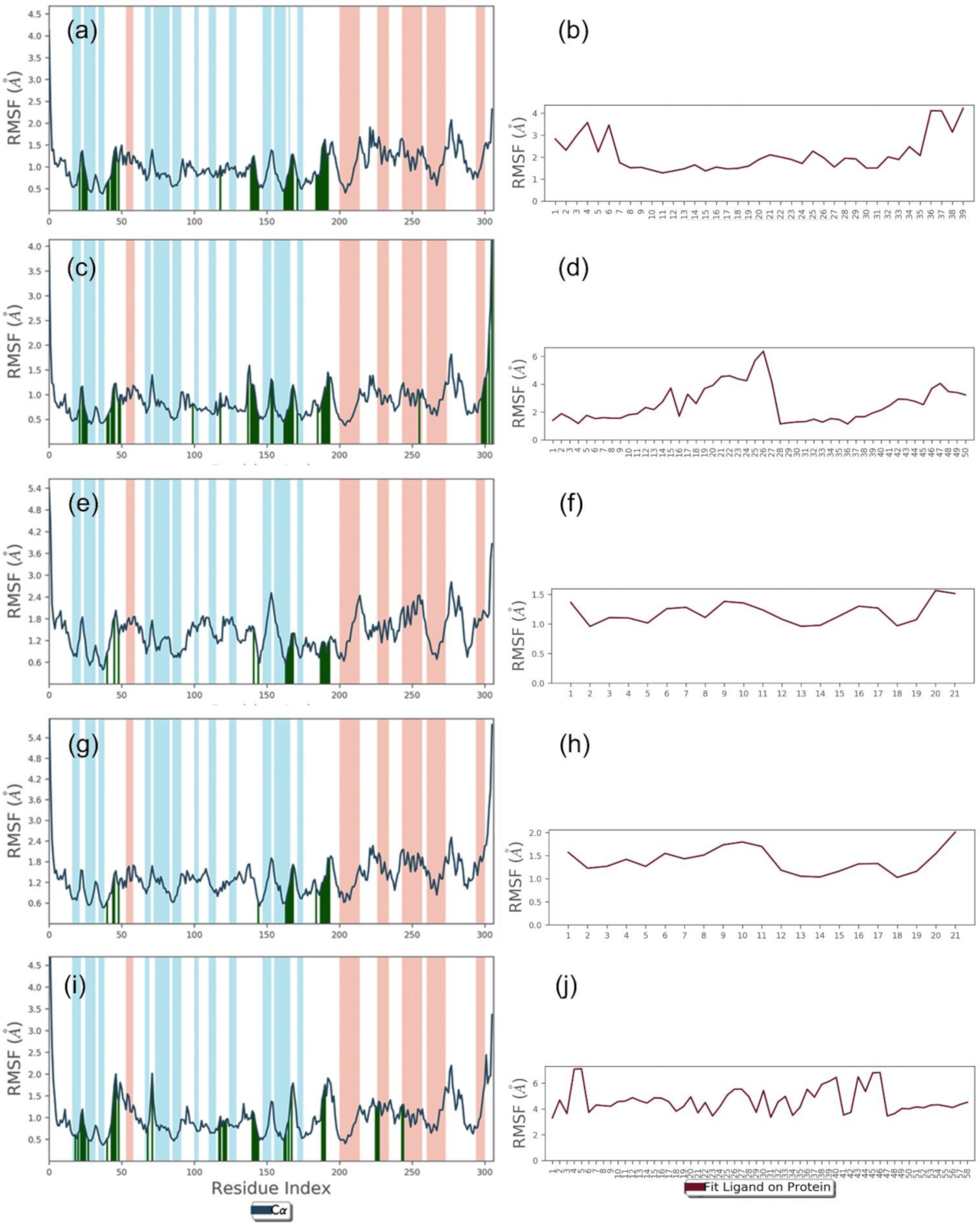
2.2.1. RMSD and RMSF Analysis
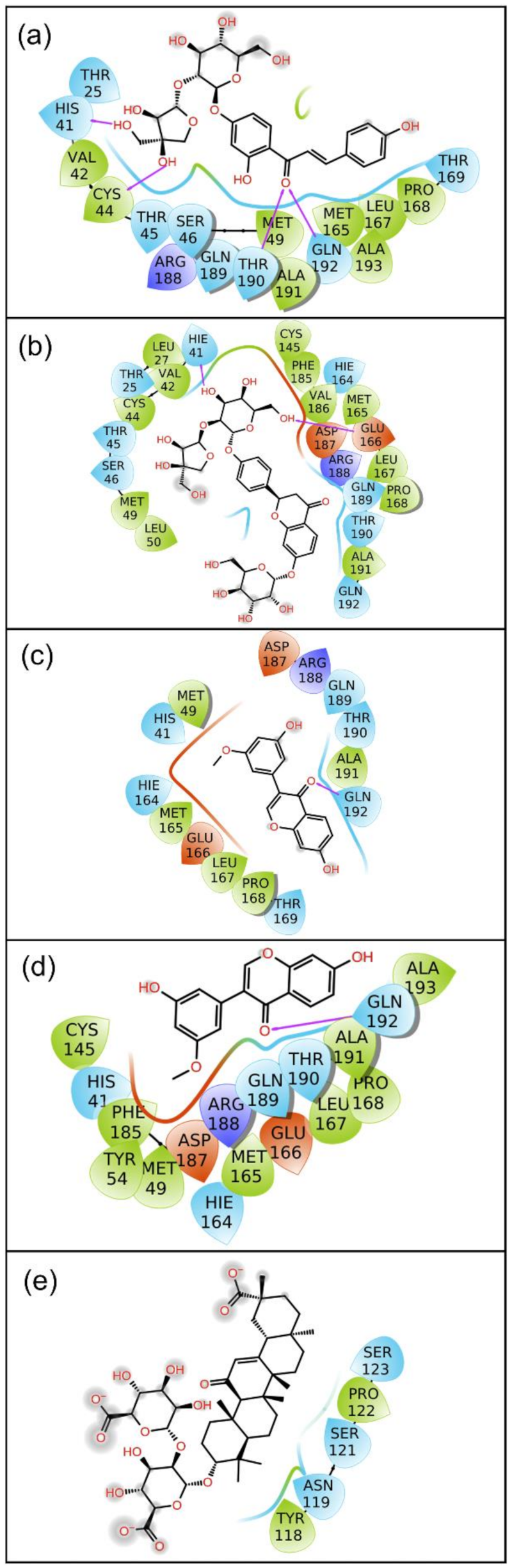
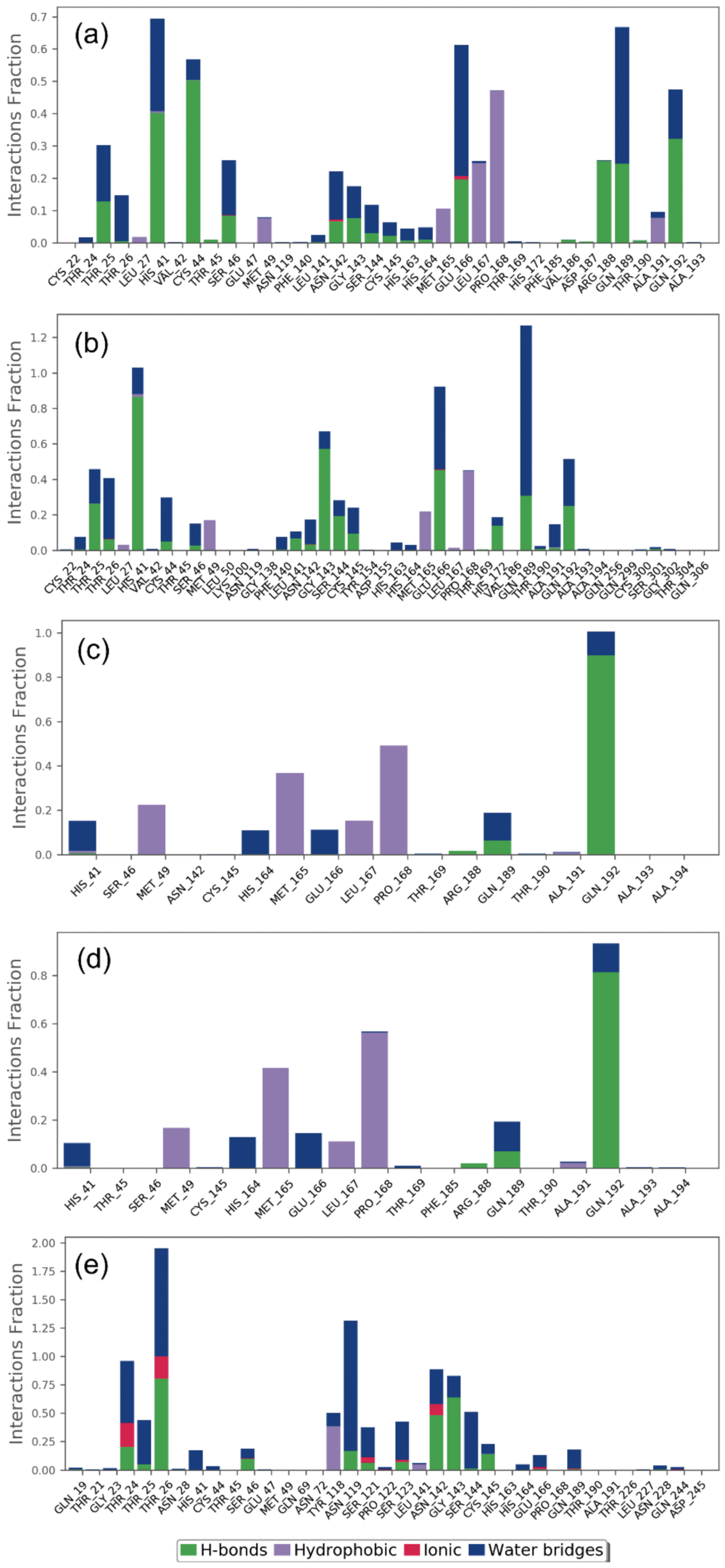
2.2.2. Protein–Ligand Contact Mapping
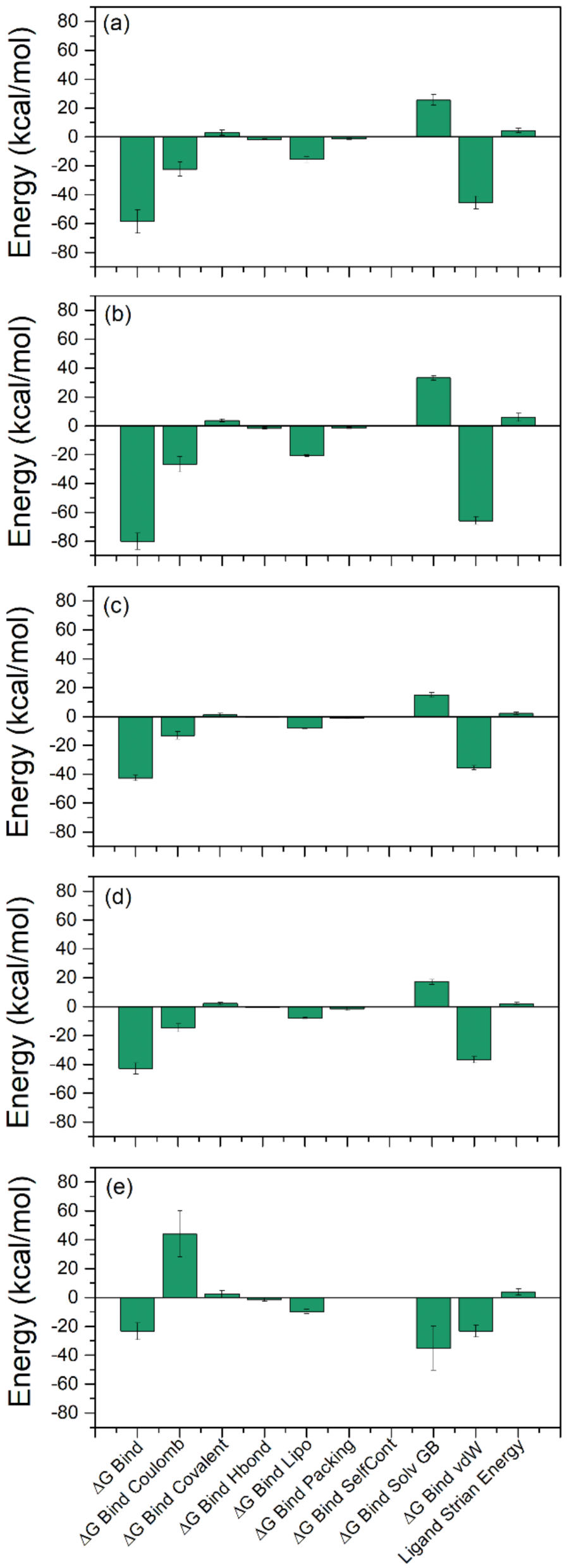
2.3. Binding Free Energy Analysis
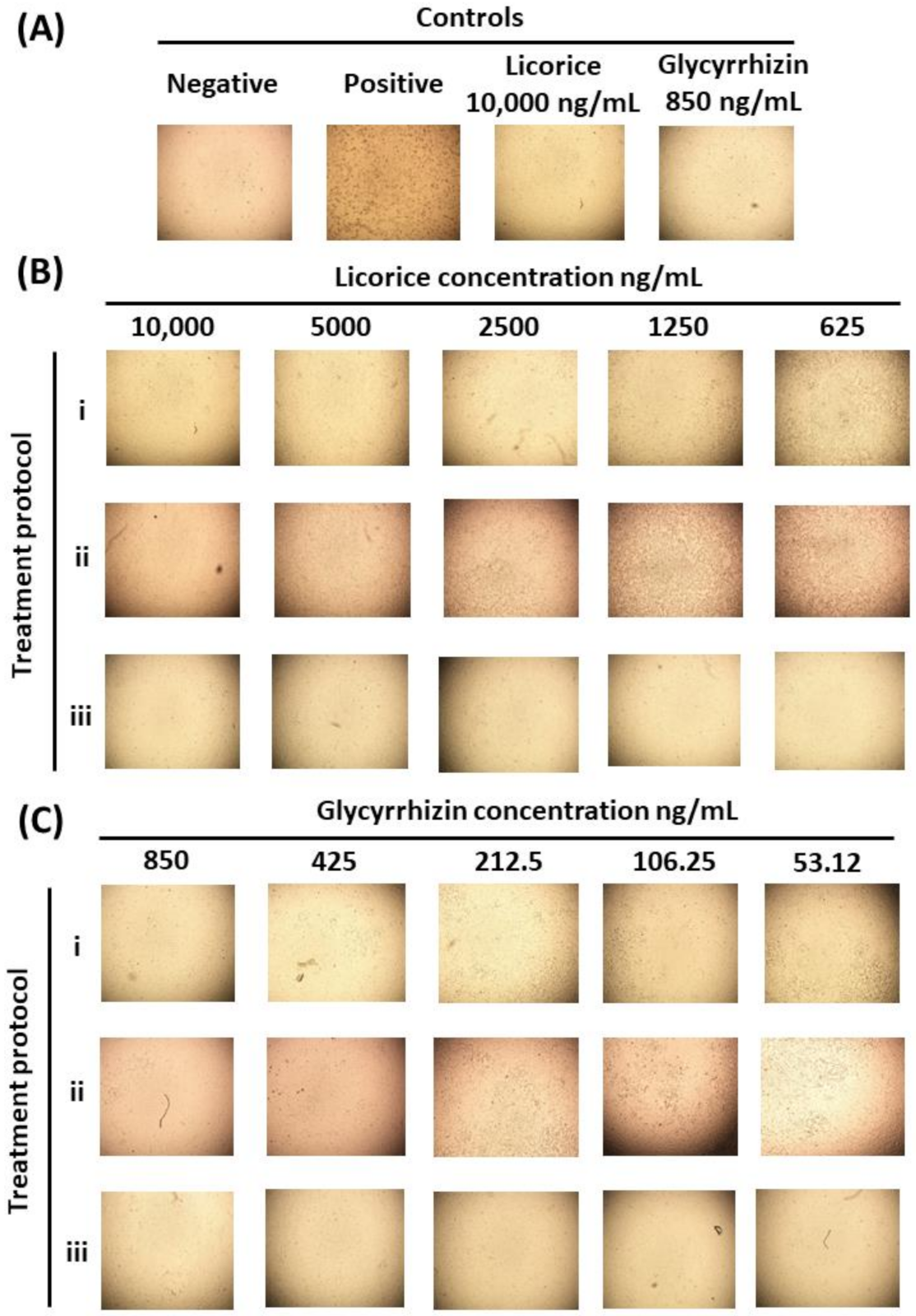
2.4. Assessment of Licorice and Glycyrrhizin Anti-SARS-CoV-2 Activity In Vitro
3. Materials and Methods
3.1. Receptor and Ligand Structure Data Collection
3.2. Structure Preparation and Virtual Screening
3.3. Re-Docking and Interaction Analysis
3.4. Molecular Dynamics Simulation and Free Binding Energy Calculations
3.5. In-Vitro Assessment of Licorice and Glycyrrhizin Antiviral Activity against SARS-CoV-2
3.5.1. Cell Line
3.5.2. Virus and Compounds
3.5.3. Cell Toxicity Assay
3.5.4. The Effect of Licorice and Glycyrrhizin on Viral Induced Cytopathic Effect (CPE) and Viral Plaque Forming Efficiency (PFE))
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; El-Kafraway, S.A.; Alandijany, T.A.; Bajrai, L.H.; Shah, A.A.; Dubey, A.; Sahoo, A.K.; Yadava, U.; Kamal, M.A.; Azhar, E.I.; et al. Structure-Based Identification of Natural Products as SARS-CoV-2 M(pro) Antagonist from Echinacea angustifolia Using Computational Approaches. Viruses 2021, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Azhar, E.I.; Kamal, M.A.; Bajrai, L.H.; Dubey, A.; Jha, K.; Yadava, U.; Kang, S.G.; Dwivedi, V.D. SARS-CoV-2 M(pro) inhibitors: Identification of anti-SARS-CoV-2 M(pro) compounds from FDA approved drugs. J. Biomol. Struct. Dyn. 2020, 38, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.C. Aerosol Transmission of SARS-CoV-2: Physical Principles and Implications. Front. Public Health 2020, 8, 590041. [Google Scholar] [CrossRef] [PubMed]
- Kembuan, G.; Lie, W.; Tumimomor, A. Potential usage of immune modulating supplements of the Echinacea genus for COVID-19 infection. Int. J. Med. Rev. Case Rep. 2020, 4, 203–217. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19-Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Arya, A.; Dwivedi, V.D. Synergistic effect of vitamin D and remdesivir can fight COVID-19. J. Biomol. Struct. Dyn. 2020, 8, 1–2. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Kang, S.G. Computational insights into tetracyclines as inhibitors against SARS-CoV-2 M(pro) via combinatorial molecular simulation calculations. Life Sci. 2020, 257, 118080. [Google Scholar] [CrossRef]
- SSalian, V.S.; Wright, J.A.; Vedell, P.T.; Nair, S.; Li, C.; Kandimalla, M.; Tang, X.; Carmona Porquera, E.M.; Kalari, K.R.; Kandimalla, K.K. COVID-19 Transmission, Current Treatment, and Future Therapeutic Strategies. Mol. Pharm. 2021, 18, 754–771. [Google Scholar] [CrossRef]
- Naqvi, A.A.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Liu, X.H.; Zhang, X.; Lu, Z.H.; Zhu, Y.S.; Wang, T. Potential molecular targets of nonstructural proteins for the development of antiviral drugs against SARS-CoV-2 infection. Biomed. Pharmacother. 2021, 133, 111035. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, V.D.; Bharadwaj, S.; Afroz, S.; Khan, N.; Ansari, M.A.; Yadava, U.; Tripathi, R.C.; Tripathi, I.P.; Mishra, S.K.; Kang, S.G. Anti-dengue infectivity evaluation of bioflavonoid from Azadirachta indica by dengue virus serine protease inhibition. J. Biomol. Struct. Dyn. 2021, 39, 1417–1430. [Google Scholar] [CrossRef]
- Armanini, D.; Fiore, C.; Bielenberg, J.; Sabbadin, C.; Bordin, L. Coronavirus-19: Possible Therapeutic Implications of Spironolactone and Dry Extract of Glycyrrhiza glabra L. (Licorice). Front. Pharmacol. 2020, 11, 558418. [Google Scholar] [CrossRef]
- Sinha, S.K.; Prasad, S.K.; Islam, M.A.; Gurav, S.S.; Patil, R.B.; AlFaris, N.A.; Aldayel, T.S.; AlKehayez, N.M.; Wabaidur, S.M.; Shakya, A. Identification of bioactive compounds from Glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: A pharmacoinformatics study. J. Biomol. Struct. Dyn. 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Gomaa, A.A.; Abdel-Wadood, Y.A. The potential of glycyrrhizin and licorice extract in combating COVID-19 and associated conditions. Phytomed. Plus 2021, 1, 100043. [Google Scholar] [CrossRef]
- Fukuchi, K.; Okudaira, N.; Adachi, K.; Odai-Ide, R.; Watanabe, S.; Ohno, H.; Yamamoto, M.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; et al. Antiviral and Antitumor Activity of Licorice Root Extracts. In Vivo 2016, 30, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Fiore, C.; Eisenhut, M.; Krausse, R.; Ragazzi, E.; Pellati, D.; Armanini, D.; Bielenberg, J. Antiviral effects of Glycyrrhiza species. Phytother. Res. 2008, 22, 141–148. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y. Potential interventions for novel coronavirus in China: A systematic review. J. Med. Virol. 2020, 92, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Zhu, Y.; Xu, J.; Yao, G.; Zhang, P.; Wang, M.; Zhao, Y.; Lin, G.; Chen, H.; Chen, L.; et al. Glycyrrhizic acid exerts inhibitory activity against the spike protein of SARS-CoV-2. Phytomedicine 2021, 85, 153364. [Google Scholar] [CrossRef]
- Tsui, V.; Case, D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Ngo, S.T.; Tam, N.M.; Quan, P.M.; Nguyen, T.H. A Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV2 Mpro. J. Chem. Inf. Model. 2020, 61, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Nees, M.; Kang, S.G. Density functional theory and molecular dynamics simulation support Ganoderma lucidum triterpenoids as broad range antagonist of matrix metalloproteinases. J. Mol. Liq. 2020, 311, 113322. [Google Scholar] [CrossRef]
- van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O.; et al. Glycyrrhizin Effectively Inhibits SARS-CoV-2 Replication by Inhibiting the Viral Main Protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Azhar, E.I.; Hindawi, S.I.; El-Kafrawy, S.A.; Hassan, A.M.; Tolah, A.M.; Alandijany, T.A.; Bajrai, L.H.; Damanhouri, G.A. Amotosalen and ultraviolet A light treatment efficiently inactivates severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in human plasma. Vox Sang. 2021, 116, 673–681. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compound Name | Docking Score | H-Bond | π–π Stacking | Hydrophobic | Polar | Negative | Positive | Glycine |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Licuraside | −8.7 | Hie164, Phe140, Cys145, Gly143, Thr25 | -- | Cys145, Leu141, Phe140, Ala173, Ala194, Ala191, Pro168, Leu167, Val186, Phe185, Cys44, Met49, Met165 | Thr24, Thr25, Thr26, Ser46, His41, His163, Hie164, Gln189, Thr190, Gln192, Hie172, Asn142, Ser144 | Asp187, Glu166 | Arg188 | Gly143 |
| 2 | Glucoliquiritin apioside | −8.8 | Asn142, Hie41 | -- | Leu50, Met49, Phe140, Leu141, Cys145, Leu27, Met165, Phe185, Val186, Leu167, Pro168, Ala191 | Gln192, Thr190, Gln189, Hie164, His163, Hie41, Thr25, Ser144, Asn142, Hie172 | Asp187, Glu166 | Arg188 | Gly143 |
| 3 | 7,3′-Dihydroxy-5′-methoxyisoflavone | −8.1 | Gln189, Gln192 | -- | Met49, Tyr54, Phe185, Val186, Ala194, Pro168, Leu167, Met165 | His41, Gln189, Gln192 | Asp187, Glu166 | Arg188 | -- |
| 4 | Licuroside | −8.1 | Gln189 | -- | Ala194, Val186, Phe185, Met49, Tyr54, Met165, Leu167, Pro168 | Gln192, Gln189, His41 | Asp187, Glu166 | Arg188 | -- |
| 5 | Kanzonol R | −7.5 | His41 | -- | Met165, Leu167,Pro168, Ala194, Ala191, Val186, Phe185, Ala173, Met49, Cys145 | His41, Hie172, Gln189, Thr190, Gln192, Hie164, His163 | Glu166 | Arg188 | -- |
| 6 | Neoisoliquiritin | −8.7 | His41, Cys145, Gly143 | -- | Phe185, Val186, Met49, Ala191, Ala194, Pro168, Leu167, Ala173, Met165, Phe140, Leu141, Cys145 | Thr25, His41, Gln189, Thr190, Gln192, Hie172, Hie164, His163, Asn142, Ser144 | Asp187, Glu166 | Arg188 | Gly143 |
| 7 | Licochalcone-A | −8.0 | Hie164, Gln192 | His41 | Met165, Leu167, Pro168, Ala194, Ala193, Ala191, Met49, Pro52, Phe185, Tyr54, Cys145, Phe140 | His163, Hie164, Gln192, Thr190, Gln189, His41, Asn142 | Asp187, Glu166 | Arg188 | -- |
| 8 | Formononetin | −7.3 | Gln192 | -- | Tyr54, Met49, Ala191, Pro168, Leu167, Met165, Val186, Phe185 | His41, Gln192, Thr190, Gln189, Hie164 | Asp187, Glu166 | Arg188 | -- |
| 9 | Isomucronulatol | −8.0 | Gln192 | -- | Ala191, Val186, Phe185, Met49, Tyr54, Cys145, Met165, Leu167, Pro168 | Gln192, Thr190, Gln189, His41, His164 | Asp187, Glu166 | Arg188 | -- |
| 10 | Licoricone | −7.4 | Phe140 | Hie41 | Cys145, Leu141, Phe140, Met165, Val186, Met49, Pro52, Tyr54 | Asn142, Hie172, His163, Hie164, Gln192, Thr190, Gln189, Hie41 | Asp187, Glu166 | Arg188 | -- |
| 11 | Reference complex | −8.0 | Thr25, Thr26, Asn142, Gly143 | -- | Leu27, Cys44, Met49, Leu167, Pro168, Ala191 | Thr24, Thr25, Thr26, His41, Thr45, Ser46, Asn142, Gln189 | Glu166 | -- | Gly143 |
| S. No. | Drug | H-Bond | Pi–Pi Stacking | Hydrophobic | Polar | Negative | Positive | Glycine |
|---|---|---|---|---|---|---|---|---|
| 1 | Licuraside | His41, Cys44, Thr190, Gln192 | - | Val42, Cys44, Met49, Ala191, Met165, Ala193, Leu167, Pro168 | Thr25, His41, Thr45, Ser46, Gln189, Thr190, Gln192, Thr169 | - | Arg188 | - |
| 2 | Glucoliquiritin apioside | Hie41, Glu166 | - | Leu27, Val42, Cys44, Met49, Leu50, Cys145, Phe185, Val186, Met165, Leu167, Pro168, Ala191 | Hie41, Thr25, Thr45, Ser46, Hie164, Gln189, Thr190, Gln192 | Glu166, Asp187 | Arg188 | - |
| 3 | 7,3′-Dihydroxy-5′-methoxyisoflavone | Gln192 | - | Met49, Met165, Leu167, Pro168, Ala191 | His41, Hie164, Thr169, Gln189, Thr190, Gln192 | Glu166, Asp187 | Arg188 | - |
| 4 | Licuroside | Gln192 | - | Ala193, Ala191, Pro168, Leu167, Met165, Met49, Tyr54, Phe185, Cys145 | Gln192, Thr190, Gln189, His41, Hie164 | Glu166, Asp187 | Arg188 | - |
| 5 | Reference | - | - | Pro122, Tyr118 | Ser123, Ser121, Asn119 | - | - | - |
| Components | Energy (kcal/mol) | ||||
|---|---|---|---|---|---|
| SARS-CoV-2 Mpro-Licuraside | SARS-CoV-2 Mpro-Glucoliquiritin | SARS-CoV-2 Mpro-7,3′-Dihydroxy-5′-Methoxyisoflavone | SARS-CoV-2 Mpro-Licuroside, | SARS-CoV-2 Mpro-Glycyrrhizin | |
| ΔGBind | −58.66 ± 8.09 | −80.0 ± 5.60 | −42.73 ± 1.94 | −42.93 ± 3.96 | −23.42 ± 5.84 |
| ΔGBind Coulomb | −22.44 ± 4.91 | −26.69 ± 5.40 | −13.27 ± 2.54 | −14.78 ± 2.82 | 43.97 ± 15.90 |
| ΔGBind Covalent | 2.86 ± 1.93 | 3.58 ± 1.10 | 1.10 ± 1.13 | 2.08 ± 0.83 | 2.34 ± 2.26 |
| ΔGBind Hbond | −1.89± 0.39 | −1.77 ± 0.59 | −0.68 ± 0.18 | −0.62 ± 0.21 | −1.57 ± 1.13 |
| ΔGBind Lipo | −15.60 ± 2.20 | −20.82 ± 0.67 | −8.13 ± 0.28 | −8.02 ± 0.47 | −9.67 ± 1.52 |
| ΔGBind Packing | −1.48 ± 0.30 | −1.62 ± 0.33 | −0.91 ± 0.56 | −1.7 ± 1.23 | 0 ± 0 |
| ΔGBind SelfCont | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| ΔGBind Solv GB | 25.56 ± 3.48 | 33.06 ± 1.44 | 14.82 ± 1.69 | 17.06 ± 1.59 | −35.14 ± 15.36 |
| ΔGBind vdW | −45.66 ± 4.54 | −65.81 ± 2.69208 | −35.64 ± 1.51 | −36.94 ± 2.25 | −23.35 ± 4.22 |
| Lig Strain Energy | 4.44 ± 1.59 | 5.77 ± 2.85 | 2.12 ± 0.80 | 2.07 ± 0.75 | 3.74 ± 2.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolah, A.M.; Altayeb, L.M.; Alandijany, T.A.; Dwivedi, V.D.; El-Kafrawy, S.A.; Azhar, E.I. Computational and In Vitro Experimental Investigations Reveal Anti-Viral Activity of Licorice and Glycyrrhizin against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals 2021, 14, 1216. https://doi.org/10.3390/ph14121216
Tolah AM, Altayeb LM, Alandijany TA, Dwivedi VD, El-Kafrawy SA, Azhar EI. Computational and In Vitro Experimental Investigations Reveal Anti-Viral Activity of Licorice and Glycyrrhizin against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals. 2021; 14(12):1216. https://doi.org/10.3390/ph14121216
Chicago/Turabian StyleTolah, Ahmed M., Lamya M. Altayeb, Thamir A. Alandijany, Vivek Dhar Dwivedi, Sherif A. El-Kafrawy, and Esam I. Azhar. 2021. "Computational and In Vitro Experimental Investigations Reveal Anti-Viral Activity of Licorice and Glycyrrhizin against Severe Acute Respiratory Syndrome Coronavirus 2" Pharmaceuticals 14, no. 12: 1216. https://doi.org/10.3390/ph14121216
APA StyleTolah, A. M., Altayeb, L. M., Alandijany, T. A., Dwivedi, V. D., El-Kafrawy, S. A., & Azhar, E. I. (2021). Computational and In Vitro Experimental Investigations Reveal Anti-Viral Activity of Licorice and Glycyrrhizin against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals, 14(12), 1216. https://doi.org/10.3390/ph14121216