
Limited Sampling Strategy for Determination of Ibrutinib Plasma Exposure: Joint Analyses with Metabolite Data

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Patients
2.1.1. Development Cohort
2.1.2. Validation Cohort
2.2. Actual AUC
2.3. Correlation between trough Ibrutinib Concentrations and Actual AUC
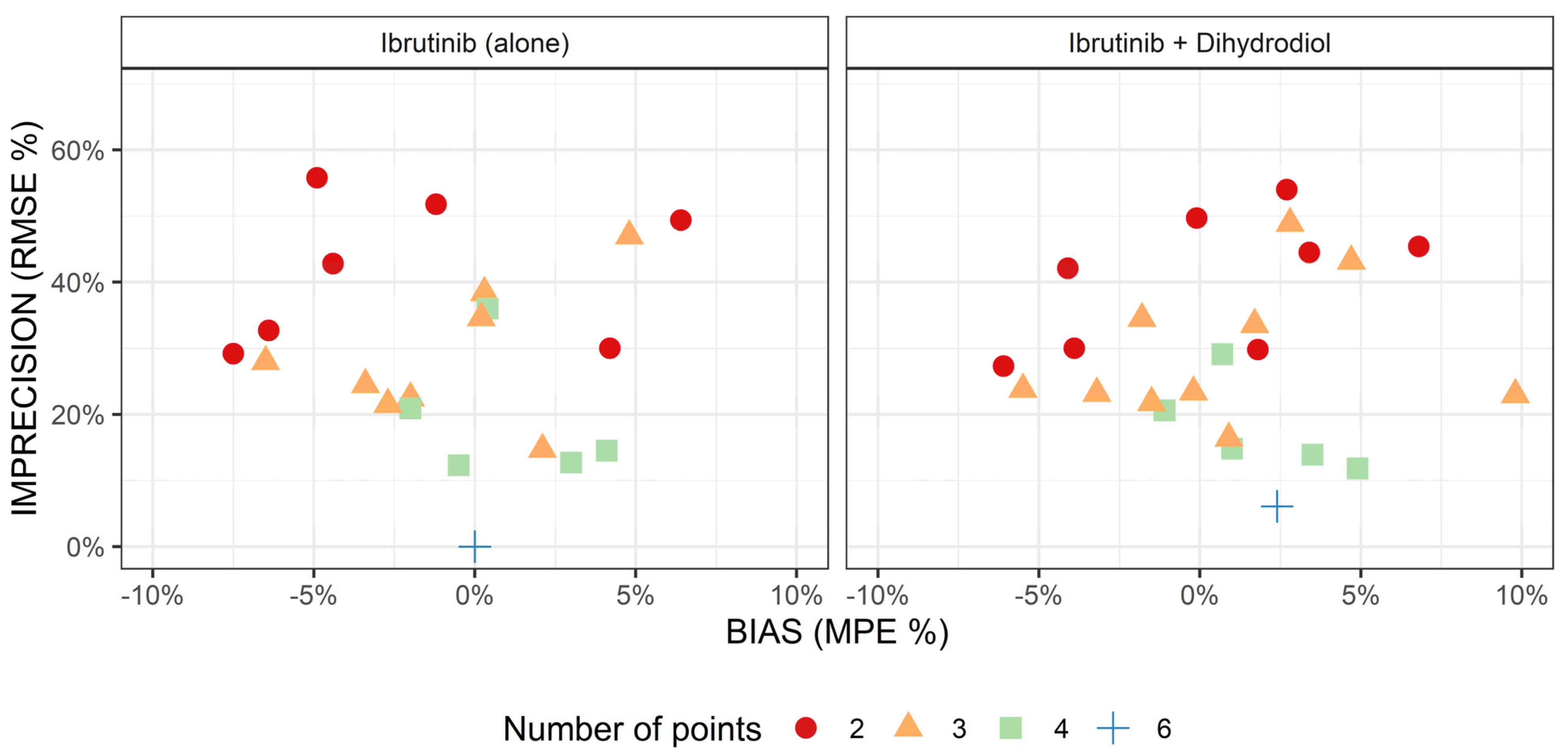
2.4. Limited Sampling Strategies for Determination of Ibrutinib AUC
2.4.1. Development
2.4.2. Prospective Validation
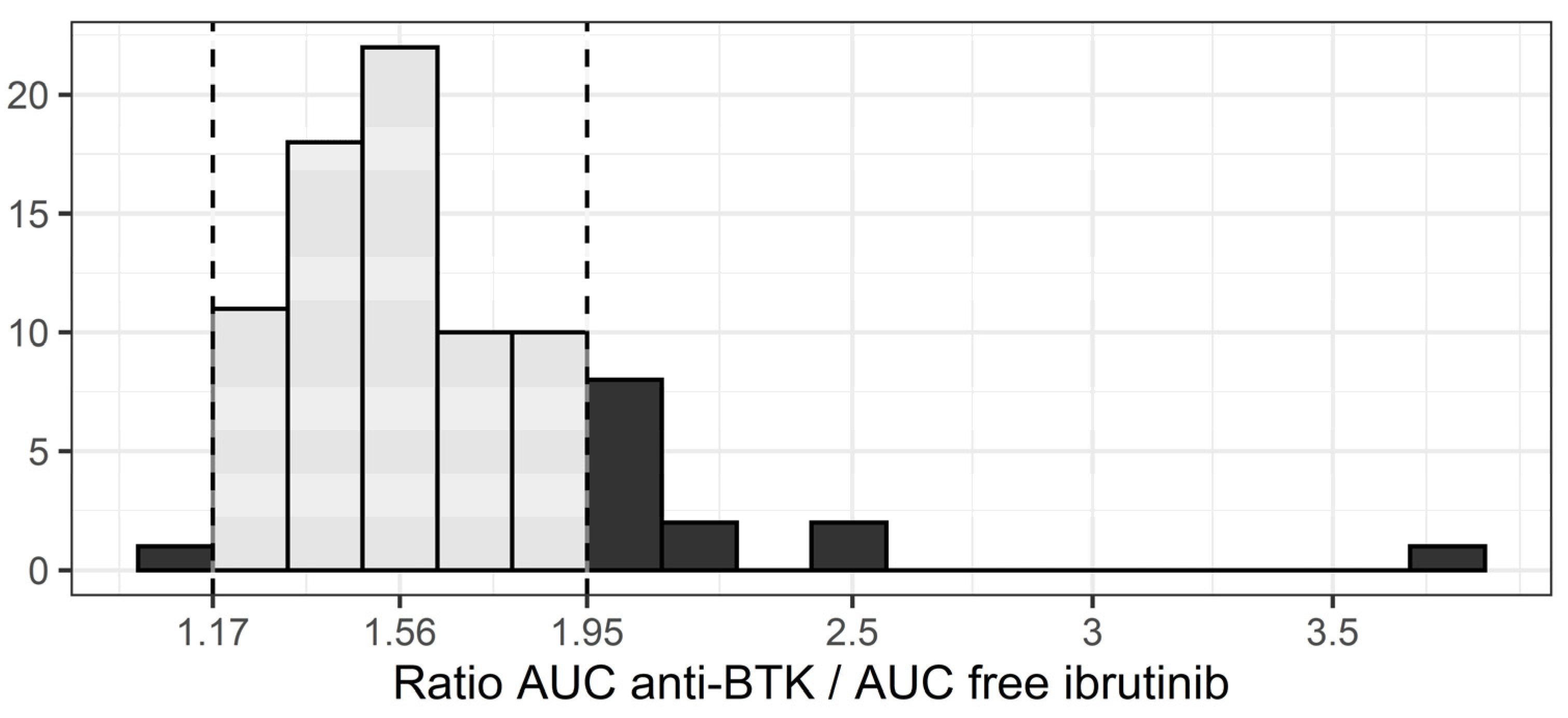
2.5. Anti-BTK AUC
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Pharmacokinetic Data
4.3. Pharmacokinetic Model
4.4. AUC Computation
4.5. Bayesian Analysis
4.6. Actual AUC
4.7. Correlation between trough Ibrutinib Concentrations and Actual AUC
4.8. Limited Sampling Strategy
4.9. Validation
4.10. Anti-BTK AUC
4.11. Software
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Picard, S.; Titier, K.; Etienne, G.; Teilhet, E.; Ducint, D.; Bernard, M.-A.; Lassalle, R.; Marit, G.; Reiffers, J.; Begaud, B.; et al. Trough Imatinib Plasma Levels Are Associated with Both Cytogenetic and Molecular Responses to Standard-Dose Imatinib in Chronic Myeloid Leukemia. Blood 2007, 109, 3496–3499. [Google Scholar] [CrossRef] [Green Version]
- Bouchet, S.; Poulette, S.; Titier, K.; Moore, N.; Lassalle, R.; Abouelfath, A.; Italiano, A.; Chevreau, C.; Bompas, E.; Collard, O.; et al. Relationship between Imatinib Trough Concentration and Outcomes in the Treatment of Advanced Gastrointestinal Stromal Tumours in a Real-Life Setting. Eur. J. Cancer 2016, 57, 31–38. [Google Scholar] [CrossRef]
- García-Ferrer, M.; Wojnicz, A.; Mejía, G.; Koller, D.; Zubiaur, P.; Abad-Santos, F. Utility of Therapeutic Drug Monitoring of Imatinib, Nilotinib, and Dasatinib in Chronic Myeloid Leukemia: A Systematic Review and Meta-Analysis. Clin. Ther. 2019, 41, 2558–2570.e7. [Google Scholar] [CrossRef]
- Suttle, A.B.; Ball, H.A.; Molimard, M.; Hutson, T.E.; Carpenter, C.; Rajagopalan, D.; Lin, Y.; Swann, S.; Amado, R.; Pandite, L. Relationships between Pazopanib Exposure and Clinical Safety and Efficacy in Patients with Advanced Renal Cell Carcinoma. Br. J. Cancer 2014, 111, 1909–1916. [Google Scholar] [CrossRef] [Green Version]
- Houk, B.E.; Bello, C.L.; Poland, B.; Rosen, L.S.; Demetri, G.D.; Motzer, R.J. Relationship between Exposure to Sunitinib and Efficacy and Tolerability Endpoints in Patients with Cancer: Results of a Pharmacokinetic/Pharmacodynamic Meta-Analysis. Cancer Chemother. Pharm. 2010, 66, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Westerdijk, K.; Desar, I.M.E.; Steeghs, N.; van der Graaf, W.T.A.; Erp, N.P. van Imatinib, Sunitinib and Pazopanib: From Flat-Fixed Dosing towards a Pharmacokinetically Guided Personalized Dose. Br. J. Clin. Pharmacol. 2020, 86, 258–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medecines Agency. IMBRUVICA®(Ibrutinib): Summary of Product Characteristics; European Medecines Agency: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Gallais, F.; Ysebaert, L.; Despas, F.; De Barros, S.; Dupré, L.; Quillet-Mary, A.; Protin, C.; Thomas, F.; Obéric, L.; Allal, B.; et al. Population Pharmacokinetics of Ibrutinib and Its Dihydrodiol Metabolite in Patients with Lymphoid Malignancies. Clin. Pharm. 2020, 59, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Gallais, F.; Ysebaert, L.; Despas, F.; De Barros, S.; Obéric, L.; Allal, B.; Chatelut, E.; White-Koning, M. Population PK-PD Modelling of Circulating Lymphocyte Dynamics in Chronic Lymphocytic Leukemia Patients under Ibrutinib Treatment. Clin. Pharmacol. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Bose, P.; Cruz, N.D.; Jiang, Y.; Wu, Q.; Thompson, P.A.; Feng, S.; Kroll, M.H.; Qiao, W.; Huang, X.; et al. A Pilot Study of Lower Doses of Ibrutinib in Patients with Chronic Lymphocytic Leukemia. Blood 2018, 132, 2249–2259. [Google Scholar] [CrossRef] [Green Version]
- Verheijen, R.B.; Yu, H.; Schellens, J.H.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. Practical Recommendations for Therapeutic Drug Monitoring of Kinase Inhibitors in Oncology. Clin. Pharm. Ther. 2017, 102, 765–776. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research [CDER]. US FDA Clinical Pharmacology and Biopharmaceutics Review(s): Ibrutinib; CDER: Silver Spring, MD, USA, 2013. [Google Scholar]
- Scheers, E.; Leclercq, L.; de Jong, J.; Bode, N.; Bockx, M.; Laenen, A.; Cuyckens, F.; Skee, D.; Murphy, J.; Sukbuntherng, J.; et al. Absorption, Metabolism, and Excretion of Oral 14 C Radiolabeled Ibrutinib: An Open-Label, Phase I, Single-Dose Study in Healthy Men. Drug Metab. Dispos. 2015, 43, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Retmana, I.A.; Beijnen, J.H.; Sparidans, R.W. Chromatographic Bioanalytical Assays for Targeted Covalent Kinase Inhibitors and Their Metabolites. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1162, 122466. [Google Scholar] [CrossRef] [PubMed]
- Ezzeldin, E.; Iqbal, M.; Herqash, R.N.; ElNahhas, T. Simultaneous Quantitative Determination of Seven Novel Tyrosine Kinase Inhibitors in Plasma by a Validated UPLC-MS/MS Method and Its Application to Human Microsomal Metabolic Stability Study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1136, 121851. [Google Scholar] [CrossRef]
- Rood, J.J.M.; Dormans, P.J.A.; van Haren, M.J.; Schellens, J.H.M.; Beijnen, J.H.; Sparidans, R.W. Bioanalysis of Ibrutinib, and Its Dihydrodiol-and Glutathione Cycle Metabolites by Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1090, 14–21. [Google Scholar] [CrossRef]
- Huynh, H.H.; Pressiat, C.; Sauvageon, H.; Madelaine, I.; Maslanka, P.; Lebbé, C.; Thieblemont, C.; Goldwirt, L.; Mourah, S. Development and Validation of a Simultaneous Quantification Method of 14 Tyrosine Kinase Inhibitors in Human Plasma Using LC-MS/MS. Ther. Drug Monit. 2017, 39, 43–54. [Google Scholar] [CrossRef]
- Fouad, M.; Helvenstein, M.; Blankert, B. Ultra High Performance Liquid Chromatography Method for the Determination of Two Recently FDA Approved TKIs in Human Plasma Using Diode Array Detection. J. Anal. Methods Chem. 2015, 2015, 215128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantasiripitak, W.; Van Daele, R.; Gijsen, M.; Ferrante, M.; Spriet, I.; Dreesen, E. Software Tools for Model-Informed Precision Dosing: How Well Do They Satisfy the Needs? Front. Pharmacol. 2020, 11, 620. [Google Scholar] [CrossRef]
- Mato, A.R.; Nabhan, C.; Thompson, M.C.; Lamanna, N.; Brander, D.M.; Hill, B.; Howlett, C.; Skarbnik, A.; Cheson, B.D.; Zent, C.; et al. Toxicities and Outcomes of 616 Ibrutinib-Treated Patients in the United States: A Real-World Analysis. Haematologica 2018, 103, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Fancher, K.M.; Pappacena, J.J. Drug Interactions with Bruton’s Tyrosine Kinase Inhibitors: Clinical Implications and Management. Cancer Chemother. Pharm. 2020, 86, 507–515. [Google Scholar] [CrossRef]
- Benkali, K.; Prémaud, A.; Picard, N.; Rérolle, J.-P.; Toupance, O.; Hoizey, G.; Turcant, A.; Villemain, F.; Le Meur, Y.; Marquet, P.; et al. Tacrolimus Population Pharmacokinetic-Pharmacogenetic Analysis and Bayesian Estimation in Renal Transplant Recipients. Clin. Pharm. 2009, 48, 805–816. [Google Scholar] [CrossRef]
- Le Guellec, C.; Bourgoin, H.; Büchler, M.; Le Meur, Y.; Lebranchu, Y.; Marquet, P.; Paintaud, G. Population Pharmacokinetics and Bayesian Estimation of Mycophenolic Acid Concentrations in Stable Renal Transplant Patients. Clin. Pharm. 2004, 43, 253–266. [Google Scholar] [CrossRef]
- Beal, S.L.; Sheiner, L.B.; Boeckmann, A.; Bauer, R.J. NONMEM Users Guides; University of California: San Francisco, CA, USA, 1992. [Google Scholar]
- Lindbom, L.; Ribbing, J.; Jonsson, E.N. Perl-Speaks-NONMEM (PsN)—A Perl Module for NONMEM Related Programming. Comput. Methods Programs Biomed. 2004, 75, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Development Cohort (n = 85) | Validation Cohort (n = 27) | |
|---|---|---|---|
| N (%) | |||
| Disease | CLL | 73 (86%) | 23 (85%) |
| MCL | 10 (12%) | 4 (15%) | |
| WM | 2 (2%) | 0 (0%) | |
| Dose (mg) the day of PK exploration | 140 | 1 (1%) | 6 (22%) |
| 280 | 5 (6%) | 7 (26%) | |
| 420 | 70 (82%) | 14 (52%) | |
| 560 | 9 * (11%) | 0 (0%) | |
| Sex | Male | 27 (31%) | 11 (41%) |
| Female | 58 (68%) | 16 (59%) | |
| Prior treatment | No | 19 (22%) | 1 (4%) |
| Yes | 66 (78%) | 26 (96%) | |
| Median (min–max) | |||
| Age | 69 (31–84) | 68 (39–88) | |
| Height | 170 (148–187) | 169 (150–186) | |
| Weight | 70 (40–106) | 71 (48–91) | |
| Number of day of treatment | 32 (27–90) | 203 (35–665) | |
| Number of Points | Sampling Strategy | Mean Actual AUC (ng/mL·h) | RMSE (%) | MPE (%) | P20 (%) |
|---|---|---|---|---|---|
| 1 | T6 | 672 | 40.4% | +8.3% | 59% |
| T4 | 672 | 44.6% | +6.0% | 81% | |
| T2 | 672 | 45.3% | −11.9% | 62% | |
| T1 | 672 | 54.8% | +28.8% | 72% | |
| T0.5 | 672 | 61.9% | +39.6% | 71% | |
| T0 (LR) * | 672 | 67.9% | +42.7% | 71% | |
| 2 | T1–4 | 672 | 22.8% | −7.2% | 36% |
| T2–4 | 672 | 25.8% | −6.0% | 48% | |
| T1–6 | 672 | 30.0% | −9.1% | 40% | |
| T2–6 | 672 | 30.0% | −12.0% | 48% | |
| T0.5–4 | 672 | 31.6% | +4.2% | 47% | |
| T0.5–6 | 672 | 34.2% | +5.1% | 52% | |
| 3 | T0–1–4 | 672 | 14.5% | +2.2% | 25% |
| T1–4–6 | 672 | 19.2% | −7.9% | 31% | |
| T1–2–4 | 672 | 19.9% | −6.1% | 28% | |
| T0.5–2–6 | 672 | 22.0% | −9.2% | 28% | |
| T0–2–4 | 672 | 22.7% | −1.9% | 29% | |
| T0.5–2–4 | 672 | 22.7% | −2.7% | 31% | |
| 4 | T0–1–2–4 | 672 | 11.0% | −0.3% | 4% |
| T0–1–4–6 | 672 | 13.1% | −0.5% | 18% | |
| T0–0.5–2–4 | 672 | 13.3% | +3.1% | 11% | |
| T0–0.5–1–4 | 672 | 15.3% | +4.1% | 24% | |
| T1–2–4–6 | 672 | 15.7% | −8.2% | 18% | |
| T0.5–2–4–6 | 672 | 17.1% | −4.5% | 16% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Louedec, F.; Gallais, F.; Thomas, F.; White-Koning, M.; Allal, B.; Protin, C.; Ysebaert, L.; Chatelut, É.; Puisset, F. Limited Sampling Strategy for Determination of Ibrutinib Plasma Exposure: Joint Analyses with Metabolite Data. Pharmaceuticals 2021, 14, 162. https://doi.org/10.3390/ph14020162
Le Louedec F, Gallais F, Thomas F, White-Koning M, Allal B, Protin C, Ysebaert L, Chatelut É, Puisset F. Limited Sampling Strategy for Determination of Ibrutinib Plasma Exposure: Joint Analyses with Metabolite Data. Pharmaceuticals. 2021; 14(2):162. https://doi.org/10.3390/ph14020162
Chicago/Turabian StyleLe Louedec, Félicien, Fanny Gallais, Fabienne Thomas, Mélanie White-Koning, Ben Allal, Caroline Protin, Loïc Ysebaert, Étienne Chatelut, and Florent Puisset. 2021. "Limited Sampling Strategy for Determination of Ibrutinib Plasma Exposure: Joint Analyses with Metabolite Data" Pharmaceuticals 14, no. 2: 162. https://doi.org/10.3390/ph14020162
APA StyleLe Louedec, F., Gallais, F., Thomas, F., White-Koning, M., Allal, B., Protin, C., Ysebaert, L., Chatelut, É., & Puisset, F. (2021). Limited Sampling Strategy for Determination of Ibrutinib Plasma Exposure: Joint Analyses with Metabolite Data. Pharmaceuticals, 14(2), 162. https://doi.org/10.3390/ph14020162