
Identification of 37 Heterogeneous Drug Candidates for Treatment of COVID-19 via a Rational Transcriptomics-Based Drug Repurposing Approach




Abstract
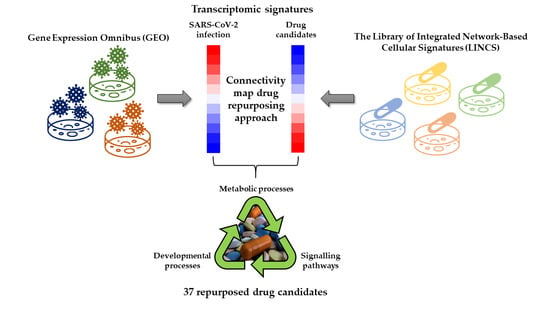
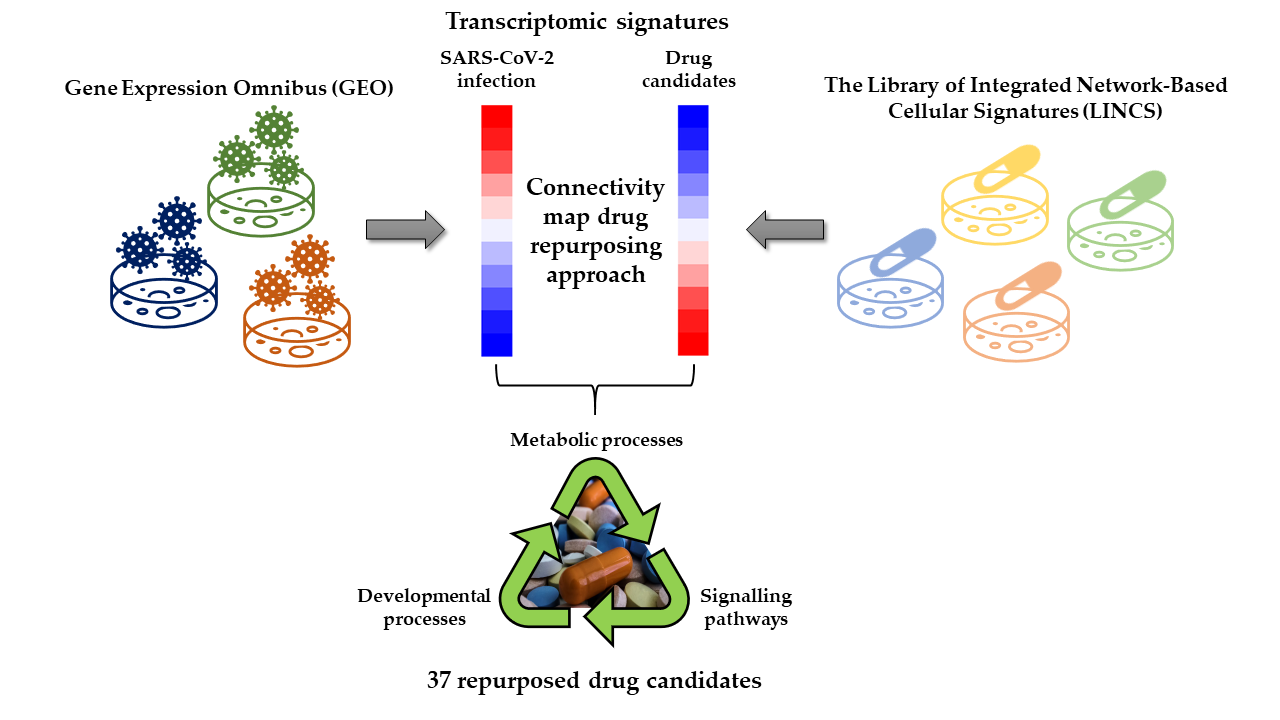
:
1. Introduction
2. Results
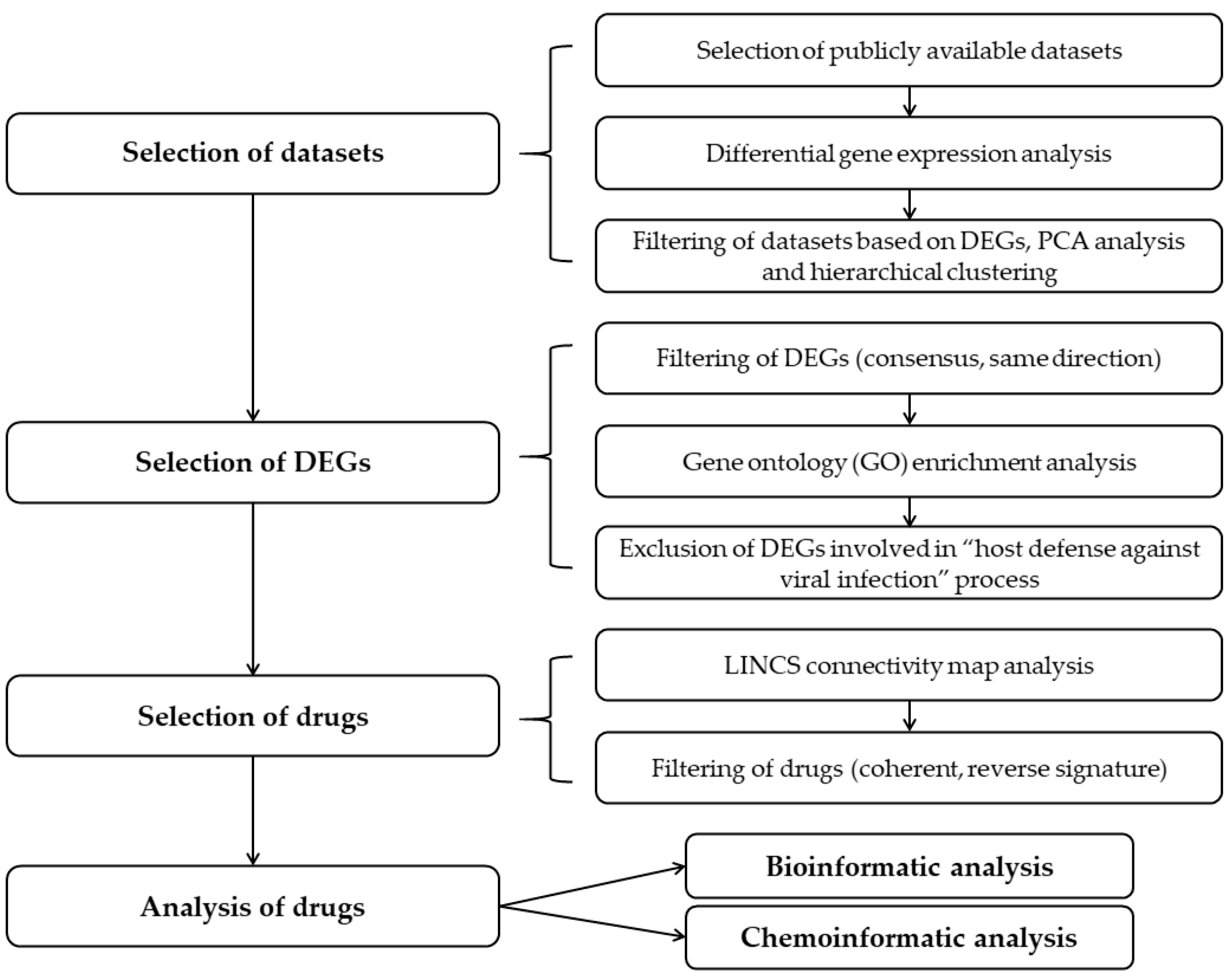
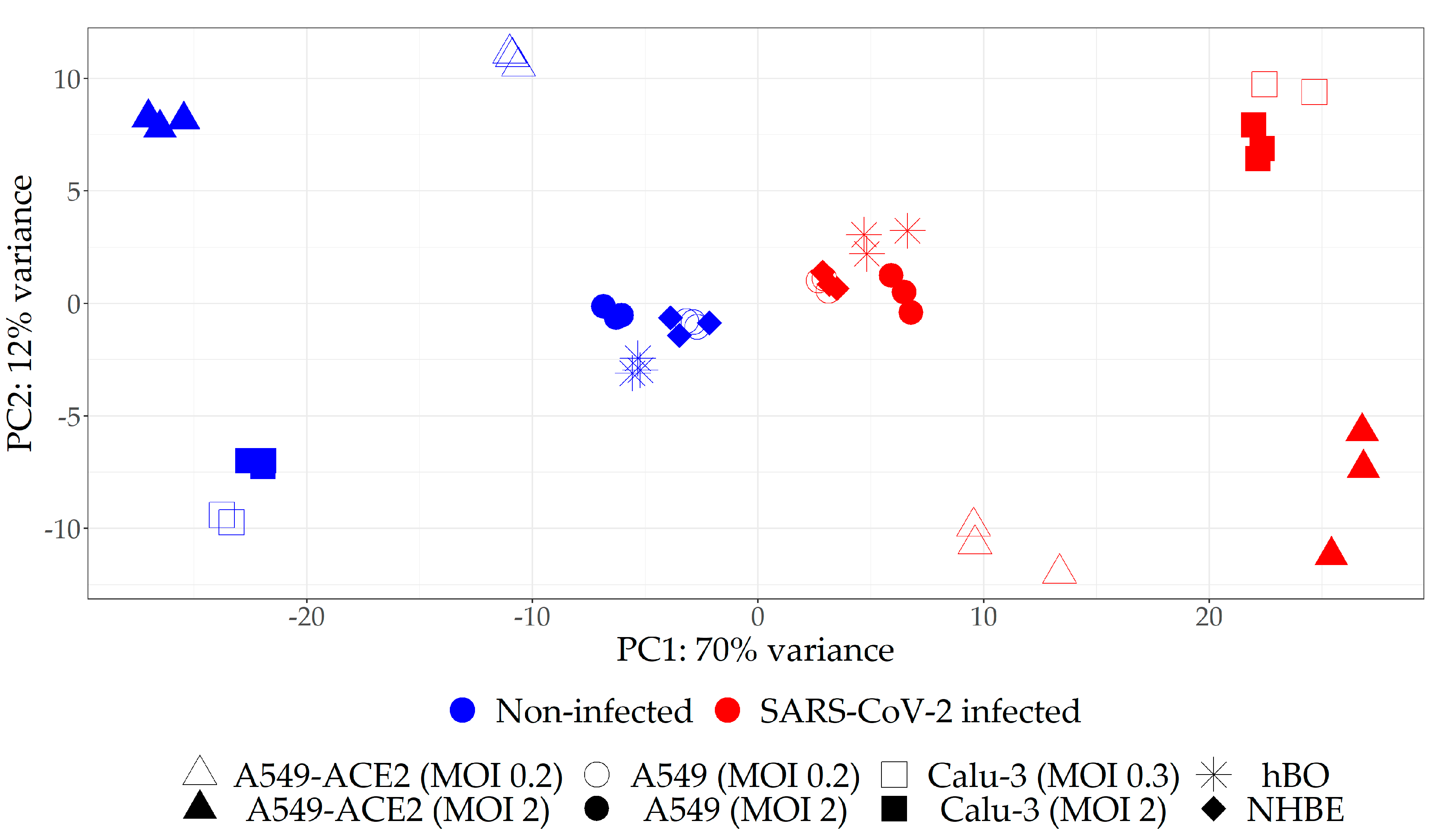
2.1. Selection of the Relevant Datasets
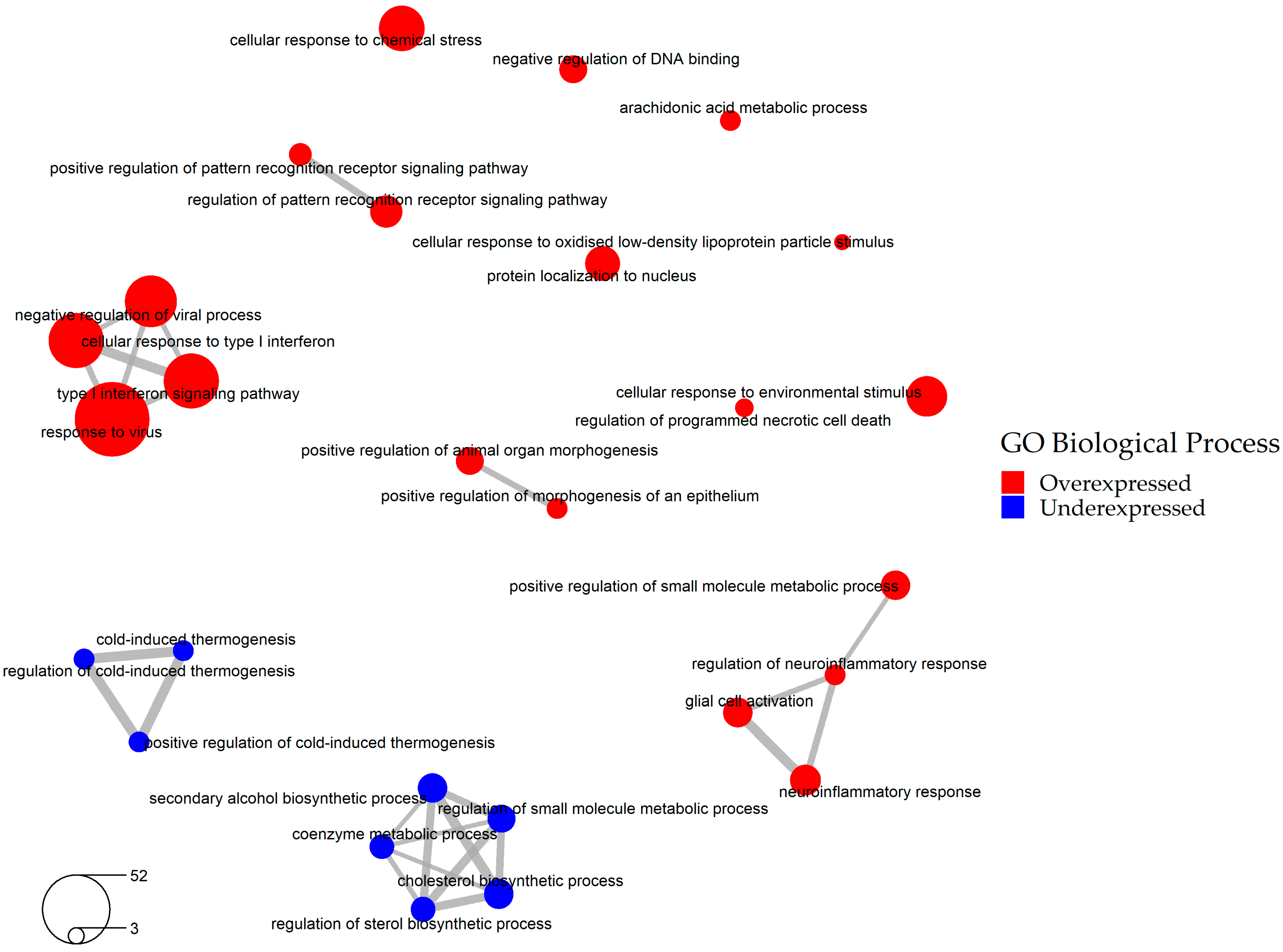
2.2. Selection of the Relevant DEGs upon SARS-CoV-2 Infection
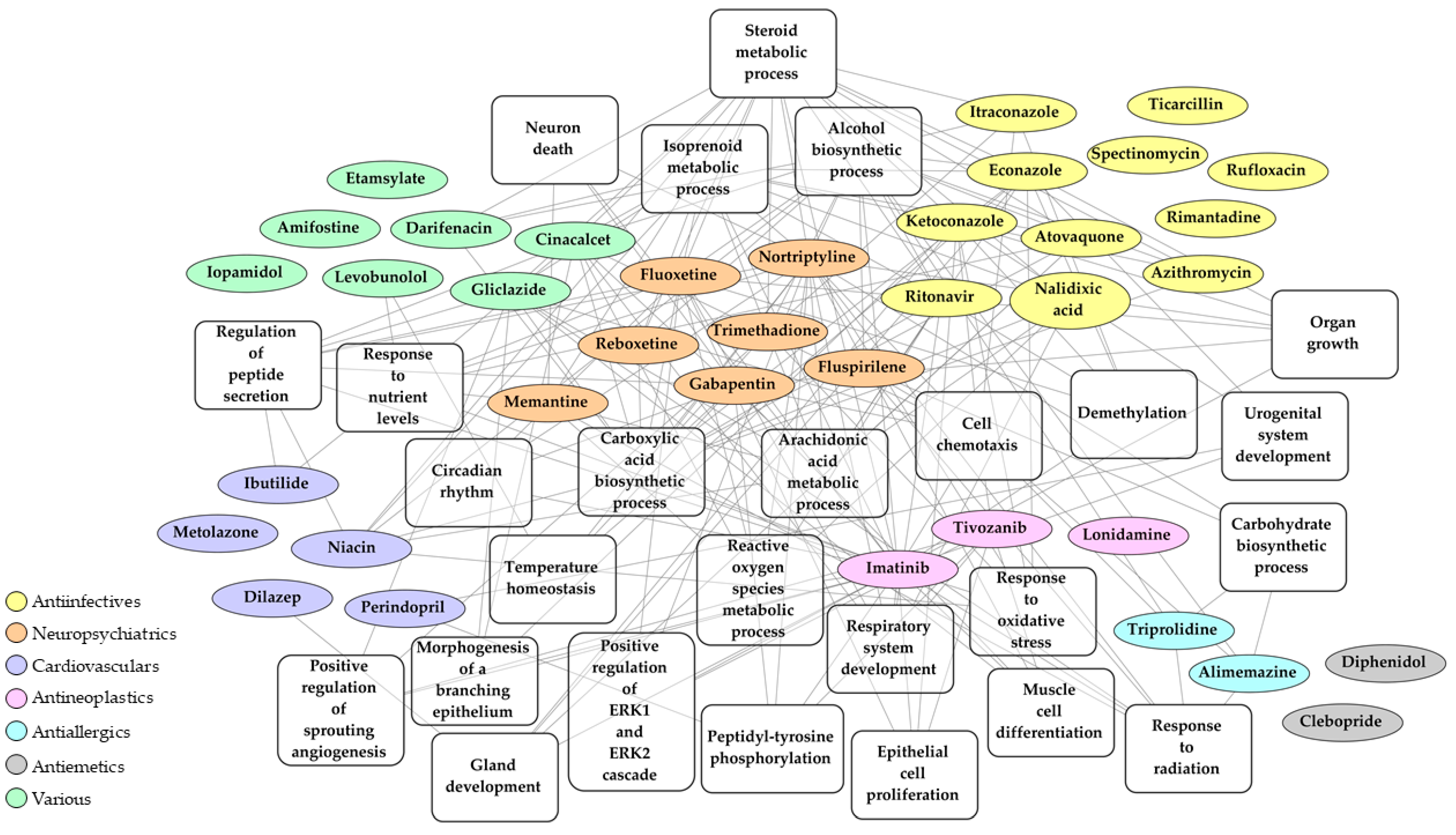
2.3. Identification of Drugs with a Potential to Reverse Transcriptomic Signature Upon SARS-CoV-2 Infection
2.4. Bio- and Chemoinformatic Characterization of the Drug Candidates for Repurposing against SARS-CoV-2 Infection
3. Discussion
4. Materials and Methods
4.1. Publicly Available Transcriptomics Datasets
4.2. Differential Gene Expression Analyses
4.3. Library of Integrated Network-Based Cellular Signatures (LINCS) Database Analysis
- (1)
- FDR adjusted p-value of weighted connectivity score was given for each perturbagen-cell line combination. Only significant combinations with FDR adjusted p-value less than 0.05 were selected.
- (2)
- Tau connectivity score was given for all significant perturbagen-cell line combinations. Wherever a perturbagen was tested in multiple cell lines, the mean Tau connectivity score and its coefficient of variation (CV, described as the standard deviation divided by the mean) were calculated. Only perturbagens with CV < 1, i.e., those that showed coherent transcriptomic signature in multiple cell lines were chosen. Finally, all perturbagens with Tau < −85 were filtered for further analysis. The recommended Tau threshold of −90 was lowered to −85 to increase the final number of identified drug candidates.
- (3)
- The list of perturbagens was additionally reduced to include only approved drugs which were used for downstream analysis. Information about drug approval status was obtained via CLUE Repurposing App (https://clue.io/repurposing-app/; selection of 2427 drugs in launched phase).
4.4. Bio- and Chemoinformatic Analyses of Candidate Drugs
4.5. Preparation of Figures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsatsakis, A.; Calina, D.; Falzone, L.; Petrakis, D.; Mitrut, R.; Siokas, V.; Pennisi, M.; Lanza, G.; Libra, M.; Doukas, S.G.; et al. SARS-CoV-2 pathophysiology and its clinical implications: An integrative overview of the pharmacotherapeutic management of COVID-19. Food Chem. Toxicol. 2020, 146, 111769. [Google Scholar] [CrossRef]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Listings of WHO’s Response to COVID-19. Available online: https://www.who.int/news/item/29-06-2020-covidtimeline (accessed on 26 November 2020).
- Center for Systems Science and Engineering at Johns Hopkins University Interactive Real-Time Web-Based COVID-19 Dashboard. Available online: https://coronavirus.jhu.edu/map.html (accessed on 23 December 2020).
- Coronavirus Update (Live): 78,475,152 Cases and 1726,535 Deaths from COVID-19 Virus Pandemic—Worldometer. Available online: https://www.worldometers.info/coronavirus/ (accessed on 23 December 2020).
- Nicola, M.; Alsafi, Z.; Sohrabi, C.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, M.; Agha, R. The socio-economic implications of the coronavirus pandemic (COVID-19): A review. Int. J. Surg. 2020, 78, 185–193. [Google Scholar] [CrossRef]
- Xiong, J.; Lipsitz, O.; Nasri, F.; Lui, L.M.W.; Gill, H.; Phan, L.; Chen-Li, D.; Iacobucci, M.; Ho, R.; Majeed, A.; et al. Impact of COVID-19 pandemic on mental health in the general population: A systematic review. J. Affect. Disord. 2020, 277, 55–64. [Google Scholar] [CrossRef]
- Milken Institute and First Person COVID-19 Vaccine Tracker. Available online: https://www.covid-19vaccinetracker.org/ (accessed on 23 December 2020).
- U.S. National Library of Medicine at the National Institutes of Health ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 23 December 2020).
- The RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Kashour, Z.; Riaz, M.; Garbati, M.A.; AlDosary, O.; Tlayjeh, H.; Gerberi, D.; Murad, M.H.; Sohail, M.R.; Kashour, T.; Tleyjeh, I.M. Efficacy of chloroquine or hydroxychloroquine in COVID-19 patients: A systematic review and meta-analysis. J. Antimicrob. Chemother. 2021, 76, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Horby, P.W.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Emberson, J.; Palfreeman, A.; Raw, J.; Elmahi, E.; Prudon, B.; et al. Lopinavir–ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- WHO. Solidarity Trial Consortium Repurposed Antiviral Drugs for Covid-19 — Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- National Institutes of Health (NIH). COVID-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 12 January 2021).
- U.S. Food and Drug Administration. FDA Approves First Treatment for COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19 (accessed on 26 November 2020).
- U.S. Food and Drug Administration. Coronavirus (COVID-19) Update: FDA Authorizes Drug Combination for Treatment of COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-drug-combination-treatment-covid-19 (accessed on 26 November 2020).
- World Health Organization. WHO Recommends against the Use of Remdesivir in COVID-19 Patients. Available online: https://www.who.int/news-room/feature-stories/detail/who-recommends-against-the-use-of-remdesivir-in-covid-19-patients (accessed on 23 December 2020).
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA J. Am. Med. Assoc. 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Koudijs, K.K.M.; Terwisscha Van Scheltinga, A.G.T.; Böhringer, S.; Schimmel, K.J.M.; Guchelaar, H.J. Transcriptome Signature Reversion as a Method to Reposition Drugs Against Cancer for Precision Oncology. Cancer J. 2019, 25, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Musa, A.; Ghoraie, L.S.; Zhang, S.D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, J.T.; Sirota, M.; Shenoy, M.; Pai, R.K.; Roedder, S.; Chiang, A.P.; Morgan, A.A.; Sarwal, M.M.; Pasricha, P.J.; Butte, A.J. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Malcomson, B.; Wilson, H.; Veglia, E.; Thillaiyampalam, G.; Barsden, R.; Donegan, S.; El Banna, A.; Elborn, J.S.; Ennis, M.; Kelly, C.; et al. Connectivity mapping (ssCMap) to predict A20-inducing drugs and their antiinflammatory action in cystic fibrosis. Proc. Natl. Acad. Sci. USA 2016, 113, E3725–E3734. [Google Scholar] [CrossRef] [Green Version]
- Loganathan, T.; Ramachandran, S.; Shankaran, P.; Nagarajan, D.; Suma Mohan, S. Host transcriptome-guided drug repurposing for COVID-19 treatment: A meta-analysis based approach. PeerJ 2020, 2020. [Google Scholar] [CrossRef]
- Li, Z.; Yang, L. Underlying Mechanisms and Candidate Drugs for COVID-19 Based on the Connectivity Map Database. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Mousavi, S.Z.; Rahmanian, M.; Sami, A. A connectivity map-based drug repurposing study and integrative analysis of transcriptomic profiling of SARS-CoV-2 infection. Infect. Genet. Evol. 2020, 86, 104610. [Google Scholar] [CrossRef]
- El-Hachem, N.; Eid, E.; Nemer, G.; Dbaibo, G.; Abbas, O.; Rubeiz, N.; Zeineldine, S.; Matar, G.M.; Bikorimana, J.P.; Shammaa, R.; et al. Integrative Transcriptome Analyses Empower the Anti-COVID-19 Drug Arsenal. iScience 2020, 23, 101697. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, P.; Raj, A.S.; Roy, S.; Kumar, N.S.; Kumar, H. Comparative transcriptome analysis of SARS-CoV, MERS-CoV, and SARS-CoV-2 to identify potential pathways for drug repurposing. Comput. Biol. Med. 2021, 128, 104123. [Google Scholar] [CrossRef] [PubMed]
- De Jong, T.V.; Moshkin, Y.M.; Guryev, V. Gene expression variability: The other dimension in transcriptome analysis. Physiol. Genomics 2019, 51, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Emanuel, W.; Kirstin, M.; Vedran, F.; Asija, D.; Theresa, G.L.; Roberto, A.; Filippos, K.; David, K.; Salah, A.; Christopher, B.; et al. Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. bioRxiv 2020. [Google Scholar] [CrossRef]
- Suzuki, T.; Itoh, Y.; Sakai, Y.; Saito, A.; Okuzaki, D.; Motooka, D.; Minami, S.; Kobayashi, T.; Yamamoto, T.; Okamoto, T.; et al. Generation of human bronchial organoids for SARS-CoV-2 research. bioRxiv 2020. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.C.; Huang, C.; Narayanan, K.; Makino, S.; Tesh, R.B.; Peters, C.J. Exogenous ACE2 Expression Allows Refractory Cell Lines To Support Severe Acute Respiratory Syndrome Coronavirus Replication. J. Virol. 2005, 79, 3846–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.; Chan, J.F.-W.; Yuen, T.T.-T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.-Y.; Tsang, J.O.-L.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B. ACE2 Receptor Expression and Severe Acute Respiratory Syndrome Coronavirus Infection Depend on Differentiation of Human Airway Epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Glende, J.; Schwegmann-Wessels, C.; Enjuanes, L.; Herrler, G.; Ren, X. Cholesterol is important for a post-adsorption step in the entry process of transmissible gastroenteritis virus. Antiviral Res. 2010, 88, 311–316. [Google Scholar] [CrossRef]
- Jeon, J.H.; Lee, C. Cellular cholesterol is required for porcine nidovirus infection. Arch. Virol. 2017, 162, 3753–3767. [Google Scholar] [CrossRef]
- Alsaadi, E.A.; Jones, I.M. Membrane binding proteins of coronaviruses. Future Virol. 2019, 14, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, S.; Marcuzzi, A.; Piscianz, E.; Tommasini, A.; Fabris, B. The complex interplay between lipids, immune system and interleukins in cardio-metabolic diseases. Int. J. Mol. Sci. 2018, 19, 4058. [Google Scholar] [CrossRef] [Green Version]
- Yaqoob, P. Lipids and the immune response: From molecular mechanisms to clinical applications. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 133–150. [Google Scholar] [CrossRef]
- Hubler, M.J.; Kennedy, A.J. Role of lipids in the metabolism and activation of immune cells. J. Nutr. Biochem. 2016, 34, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hou, Y.; Shen, J.; Mehra, R.; Kallianpur, A.; Culver, D.A.; Gack, M.U.; Farha, S.; Zein, J.; Comhair, S.; et al. A network medicine approach to investigation and population-based validation of disease manifestations and drug repurposing for COVID-19. PLoS Biol. 2020, 18, e3000970. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.r-project.org/ (accessed on 23 December 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/ Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Yu, G.; Li, F.; Qin, Y.; Bo, X.; Wu, Y.; Wang, S. GOSemSim: An R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 2010, 26, 976–978. [Google Scholar] [CrossRef]
- Duan, Y.; Evans, D.S.; Miller, R.A.; Schork, N.J.; Cummings, S.R.; Girke, T. signatureSearch: Environment for gene expression signature searching and functional interpretation. Nucleic Acids Res. 2020, 48, e124. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowless, M.S.; Waldman, M.; Franczkiewicz, R.; Clark, R.D. Using chemoinformatics in drug discovery. In New Approaches to Drug Discovery, Handbook of Experimental Pharmacology; Nielsch, U., Fuhrmann, U., Jaroch, S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 232, pp. 139–170. ISBN 978-3-319-28912-0. [Google Scholar]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34. [Google Scholar] [CrossRef]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Fleischmann, A.; Darsow, M.; Degtyarenko, K.; Fleischmann, W.; Boyce, S.; Axelsen, K.B.; Bairoch, A.; Schomburg, D.; Tipton, K.F.; Apweiler, R. IntEnz, the integrated relational enzyme database. Nucleic Acids Res. 2004, 32, D434–D437. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, 21800. [Google Scholar] [CrossRef] [Green Version]
- Guha, R. Chemical informatics functionality in R. J. Stat. Softw. 2007, 18, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Guha, R. fingerprint: Functions to Operate on Binary Fingerprint Data. R package version 3.5.7. Available online: https://CRAN.R-project.org/package=fingerprint2018 (accessed on 23 December 2020).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Label | Description | No. Samples (H/I *) | GEO Accession | Reference |
|---|---|---|---|---|
| Cells collected for RNA-seq 24 h post-infection | ||||
| A549 (MOI 0.2) | Human lung adenocarcinoma alveolar epithelial cells | 3/3 | GSE147507 | [34] |
| A549 (MOI 2) | Human lung adenocarcinoma alveolar epithelial cells | 3/3 | ||
| A549-ACE2 (MOI 0.2) | Human lung adenocarcinoma alveolar epithelial cells with overexpressed ACE2 | 3/3 | ||
| A549-ACE2 (MOI 2) | Human lung adenocarcinoma alveolar epithelial cells with overexpressed ACE2 | 3/3 | ||
| Calu-3 (MOI 2) | Human lung adenocarcinoma airway epithelial cells | 3/3 | ||
| NHBE (MOI 2) | Normal human bronchial epithelial cells | 3/3 | ||
| Calu-3 (MOI 0.3) | Human lung adenocarcinoma airway epithelial cells | 2/2 | GSE148729 | [35] |
| Organoids collected for RNA-seq 5 days post-infection | ||||
| hBO | Human bronchial organoids generated from NHBE cells | 3/2 | GSE150819 | [36] |
| Drug | Mean Tau | N Cell Lines with Same Effect | Pharmacological Class (Current Indication) | Mechanism of Action (MOA) |
|---|---|---|---|---|
| Alimemazine | −89.98 | 4 | Antiallergic agent | Histamine receptor antagonist |
| Amifostine | −90.18 | 1 | Radiation protective agent | Free radical scavenging activity |
| Atovaquone | −89.15 | 4 | Antiinfective agent (antiprotozoal) | Protozoal mitochondrial electron transport inhibitor |
| Azithromycin | −96.18 | 1 | Antiinfective agent (antibacterial) | Bacterial 50S ribosomal subunit inhibitor |
| Cinacalcet | −88.68 | 6 | Calcimimetic agent | Calcium-sensing receptor agonist |
| Clebopride | −93.96 | 1 | Antiemetic agent | Dopamine receptor antagonist |
| Darifenacin | −91.02 | 1 | Anticholinergic agent | Cholinergic muscarinic antagonist |
| Dilazep | −88.20 | 4 | Antihypertensive agent (vasodilatator) | Adenosine reuptake inhibitor |
| Diphenidol | −88.98 | 1 | Antiemetic agent | Acetylcholine receptor inhibitor |
| Econazole | −91.43 | 1 | Antiinfective agent (antifungal) | Fungal cytochrome P450 inhibitor (14-alpha demethylase inhibitors) |
| Etamsylate | −91.82 | 1 | Hemostatic agent | Hemostatic |
| Fluoxetine | −92.79 | 2 | Antidepressant agent | Selective serotonin reuptake inhibitor |
| Fluspirilene | −93.58 | 6 | Antipsychotic agent | Dopamine receptor antagonists |
| Gabapentin | −86.68 | 2 | Anticonvulsant agent | Excitatory neuron activity inhibitor |
| Gliclazide | −85.24 | 1 | Hypoglycemic agent | ATP sensitive potassium channel inhibitor |
| Ibutilide | −96.53 | 1 | Antiarrhythmia agent | Potassium channel blocker |
| Imatinib | −99.33 | 1 | Antineoplastic agent | Tyrosine kinase inhibitor |
| Iopamidol | −90.05 | 1 | Radiographic contrast agent | X-ray contrast activity |
| Itraconazole | −95.47 | 5 | Antiinfective agent (antifungal) | Fungal cytochrome P450 inhibitor (14-alpha demethylase inhibitors) |
| Ketoconazole | −87.97 | 2 | Antiinfective agent (antifungal) | Fungal cytochrome P450 inhibitor (14-alpha demethylase inhibitors) |
| Levobunolol | −86.31 | 1 | Sympatholytic agent | Beta-adrenergic receptor antagonist |
| Lonidamine | −89.97 | 1 | Antineoplastic agent | Glucokinase inhibitor |
| Memantine | −97.66 | 1 | Neuroprotective agent | N-methyl-d-aspartate glutamate receptor antagonist |
| Metolazone | −85.72 | 3 | Antihypertensive agent (diuretic) | Sodium chloride symporter inhibitor |
| Nalidixic acid | −89.81 | 1 | Antiinfective agent (antibacterial) | Bacterial topoisomerase II inhibitor |
| Niacin | −92.13 | 1 | Antihypertensive agent (vasodilatator, hypolipidemic) | Lowering cholesterol |
| Nortriptyline | −92.84 | 2 | Antidepressant agent | Adrenergic uptake inhibitor |
| Perindopril | −88.63 | 2 | Antihypertensive agent | Angiotensin converting enzyme inhibitor |
| Reboxetine | −89.55 | 2 | Antidepressant agent | Selective noradrenaline reuptake inhibitor |
| Rimantadine | −89.63 | 1 | Antiinfective agent (antiviral) | Viral (influenza A) nucleic acid synthesis inhibitor |
| Ritonavir | −88.75 | 1 | Antiinfective agent (antiviral) | Viral (HIV) protease inhibitor |
| Rufloxacin | −93.98 | 1 | Antiinfective agent (antibacterial) | Bacterial topoisomerase II inhibitor |
| Spectinomycin | −93.99 | 1 | Antiinfective agent (antibacterial) | Bacterial 30S ribosomal subunit inhibitor |
| Ticarcillin | −88.28 | 1 | Antiinfective agent (antibacterial) | Inhibitor of bacterial cell wall synthesis |
| Tivozanib | −87.17 | 2 | Antineoplastic agent | Vascular endothelial growth factor receptors inhibitor |
| Trimethadione | −96.08 | 1 | Anticonvulsant agent | Inhibitor of voltage dependent T-type calcium channels |
| Triprolidine | −88.42 | 3 | Antiallergic agent | Histamine receptor antagonist |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelemanović, A.; Vidović, T.; Stepanić, V.; Trajković, K. Identification of 37 Heterogeneous Drug Candidates for Treatment of COVID-19 via a Rational Transcriptomics-Based Drug Repurposing Approach. Pharmaceuticals 2021, 14, 87. https://doi.org/10.3390/ph14020087
Gelemanović A, Vidović T, Stepanić V, Trajković K. Identification of 37 Heterogeneous Drug Candidates for Treatment of COVID-19 via a Rational Transcriptomics-Based Drug Repurposing Approach. Pharmaceuticals. 2021; 14(2):87. https://doi.org/10.3390/ph14020087
Chicago/Turabian StyleGelemanović, Andrea, Tinka Vidović, Višnja Stepanić, and Katarina Trajković. 2021. "Identification of 37 Heterogeneous Drug Candidates for Treatment of COVID-19 via a Rational Transcriptomics-Based Drug Repurposing Approach" Pharmaceuticals 14, no. 2: 87. https://doi.org/10.3390/ph14020087
APA StyleGelemanović, A., Vidović, T., Stepanić, V., & Trajković, K. (2021). Identification of 37 Heterogeneous Drug Candidates for Treatment of COVID-19 via a Rational Transcriptomics-Based Drug Repurposing Approach. Pharmaceuticals, 14(2), 87. https://doi.org/10.3390/ph14020087