
Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies



Abstract
:1. Introduction
2. Results and Discussion
2.1. Validation of the Crystal Structures and Generation of a Virtual Library
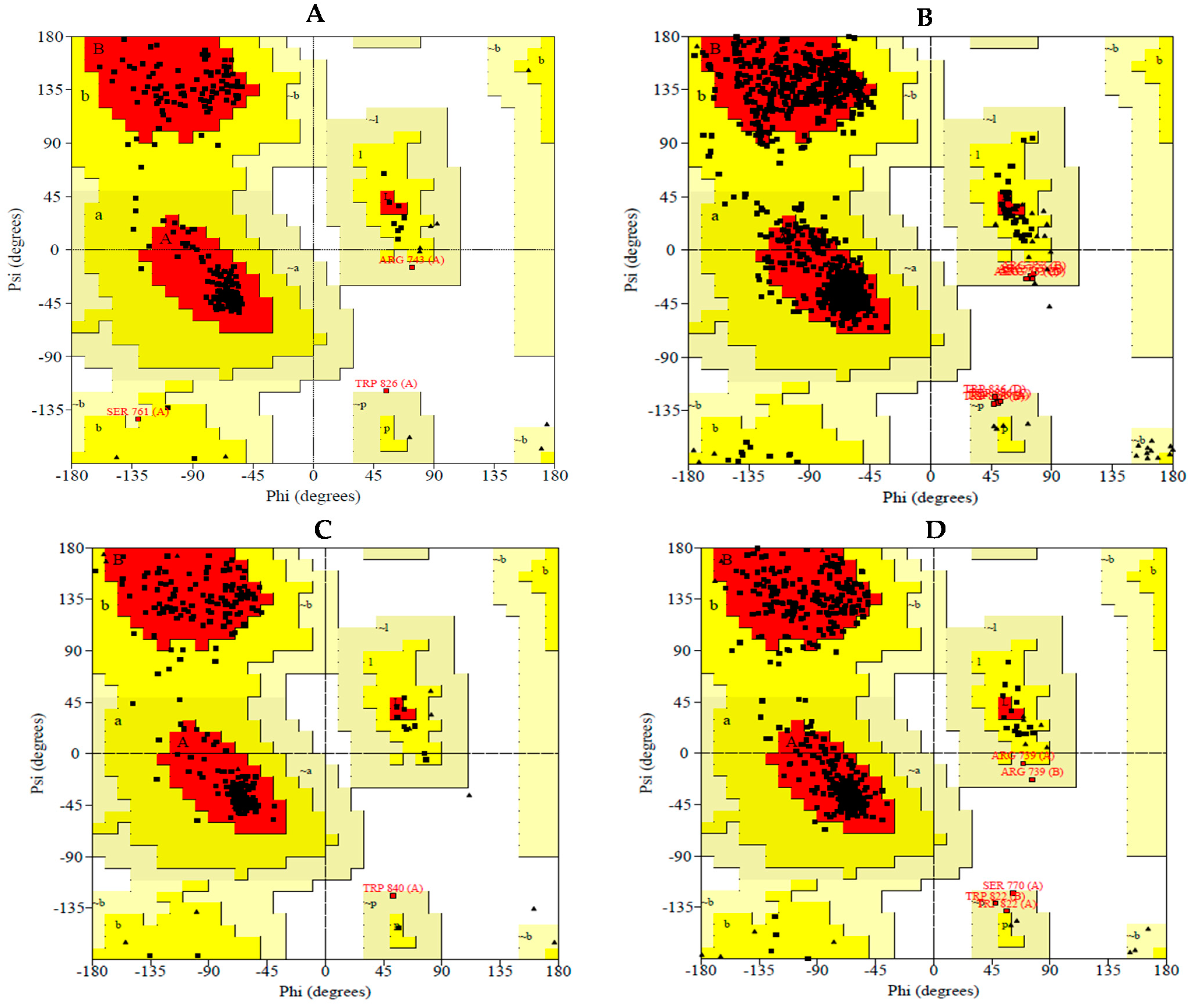
2.1.1. Validation of the Protein Structure
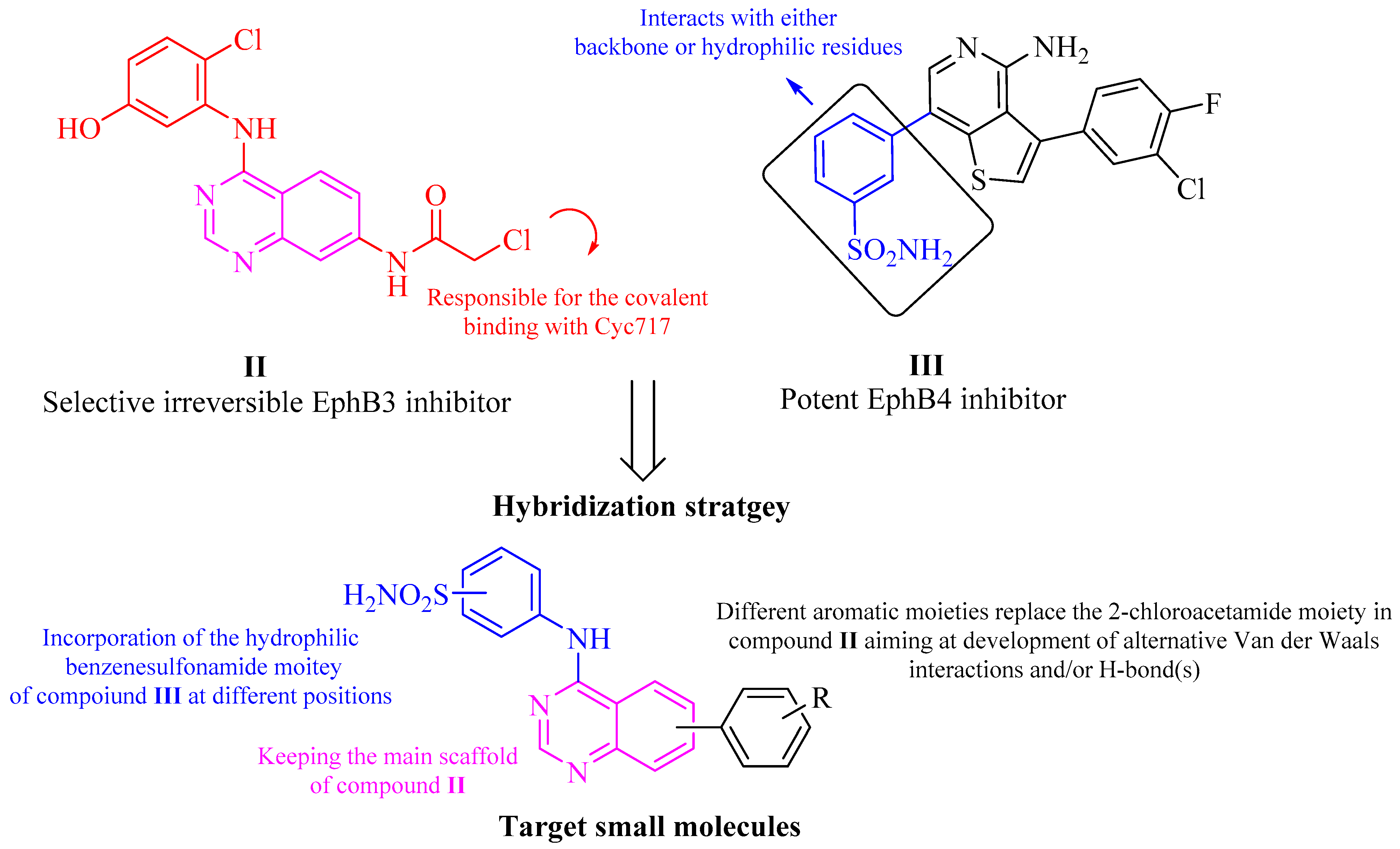
2.1.2. Generation of Virtual Combinatorial Library and Compounds Selection
2.2. Molecular Docking
2.3. MM-GBSA Calculations
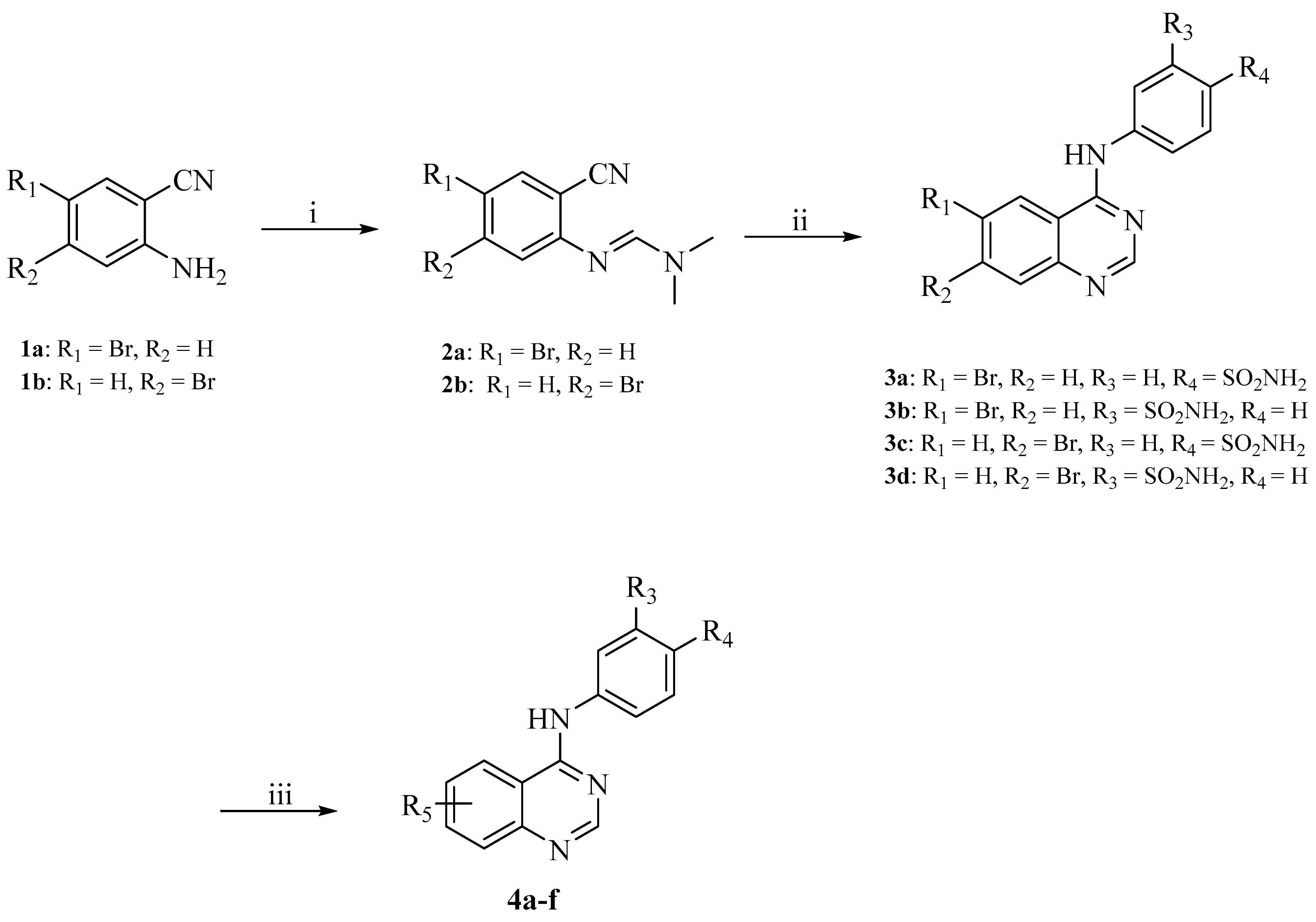
2.4. Chemical Synthesis
2.5. Biological Studies
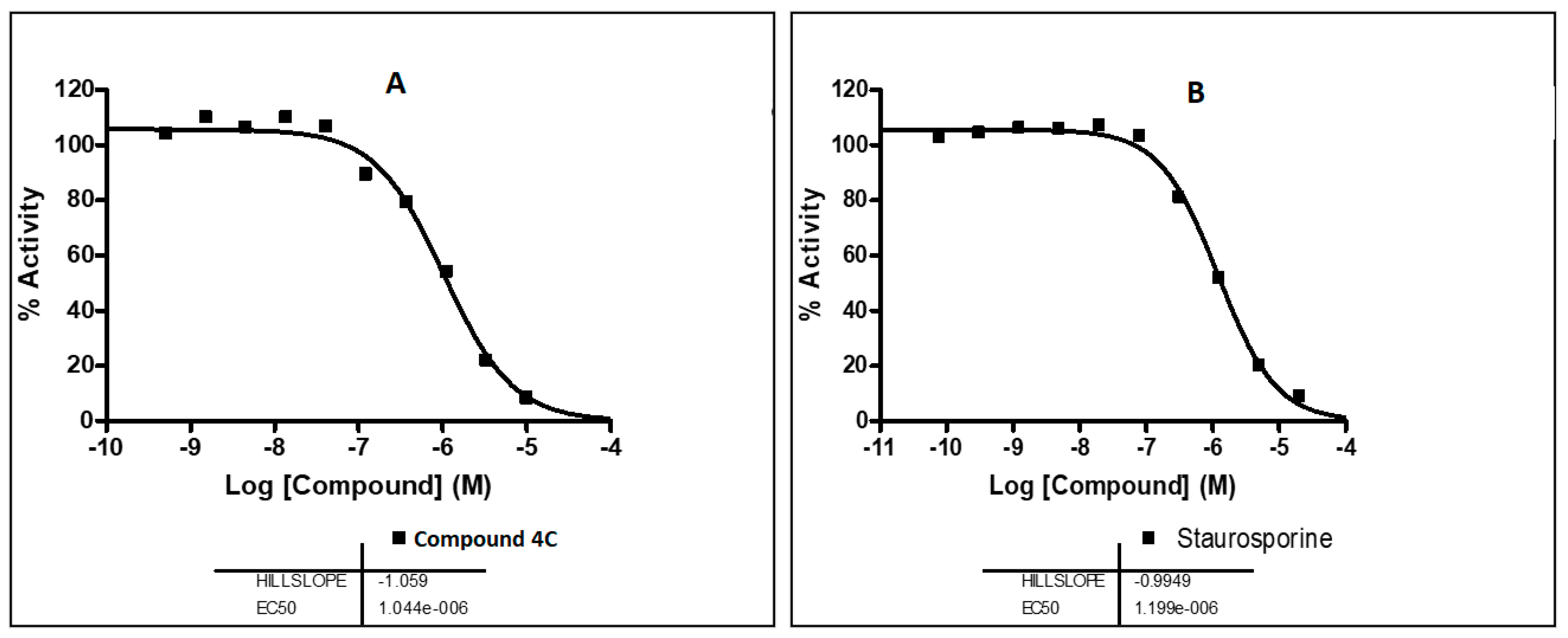
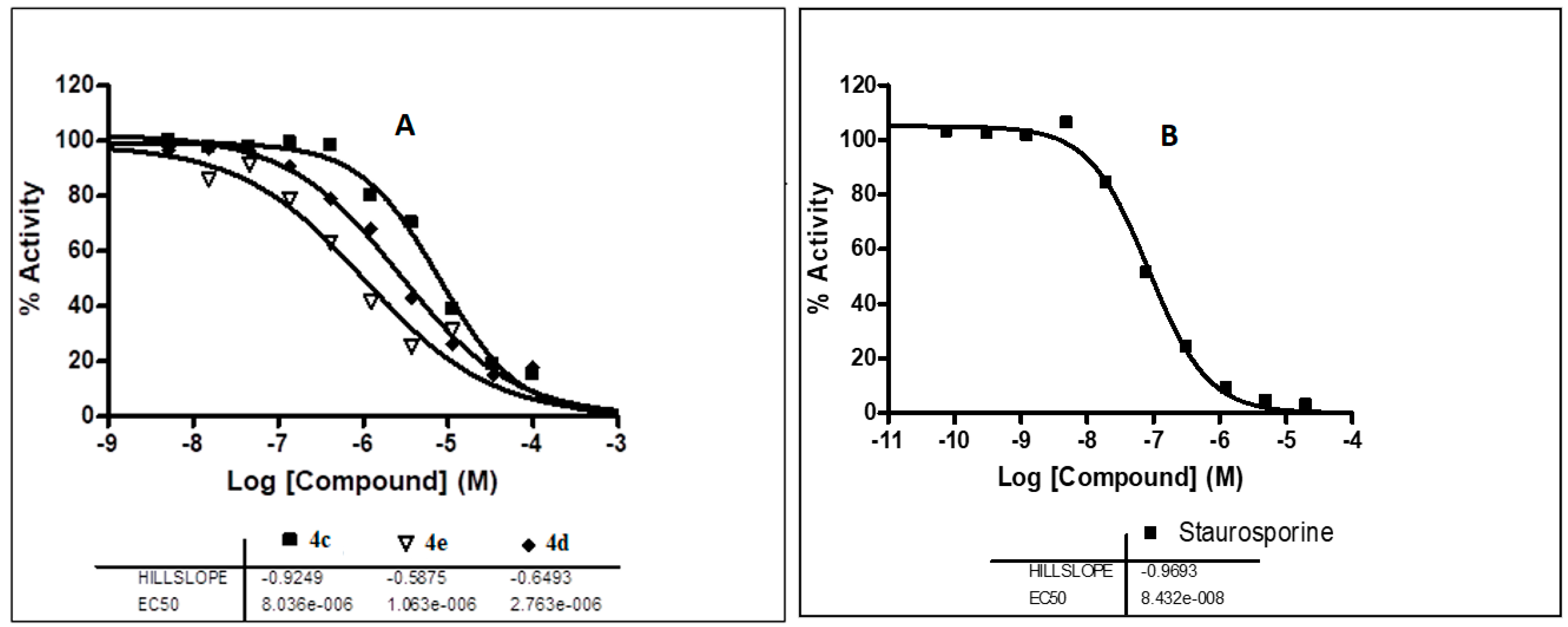
2.5.1. Assessment of the EphB3 Inhibitory Activity
2.5.2. Assessment of the EGFR Inhibitory Activity
2.5.3. Selectivity Assay of Compound 4c over a Kinase Panel
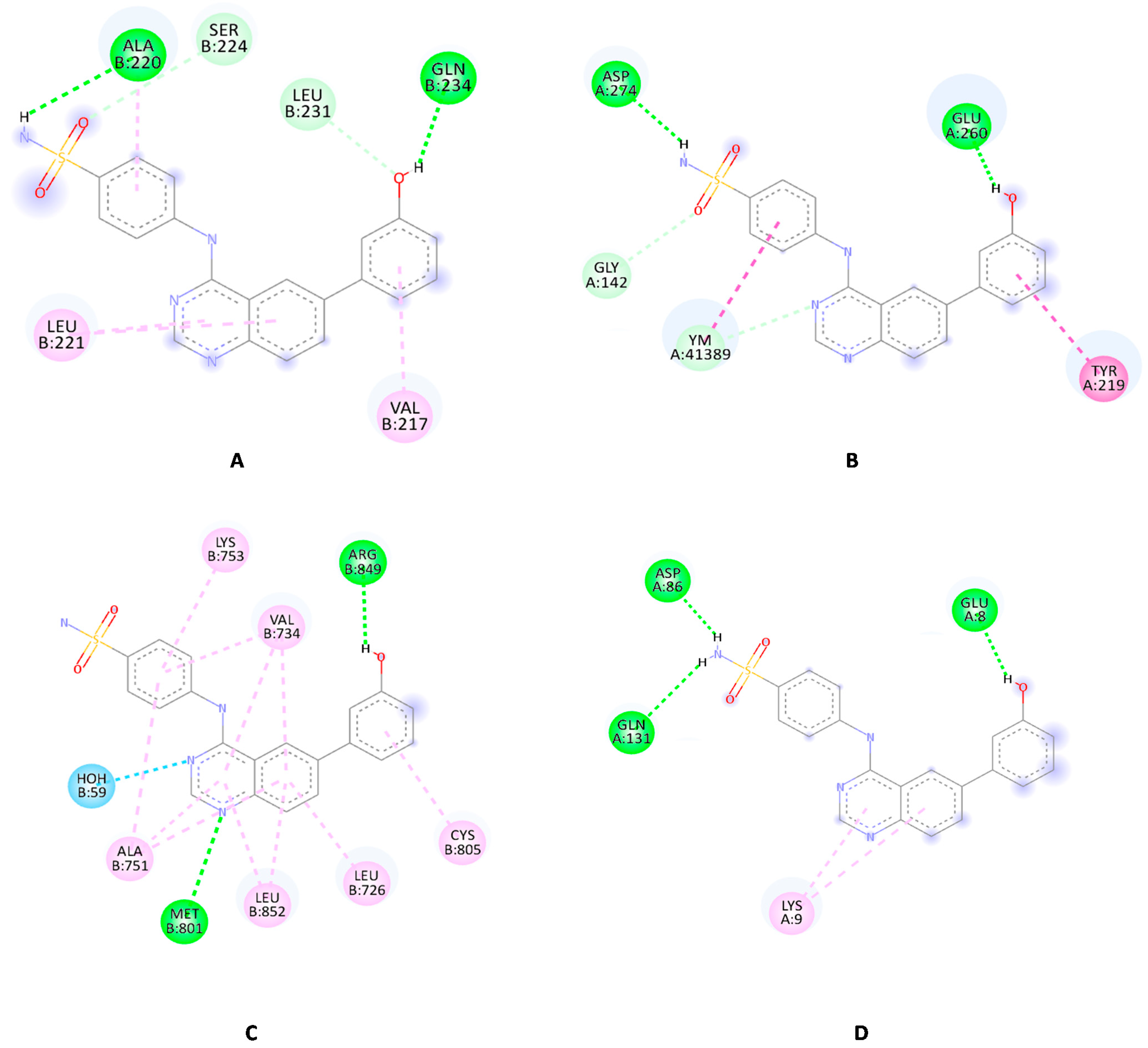
2.6. Molecular Docking Study
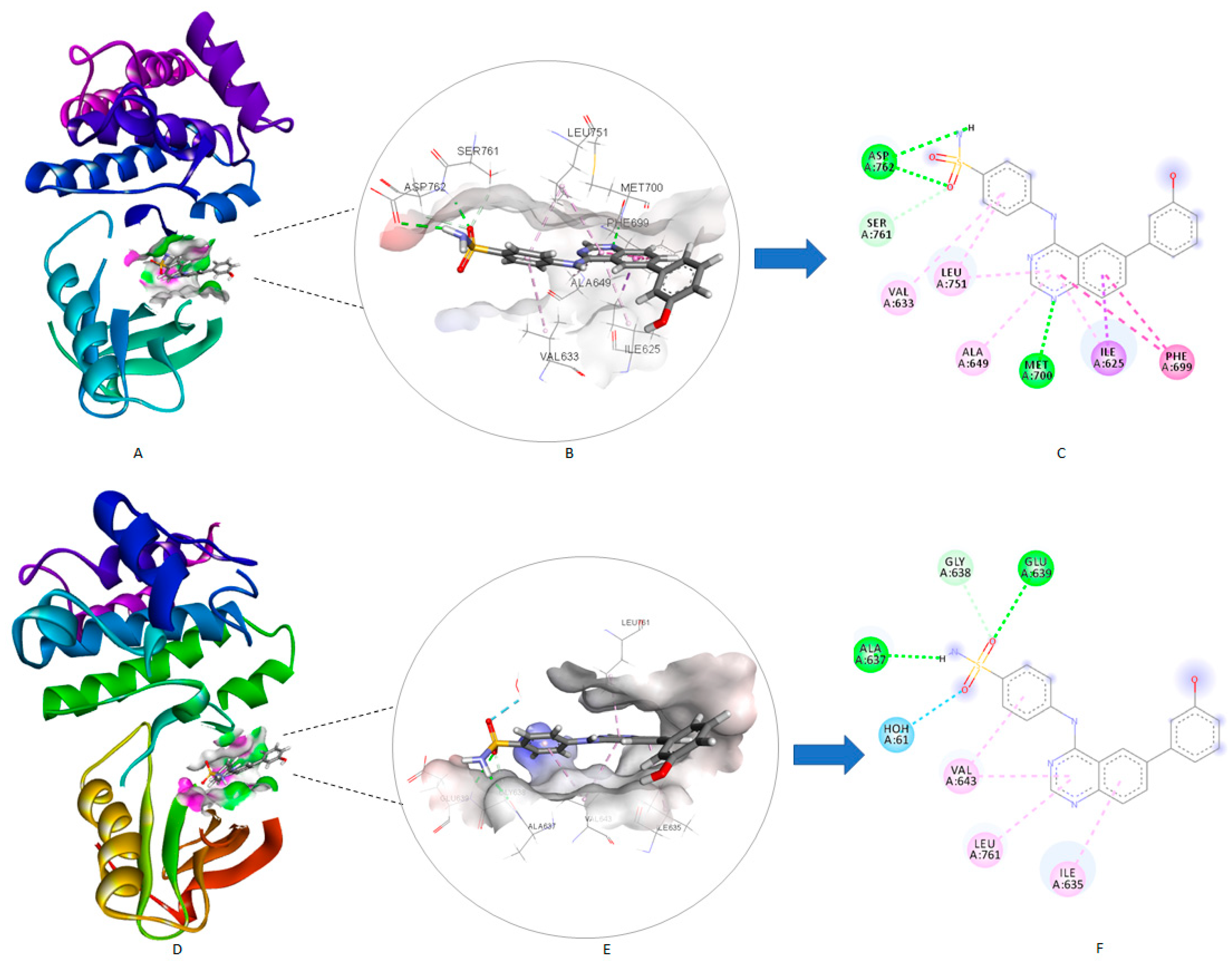
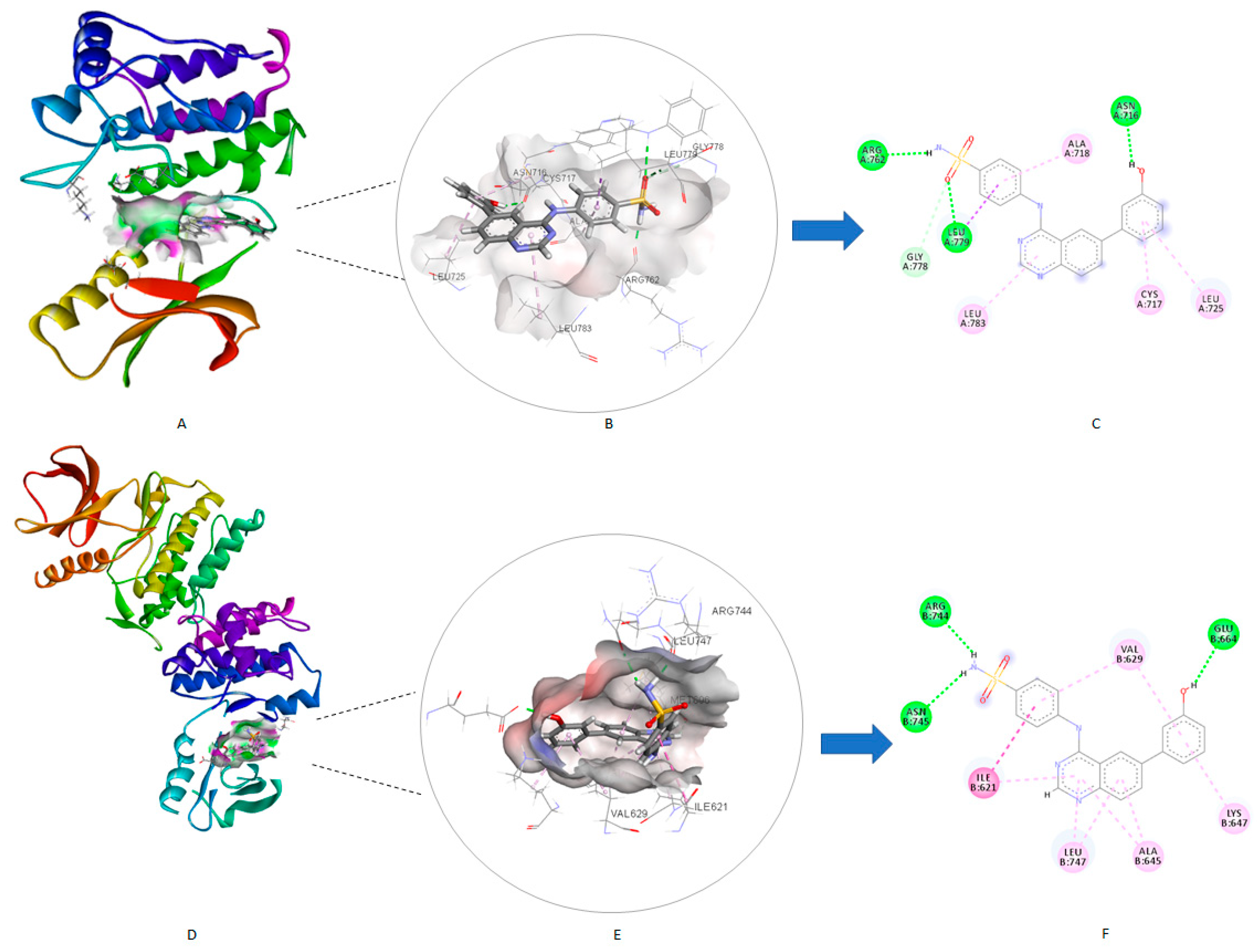
2.6.1. Molecular Docking Study for Compound 4c over the EphB Isoforms
2.6.2. Molecular Docking Study for Compound 4c over the Tested Kinase Panel
3. Materials and Methods
3.1. Protein Preparation
3.2. Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) Calculations
3.3. Chemistry
3.3.1. General Procedure for Synthesis of Compounds 2a–b
3.3.2. General Procedure for Synthesis of Compounds 3a–d
4-((6-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3a)
3-((6-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3b)
4-((7-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3c)
3-((7-Bromoquinazolin-4-yl)amino)benzenesulfonamide (3d)
3.3.3. General Procedure for Synthesis of Compounds 4a–f
4-((6-(Pyridin-3-yl)quinazolin-4-yl)amino)benzenesulfonamide (4a)
4-((6-(5-Formylfuran-2-yl)quinazolin-4-yl)amino)benzenesulfonamide (4b)
4-((6-(3-Hydroxyphenyl)quinazolin-4-yl)amino)benzenesulfonamide (4c)
4-((7-(4-(Morpholinomethyl)phenyl)quinazolin-4-yl)amino)benzenesulfonamide (4d)
3-((7-(3-(Morpholinomethyl)phenyl)quinazolin-4-yl)amino)benzenesulfonamide (4e)
3-((6-(Pyridin-3-yl)quinazolin-4-yl)amino)benzenesulfonamide (4f)
3.4. In Vitro Kinase Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Vaught, D.; Brantley-Sieders, D.M.; Chen, J. Eph receptors in breast cancer: Roles in tumor promotion and tumor suppression. Breast Cancer Res. 2008, 10, 217. [Google Scholar] [CrossRef] [Green Version]
- Willson, C.A.; Foster, R.D.; Onifer, S.M.; Whittemore, S.R.; Miranda, J.D. EphB3 receptor and ligand expression in the adult rat brain. J. Mol. Histol. 2006, 37, 369–380. [Google Scholar] [CrossRef]
- Darling, T.K.; Lamb, T.J. Emerging Roles for Eph Receptors and Ephrin Ligands in Immunity. Front. Immunol. 2019, 10, 1473. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.D.; Li, G.; Feng, Y.X.; Zhao, J.S.; Li, J.J.; Sun, Z.J.; Shi, S.; Deng, Y.Z.; Xu, J.F.; Zhu, Y.Q.; et al. EphB3 is overexpressed in non-small-cell lung cancer and promotes tumor metastasis by enhancing cell survival and migration. Cancer Res. 2011, 71, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Na, Y.J.; Jeong, Y.A.; Kim, J.L.; Oh, S.C.; Lee, D.H. Upregulation of EphB3 in gastric cancer with acquired resistance to a FGFR inhibitor. Int. J. Biochem. Cell Biol. 2018, 102, 128–137. [Google Scholar] [CrossRef]
- Jang, B.G.; Kim, H.S.; Bae, J.M.; Kim, W.H.; Hyun, C.L.; Kang, G.H. Expression Profile and Prognostic Significance of EPHB3 in Colorectal Cancer. Biomolecules 2020, 10, 602. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Griego, A.; Lennon, S.; Oweida, A.; Sharma, J.; Rohmer, C.; Uyanga, N.; Bukkapatnam, S.; Van Court, B.; Raben, D.; et al. Role of EphB3 Receptor in Mediating Head and Neck Tumor Growth, Cell Migration, and Response to PI3K Inhibitor. Mol. Cancer Ther. 2018, 17, 2049–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; He, J.; Wang, Y.F.; Jin, S.D.; Fan, Y.; Fang, N.; Qian, J.; Xu, T.P.; Guo, R.H. EZH2-mediated epigenetic suppression of EphB3 inhibits gastric cancer proliferation and metastasis by affecting E-cadherin and vimentin expression. Gene 2019, 686, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Sun, Z.J.; Yuan, Y.M.; Yin, F.F.; Bian, Y.G.; Long, L.Y.; Zhang, X.L.; Xie, D. EphB3 Stimulates Cell Migration and Metastasis in a Kinase-dependent Manner through Vav2-Rho GTPase Axis in Papillary Thyroid Cancer. J. Biol. Chem. 2017, 292, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, X.; Li, Y.; Wang, Y.; Liang, H.; Li, K.; Li, L.; Chen, C.; Sun, W.; Ren, S.; et al. EphB3-targeted regulation of miR-149 in the migration and invasion of human colonic carcinoma HCT116 and SW620 cells. Cancer Sci. 2017, 108, 408–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herath, N.I.; Boyd, A.W. The role of Eph receptors and ephrin ligands in colorectal cancer. Int. J. Cancer 2010, 126, 2003–2011. [Google Scholar] [CrossRef]
- Chiu, S.-T.; Chang, K.-J.; Ting, C.-H.; Shen, H.-C.; Li, H.; Hsieh, F.-J. Over-expression of EphB3 enhances cell–cell contacts and suppresses tumor growth in HT-29 human colon cancer cells. Carcinogenesis 2009, 30, 1475–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Ji, X.-D.; Gao, H.; Zhao, J.-S.; Xu, J.-F.; Sun, Z.-J.; Deng, Y.-Z.; Shi, S.; Feng, Y.-X.; Zhu, Y.-Q.; et al. EphB3 suppresses non-small-cell lung cancer metastasis via a PP2A/RACK1/Akt signalling complex. Nat. Commun. 2012, 3, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
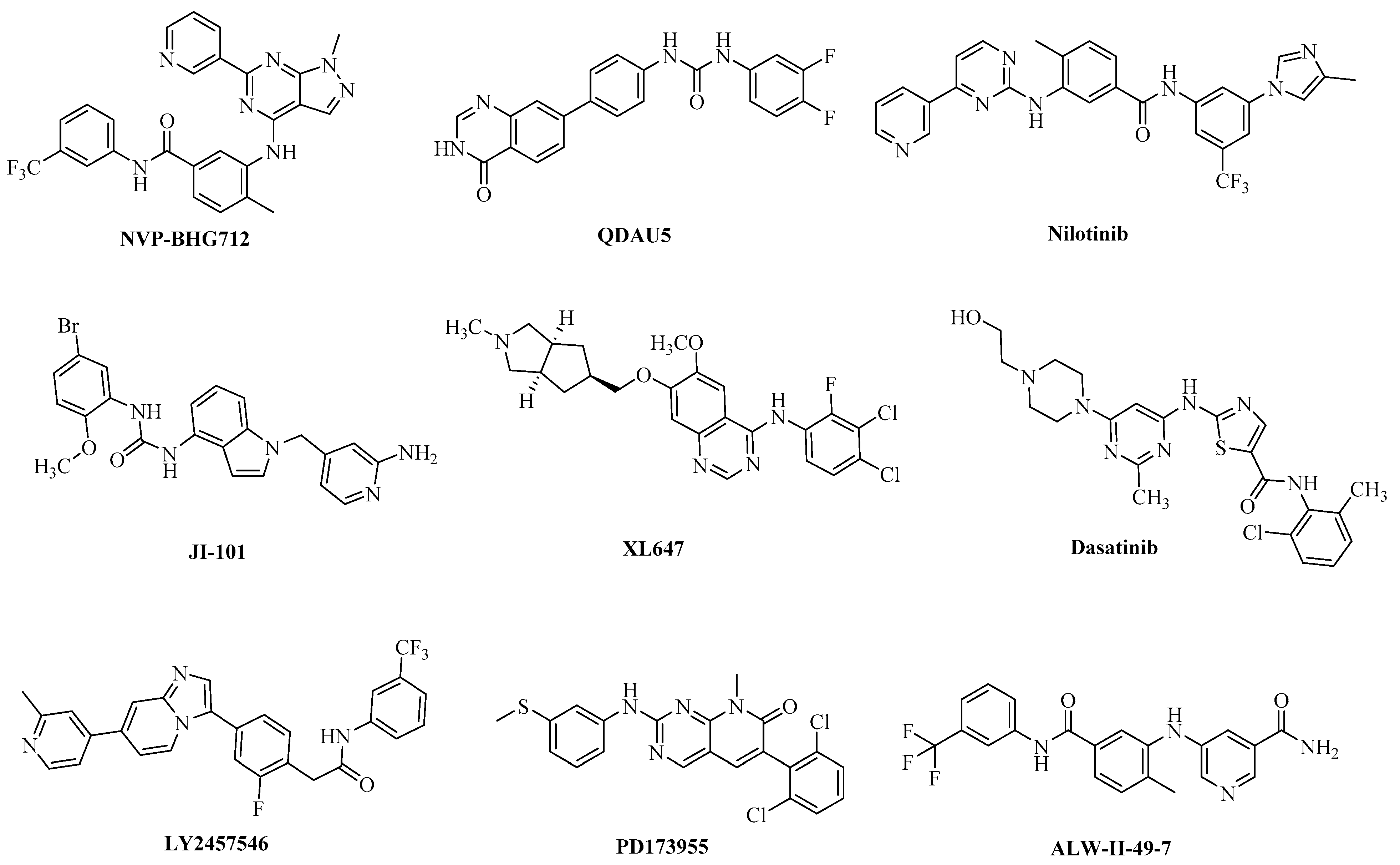
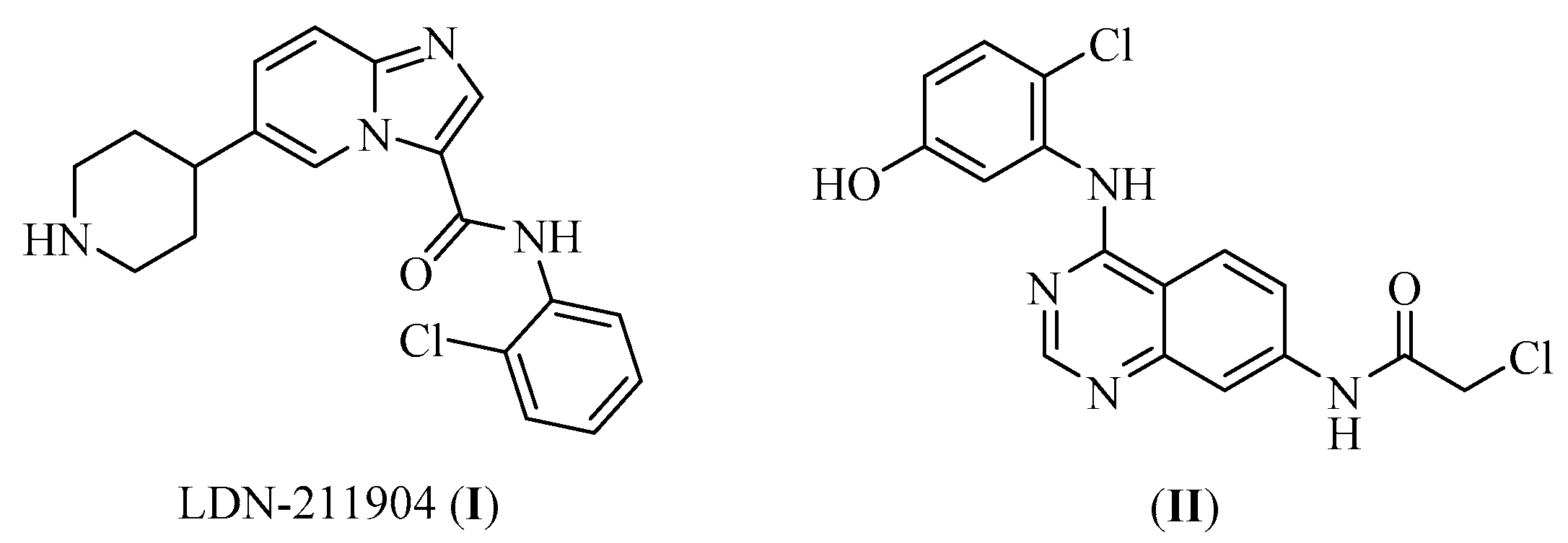
- Kung, A.; Chen, Y.-C.; Schimpl, M.; Ni, F.; Zhu, J.; Turner, M.; Molina, H.; Overman, R.; Zhang, C. Development of Specific, Irreversible Inhibitors for a Receptor Tyrosine Kinase EphB3. J. Am. Chem. Soc. 2016, 138, 10554–10560. [Google Scholar] [CrossRef]
- Montero-Herradón, S.; García-Ceca, J.; Zapata, A.G. EphB receptors, mainly EphB3, contribute to the proper development of cortical thymic epithelial cells. Organogenesis 2017, 13, 192–211. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, R.; Xiong, L.; Miao, X.; Li, D.; Zou, Q.; Yuan, Y.; Yang, Z. Prognostic and Clinicopathological Significance of EphB3 and Dysadherin Expression in Extrahepatic Cholangiocarcinoma. Cancer Manag. Res. 2020, 12, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Martiny-Baron, G.; Holzer, P.; Billy, E.; Schnell, C.; Brueggen, J.; Ferretti, M.; Schmiedeberg, N.; Wood, J.M.; Furet, P.; Imbach, P. The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis 2010, 13, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Shan, Y.; Ji, X.; Zhu, M.; Li, C.; Sun, Y.; Si, R.; Pan, X.; Wang, J.; Ma, W.; et al. Discovery and evaluation of triple inhibitors of VEGFR-2, TIE-2 and EphB4 as anti-angiogenic and anti-cancer agents. Oncotarget 2017, 8, 104745–104760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, T.L.; Wade, M.L.; Agarwal, N.; Boucher, K.; Patel, J.; Luebke, A.; Sharma, S. A pilot study of JI-101, an inhibitor of VEGFR-2, PDGFR-β, and EphB4 receptors, in combination with everolimus and as a single agent in an ovarian cancer expansion cohort. Investig. New Drugs 2015, 33, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Lynch, T.J., Jr.; Lara, P.N., Jr.; Cho, J.; Yanagihara, R.H.; Vrindavanam, N.; Chowhan, N.M.; Gadgeel, S.M.; Pennell, N.A.; Funke, R.; et al. XL647—A multitargeted tyrosine kinase inhibitor: Results of a phase II study in subjects with non-small cell lung cancer who have progressed after responding to treatment with either gefitinib or erlotinib. J. Thorac. Oncol. 2012, 7, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Du, E.; Li, X.; He, S.; Li, X.; He, S. The critical role of the interplays of EphrinB2/EphB4 and VEGF in the induction of angiogenesis. Mol. Biol. Rep. 2020, 47, 4681–4690. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, T.P.; Clayton, J.R.; Rempala, M.E.; Henry, J.R.; Knobeloch, J.M.; Mendel, D.; McLean, J.A.; Hao, Y.; Barda, D.A.; Considine, E.L.; et al. Discovery of LY2457546: A multi-targeted anti-angiogenic kinase inhibitor with a novel spectrum of activity and exquisite potency in the acute myelogenous leukemia-Flt-3-internal tandem duplication mutant human tumor xenograft model. Investig. New Drugs 2012, 30, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.; Molz, L.; Liu, Q.; Kaplan, F.; Xu, J.P.; Majeti, J.Z.; Ramos-Kelsey, R.; Murthi, K.; Lievens, S.; Tavernier, J.; et al. MASPIT: Three-hybrid trap for quantitative proteome fingerprinting of small molecule-protein interactions in mammalian cells. Chem. Biol. 2006, 13, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Syeda, F.; Walker, J.R.; Finerty, P.J., Jr.; Cuerrier, D.; Wojciechowski, A.; Liu, Q.; Dhe-Paganon, S.; Gray, N.S. Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4467–4470. [Google Scholar] [CrossRef] [Green Version]
- Noberini, R.; Lamberto, I.; Pasquale, E.B. Targeting Eph receptors with peptides and small molecules: Progress and challenges. Semin. Cell Dev. Biol. 2012, 23, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Boyd, A.W.; Bartlett, P.F.; Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2014, 13, 39–62. [Google Scholar] [CrossRef]
- Qiao, L.; Choi, S.; Case, A.; Gainer, T.G.; Seyb, K.; Glicksman, M.A.; Lo, D.C.; Stein, R.L.; Cuny, G.D. Structure–activity relationship study of EphB3 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6122–6126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, Y.; Nakano, M.; Sato, H.; Truesdale, A.T.; Stuart, J.D.; Nartey, E.N.; Hightower, K.E.; Kane-Carson, L. Design and effective synthesis of novel templates, 3,7-diphenyl-4-amino-thieno and furo-[3,2-c]pyridines as protein kinase inhibitors and in vitro evaluation targeting angiogenetic kinases. Bioorg. Med. Chem. Lett. 2007, 17, 250–254. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Sirous, H.; Chemi, G.; Campiani, G.; Brogi, S. An integrated in silico screening strategy for identifying promising disruptors of p53-MDM2 interaction. Comput. Biol. Chem. 2019, 83, 107105. [Google Scholar] [CrossRef]
- Ravez, S.; Castillo-Aguilera, O.; Depreux, P.; Goossens, L. Quinazoline derivatives as anticancer drugs: A patent review (2011–present). Expert Opin. Ther. Pat. 2015, 25, 789–804. [Google Scholar] [CrossRef]
- Li, H.-Q.; Li, D.-D.; Lu, X.; Xu, Y.-Y.; Zhu, H.-L. Design and synthesis of 4,6-substituted-(diaphenylamino)quinazolines as potent EGFR inhibitors with antitumor activity. Bioorg. Med. Chem. 2012, 20, 317–323. [Google Scholar] [CrossRef]
- Cruz-Lopez, O.; Conejo-Garcia, A.; Nunez, M.C.; Kimatrai, M.; Garcia-Rubino, M.E.; Morales, F.; Gomez-Perez, V.; Campos, J.M. Novel Substituted Quinazolines for Potent EGFR Tyrosine Kinase Inhibitors. Curr. Med. Chem. 2011, 18, 943–963. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Hassan, A.H.E.; Paik, S.; Sup Lee, Y.; Lee, H.H.; Shin, J.S.; Lee, K.T.; Roh, E.J. EGFR inhibitors from cancer to inflammation: Discovery of 4-fluoro-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide as a novel anti-inflammatory EGFR inhibitor. Bioorg. Chem. 2019, 86, 112–118. [Google Scholar] [CrossRef]
- Karaman, M.; Herrgard, S.; Treiber, D.; Gallant, P.; Atteridge, C.; Campbell, B.; Chan, K.; Ciceri, P.; Davis, M.; Edeen, P.J.N.B.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Bembenek, S.D.; Hirst, G.; Mirzadegan, T. Determination of a Focused Mini Kinase Panel for Early Identification of Selective Kinase Inhibitors. J. Chem. Inf. Model. 2018, 58, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Treiber, D.K.; Zuercher, W.J. Utilizing comprehensive and mini-kinome panels to optimize the selectivity of quinoline inhibitors for cyclin G associated kinase (GAK). Bioorg. Med. Chem. Lett. 2019, 29, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Brandt, P.; Jensen, A.J.; Nilsson, J. Small kinase assay panels can provide a measure of selectivity. Bioorg. Med. Chem. Lett. 2009, 19, 5861–5863. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.; Biggs, W.; Treiber, D.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Wang, P.; Nguyen, N.U.N.; Nakada, Y.; Menendez-Montes, I.; Ismail, M.; Bachoo, R.; Henkemeyer, M.; Sadek, H.A.; Kandil, E.S. Identification of tetracycline combinations as EphB1 tyrosine kinase inhibitors for treatment of neuropathic pain. Proc. Natl. Acad. Sci. USA 2021, 118, e2016265118. [Google Scholar] [CrossRef]
- Wiesner, S.; Wybenga-Groot, L.E.; Warner, N.; Lin, H.; Pawson, T.; Forman-Kay, J.D.; Sicheri, F. A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 2006, 25, 4686–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, R.C.; Debreczeni, J.E.; Truman, C.M.; McAlister, M.S.; Attwood, T.K. Completing the structural family portrait of the human EphB tyrosine kinase domains. Protein Sci. 2014, 23, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguro, Y.; Miyamoto, N.; Okada, K.; Takagi, T.; Iwata, H.; Awazu, Y.; Miki, H.; Hori, A.; Kamiyama, K.; Imamura, S. Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5H-pyrrolo [3,2-d]pyrimidine derivatives: Novel VEGFR2 kinase inhibitors binding to inactive kinase conformation. Bioorg. Med. Chem. 2010, 18, 7260–7273. [Google Scholar] [CrossRef]
- Bavetsias, V.; Large, J.M.; Sun, C.; Bouloc, N.; Kosmopoulou, M.; Matteucci, M.; Wilsher, N.E.; Martins, V.; Reynisson, J.; Atrash, B.; et al. Imidazo[4,5-b]pyridine Derivatives As Inhibitors of Aurora Kinases: Lead Optimization Studies toward the Identification of an Orally Bioavailable Preclinical Development Candidate. J. Med. Chem. 2010, 53, 5213–5228. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Shi, W.-W.; He, Y.-X.; Yang, Y.-H.; Zhou, C.-Z.; Chen, Y. Structures of the substrate-binding protein provide insights into the multiple compatible solute binding specificities of the Bacillus subtilis ABC transporter OpuC. Biochem. J. 2011, 436, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.-H. Inhibition of Cyclin-Dependent Kinases by Purine Analogues. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.-C.; Wolf, R.M. MM/GBSA Binding Energy Prediction on the PDBbind Data Set: Successes, Failures, and Directions for Further Improvement. J. Chem. Inf. Model. 2013, 53, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Kim, N.Y.; Hassan, A.H.E.; Park, J.E.; Yang, J.E.; Oh, K.S.; Lee, B.H.; Lee, M.Y.; Shin, K.J.; Lee, K.T.; et al. Design, synthesis and biological evaluation of novel thiazolidinedione derivatives as irreversible allosteric IKK-β modulators. Eur. J. Med. Chem. 2018, 157, 691–704. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Farag, A.K.; Viswanath, A.N.I.; Bedair, T.M.; Leem, D.G.; Lee, K.-T.; Pae, A.N.; Roh, E.J. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: Design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg. Med. Chem. Lett. 2015, 25, 5147–5154. [Google Scholar] [CrossRef]
- Yoshida, S.; Morita, T.; Hosoya, T. Synthesis of Diverse Benzotriazoles from Aryne Precursors Bearing an Azido Group via Inter- and Intramolecular Cycloadditions. Chem. Lett. 2016, 45, 726–728. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hit Cpd | GlideScore (kcal/mol) | Hit Cpd | GlideScore (kcal/mol) |
|---|---|---|---|
| 1 | −7.30 | 18 | −5.78 |
| 2 | −7.16 | 19 | −5.63 |
| 3 | −6.65 | 20 | −5.50 |
| 4 | −6.47 | 21 | −5.49 |
| 5 | −6.23 | 22 | −5.47 |
| 6 | −6.21 | 23 | −5.46 |
| 7 | −6.20 | 24 | −5.35 |
| 8 | −6.18 | 25 | −5.35 |
| 9 | −6.13 | 26 | −5.32 |
| 10 | −6.12 | 27 | −5.31 |
| 11 | −6.11 | 28 | −5.16 |
| 12 | −6.10 | 29 | −5.09 |
| 13 | −6.07 | 30 | −4.80 |
| 14 | −6.06 | 31 | −4.59 |
| 15 | −5.90 | 32 | −4.49 |
| 16 | −5.84 | 33 | −4.24 |
| 17 | −5.84 | 34 | −4.22 |
| Cpd | GlideScore (kcal/mol) | 2D Diagram | Amino Acids | Binding Group | Molecular Interactions |
|---|---|---|---|---|---|
| 3a | −6.20 |  | Arg762 Arg781 Ser721 Leu779 Ala718 Leu783 | NH (SO2NH2) NH (amide) Benzene Benzene Benzene Benzene | H-bond H-bond π-donor H-bond π-Alkyl π-Alkyl π-Alkyl |
| 4a | −6.47 |  | Asp776 Asp758 Leu779 Leu765 Ala718 Leu783 | NH (SO2NH2) NH (SO2NH2) Benzene Benzene Benzene Benzene | H-bond H-bond C-H-bond π-Alkyl π-Alkyl π-Alkyl |
| 4b | −6.21 |  | Asp776 Asp758 Leu779 Leu765 Ala718 Leu783 | NH (SO2NH2) O (SO2NH2) Benzene Benzene Benzene Benzene | H-bond H-bond π-donor H-bond π-Alkyl π-Alkyl π-Alkyl |
| 4c | −7.16 |  | Arg762 Asn716 Leu779 Gly778 Ala718 Leu783 Cys717 Leu725 | NH (SO2NH2) OH O (SO2NH2) O (SO2NH2) Benzene Benzene Benzene Benzene | H-bond H-bond H-bond C-H-bond π-Alkyl π-Alkyl π-Alkyl π-Alkyl |
| 4d | −6.65 |  | Ser721 Leu779 Asp776 Gly778 Asp758 Leu783 Arg762 | NH (SO2NH2) H (morpholine) H (morpholine) N (quinazoline) H (morpholine) Benzene Benzene | H-bond H-bond H-bond C-H-bond C-H-bond π-Alkyl π-Alkyl |
| 4e | −7.30 |  | Asp758 Asp776 Gly778 Leu779 Ser775 Ala718 Arg781 | NH (SO2NH2) NH (SO2NH2) N (quinazoline) NH (amide) H (quinazoline) Benzene Benzene | H-bond H-bond H-bond H-bond C-H-bond π-Alkyl π-Alkyl |
| 4f | −6.23 |  | Asp776 Gly778 Phe777 Leu779 Leu765 Leu783 | NH (SO2NH2) O (SO2NH2) O (SO2NH2) Benzene Benzene Benzene | H-bond H-bond H-bond π-donor H-bond π-Alkyl π-Alkyl |
| Cpd | MM-GBSA (kcal/mol) |
|---|---|
| 3a | −53.14 |
| 4a | −52.09 |
| 4b | −35.79 |
| 4c | −74.13 |
| 4d | −57.47 |
| 4e | −63.69 |
| 4f | −40.08 |
| Cpd | Structure | Average % Enzyme Inhibition at 10 µM (Relative to DMSO Control) |
|---|---|---|
| 3a |  | 26.97 ± 0.24 |
| 4a |  | 25.81 ± 0.24 |
| 4b |  | 26.71 ± 0.45 |
| 4c |  | 93.27 ± 0.06 |
| 4d |  | 18.86 ± 1.09 |
| 4e |  | −21.35 ± 0.09 |
| 4f |  | 23.48 ± 5.36 |
| Cpd | Average % Enzyme Inhibition (Relative to DMSO Controls) at 10 µM |
|---|---|
| 3a | −13.16 ± 4.31 |
| 4a | 0.56 ± 4.32 |
| 4b | 25.25 ± 1.25 |
| 4c | 46.78 ± 0.54 |
| 4d | 65.76 ± 0.98 |
| 4e | 63.28 ± 0.21 |
| 4f | 4.57 ± 2.16 |
| Staurosporine | - |
| Kinase | Average % Enzyme Inhibition (Relative to DMSO Controls) at 10 µM |
|---|---|
| Aurora A | 67.86 ± 4.93 |
| CDK2/cyclin A | 74.96 ± 2.07 |
| EphB1 | 58.89 ± 1.98 |
| EphB2 | 76.32 ± 0.61 |
| EphB3 | 93.27 ± 0.06 |
| EphB4 | 70.15 ± 3.64 |
| ERBB2/HER2 | 67.75 ± 0.99 |
| KDR/VEGFR2 | ±0.66 |
| Kinase | 4c IC50 (µM) | Staurosporine IC50 (µM) |
|---|---|---|
| EphB1 | 5.86 | 0.03 |
| EphB2 | 3.95 | 0.08 |
| EphB3 | 1.04 | 1.20 |
| EphB4 | 3.98 | 0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.; Nada, H.; Byun, H.J.; Lee, C.H.; Elkamhawy, A. Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies. Pharmaceuticals 2021, 14, 1247. https://doi.org/10.3390/ph14121247
Lee K, Nada H, Byun HJ, Lee CH, Elkamhawy A. Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies. Pharmaceuticals. 2021; 14(12):1247. https://doi.org/10.3390/ph14121247
Chicago/Turabian StyleLee, Kyeong, Hossam Nada, Hyun Jung Byun, Chang Hoon Lee, and Ahmed Elkamhawy. 2021. "Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies" Pharmaceuticals 14, no. 12: 1247. https://doi.org/10.3390/ph14121247
APA StyleLee, K., Nada, H., Byun, H. J., Lee, C. H., & Elkamhawy, A. (2021). Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies. Pharmaceuticals, 14(12), 1247. https://doi.org/10.3390/ph14121247