
TIGIT/CD226 Axis Regulates Anti-Tumor Immunity


Abstract
:1. Introduction
2. TIGIT
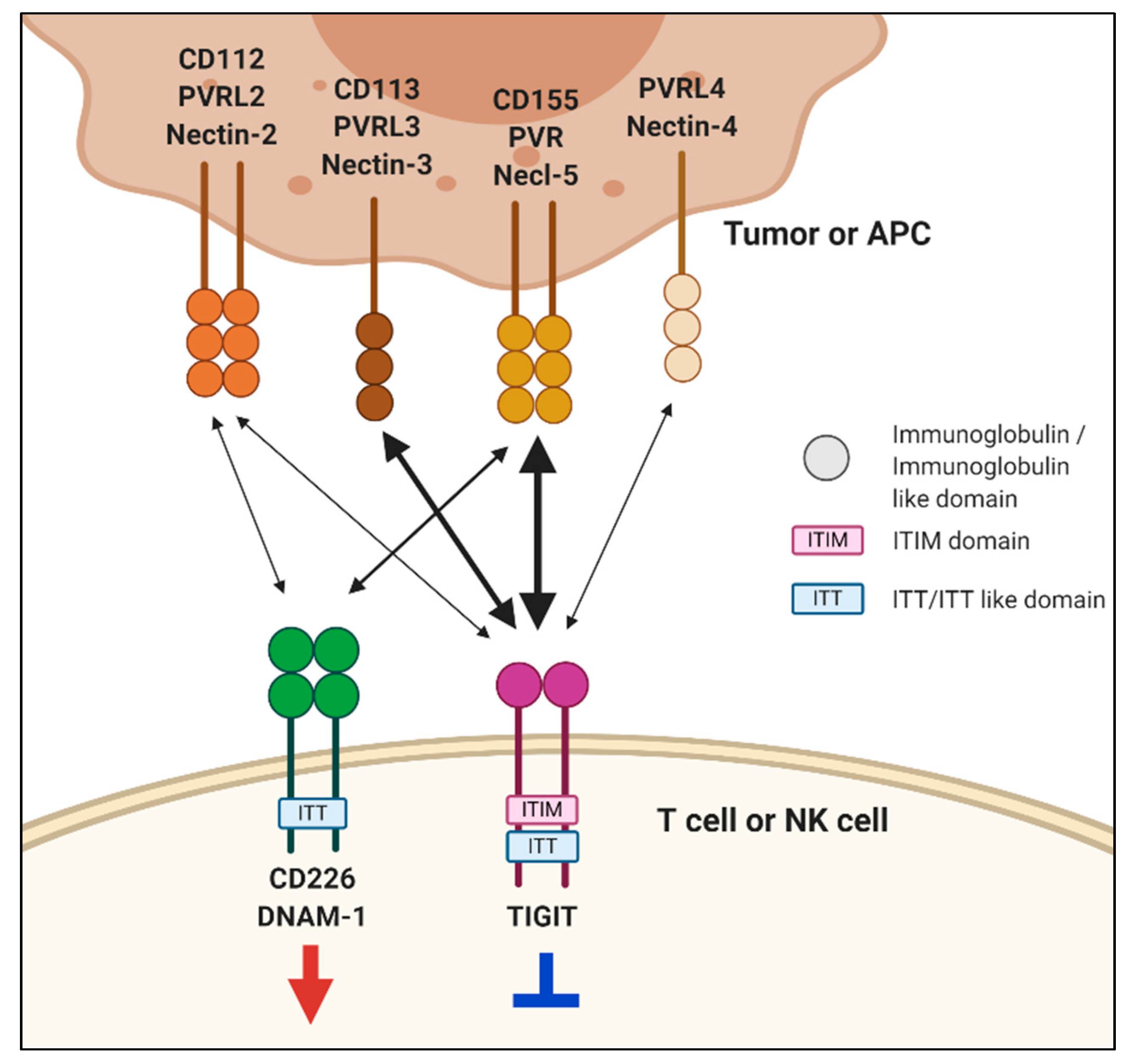
2.1. TIGIT Structure and Its Ligands
2.2. Role of TIGIT in Immune Cell Regulation
2.3. Targeting TIGIT for Cancer Immunotherapy
2.3.1. TIGIT as a Potential Prognostic Marker for Cancer
2.3.2. TIGIT Blockade in Anti-Tumor Immunity
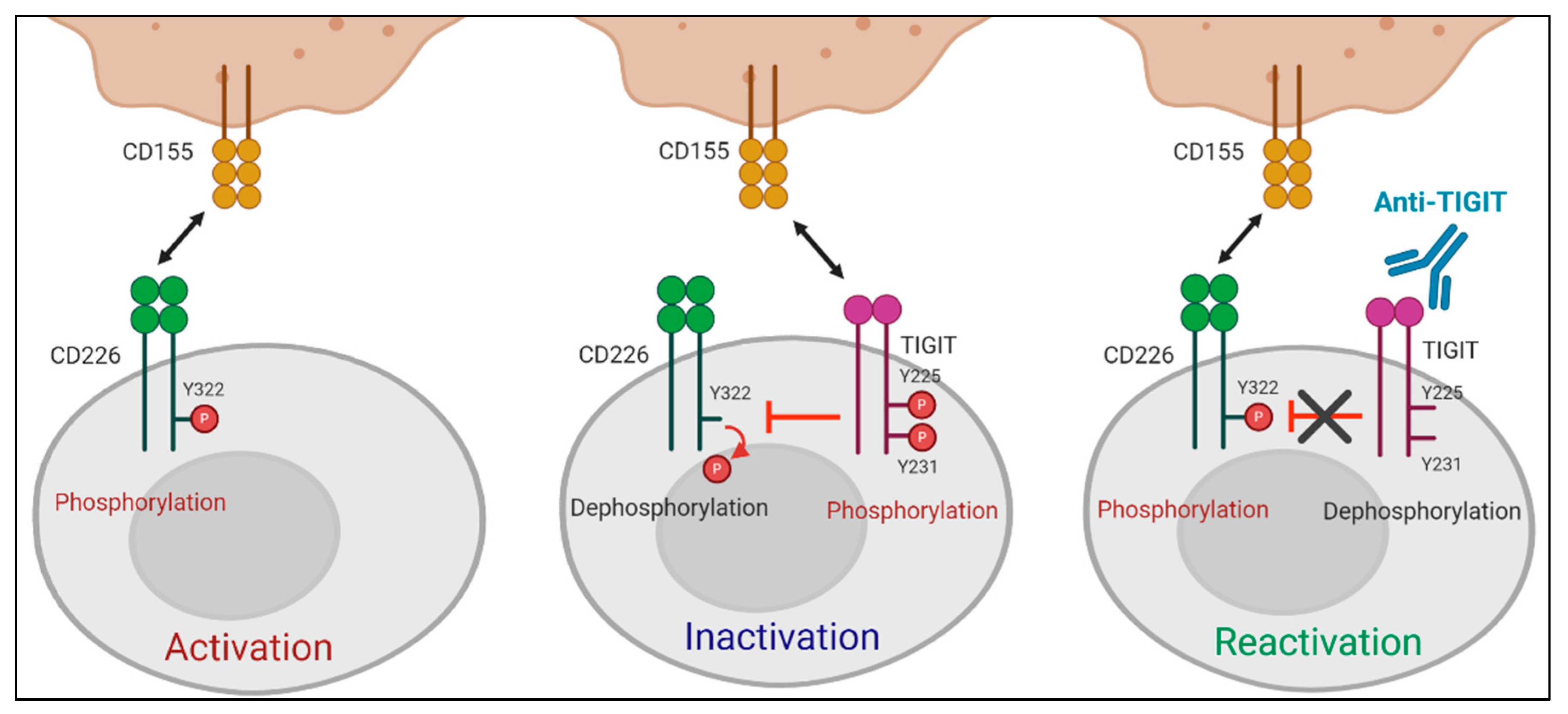
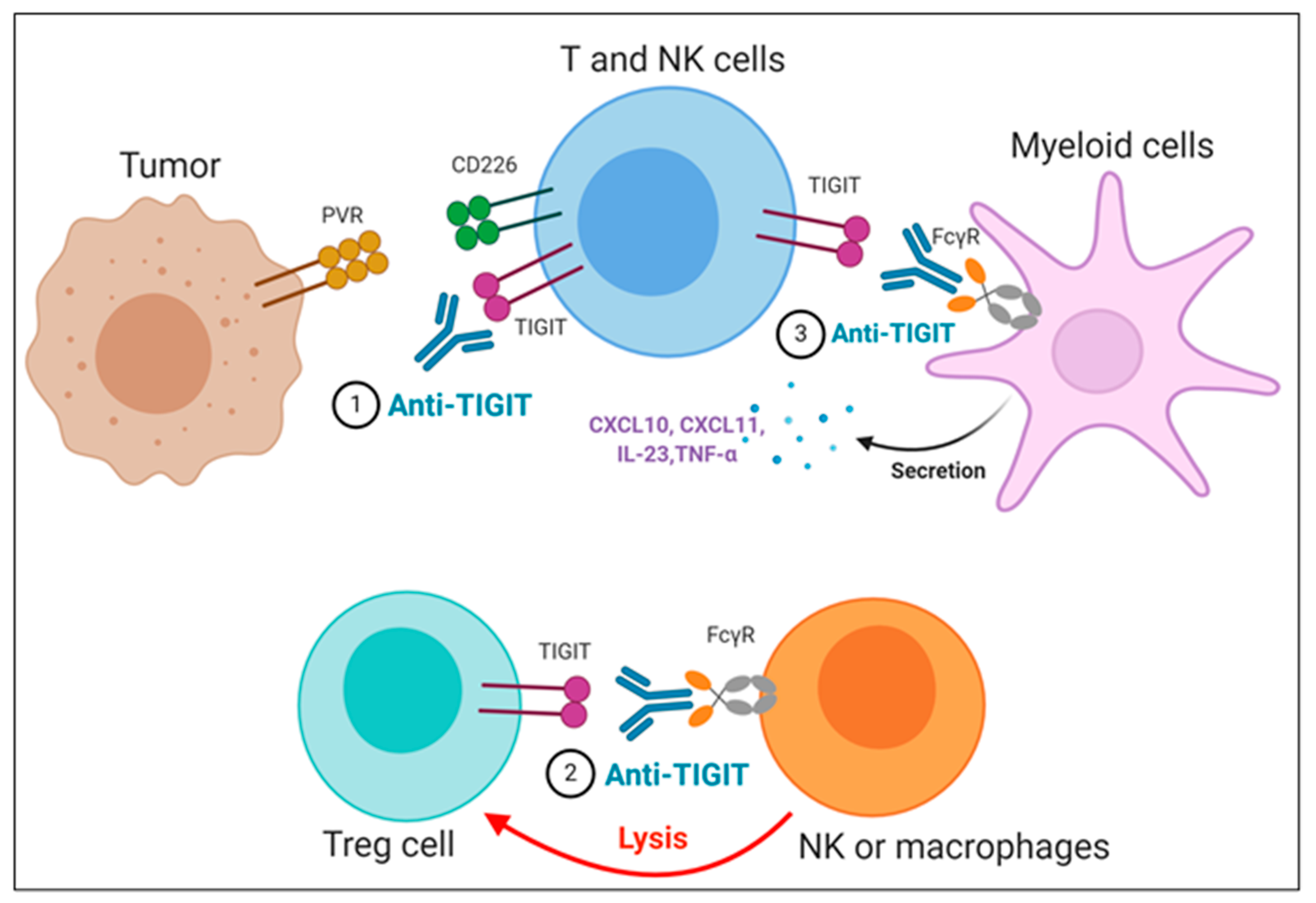
2.3.3. Mode of Action of Anti-TIGIT Therapy
- Intracellular Regulation by Anti-TIGIT mAbs
- Isotype Selection of Anti-TIGIT mAbs
2.4. Anti-TIGIT Antibodies in Clinical Trials
3. CD226
3.1. CD226 Structure and Its Ligands
3.2. CD226 Signaling
3.3. CD226 in Tumor Immunity
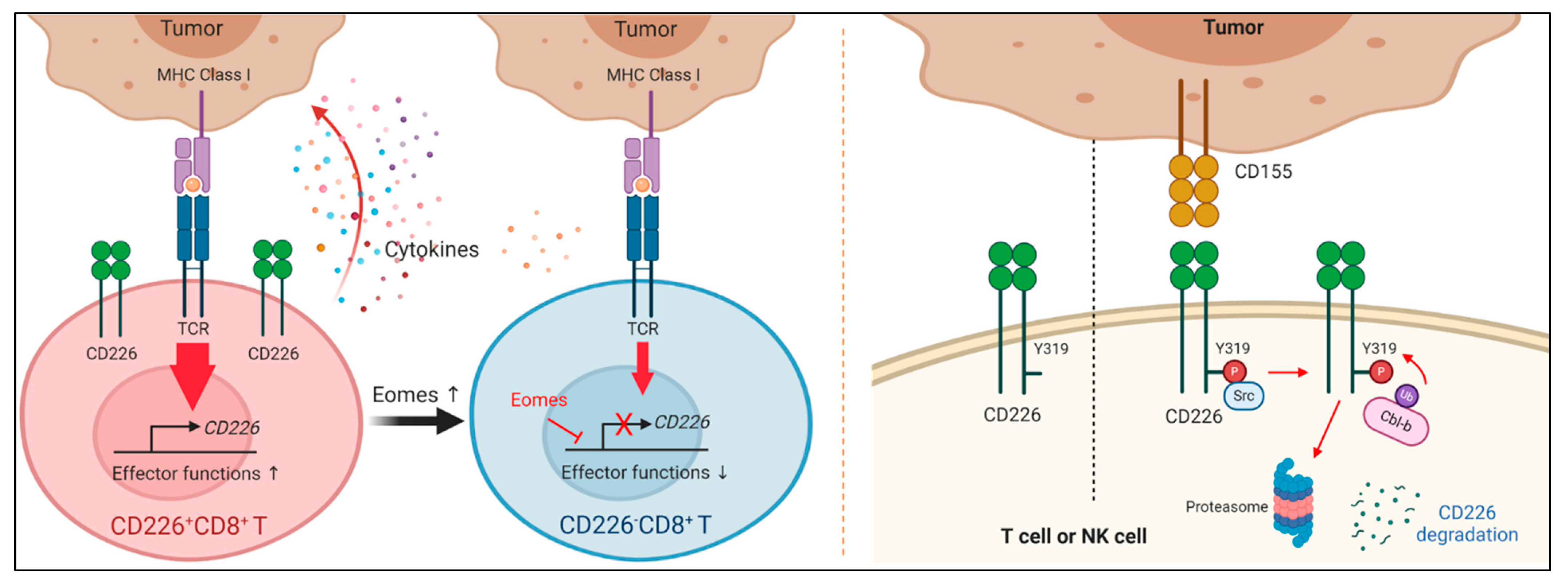
3.3.1. CD226 Downregulation in Dysfunctional T Cells
3.3.2. Mechanisms of CD226 Downregulation
3.3.3. Predictive Value of CD226 for Immune Checkpoint Blockade Therapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fritsch, E.F.; Burkhardt, U.E.; Hacohen, N.; Wu, C.J. Personal Neoantigen Cancer Vaccines: A Road Not Fully Paved. Cancer Immunol. Res. 2020, 8, 1465–1469. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [Green Version]
- Kreileder, M.; Barrett, I.; Bendtsen, C.; Brennan, D.; Kolch, W. Signaling Dynamics Regulating Crosstalks between T-Cell Activation and Immune Checkpoints. Trends Cell Biol. 2020, 31, 224–235. [Google Scholar] [CrossRef]
- Lonberg, N.; Korman, A.J. Masterful Antibodies: Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Tray, N.; Weber, J.S.; Adams, S. Predictive Biomarkers for Checkpoint Immunotherapy: Current Status and Challenges for Clinical Application. Cancer Immunol. Res. 2018, 6, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Bal, S.; Badros, A.Z. Lessons Learned from Checkpoint Blockade Targeting PD-1 in Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Arens, R.; Scheeren, F.A. Genetic Screening for Novel Regulators of Immune Checkpoint Molecules. Trends Immunol. 2020, 41, 692–705. [Google Scholar] [CrossRef]
- Zebley, C.C.; Gottschalk, S.; Youngblood, B. Rewriting History: Epigenetic Reprogramming of CD8(+) T Cell Differentiation to Enhance Immunotherapy. Trends Immunol. 2020, 41, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Harjunpaa, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, S.; Ophir, E.; Kotturi, M.F.; Levy, O.; Ganguly, S.; Leung, L.; Vaknin, I.; Kumar, S.; Dassa, L.; Hansen, K.; et al. PVRIG and PVRL2 Are Induced in Cancer and Inhibit CD8(+) T-cell Function. Cancer Immunol. Res. 2019, 7, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Marin-Acevedo, J.A.; Soyano, A.E.; Dholaria, B.; Knutson, K.L.; Lou, Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 8. [Google Scholar] [CrossRef]
- Gorvel, L.; Olive, D. Targeting the “PVR-TIGIT axis” with immune checkpoint therapies. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Boles, K.S.; Vermi, W.; Facchetti, F.; Fuchs, A.; Wilson, T.J.; Diacovo, T.G.; Cella, M.; Colonna, M. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur. J. Immunol. 2009, 39, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D.; et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casas-Aviles, I.; Duran, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [Green Version]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef]
- Reches, A.; Ophir, Y.; Stein, N.; Kol, I.; Isaacson, B.; Charpak Amikam, Y.; Elnekave, A.; Tsukerman, P.; Kucan Brlic, P.; Lenac, T.; et al. Nectin4 is a novel TIGIT ligand which combines checkpoint inhibition and tumor specificity. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.G.; Kiessling, R.; Malmberg, K.J. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 2009, 183, 4921–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, A.; Cella, M.; Giurisato, E.; Shaw, A.S.; Colonna, M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J. Immunol. 2004, 172, 3994–3998. [Google Scholar] [CrossRef] [Green Version]
- Kucan Brlic, P.; Lenac Rovis, T.; Cinamon, G.; Tsukerman, P.; Mandelboim, O.; Jonjic, S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell. Mol. Immunol. 2019, 16, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Das, I.; Lepletier, A.; Addala, V.; Bald, T.; Stannard, K.; Barkauskas, D.; Liu, J.; Aguilera, A.R.; Takeda, K.; et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J. Clin. Investig. 2018, 128, 2613–2625. [Google Scholar] [CrossRef] [Green Version]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Clinical significance of CD155 expression in human pancreatic cancer. Anticancer Res. 2015, 35, 2287–2297. [Google Scholar]
- Triki, H.; Charfi, S.; Bouzidi, L.; Ben Kridis, W.; Daoud, J.; Chaabane, K.; Sellami-Boudawara, T.; Rebai, A.; Cherif, B. CD155 expression in human breast cancer: Clinical significance and relevance to natural killer cell infiltration. Life Sci. 2019, 231, 116543. [Google Scholar] [CrossRef]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.F.; Joller, N.; Tan, D.J.; Teng, M.W.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Harjunpaa, H.; Carrie, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.H.; Cai, M.; Grein, J.; Perera, S.; Wang, H.; Bigler, M.; Ueda, R.; Rosahl, T.W.; Pinheiro, E.; LaFace, D.; et al. Effective Anti-tumor Response by TIGIT Blockade Associated With FcgammaR Engagement and Myeloid Cell Activation. Front. Immunol. 2020, 11, 573405. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.S.; Ko, M.; Choi, D.S.; Kim, J.H.; Lee, D.H.; Kang, S.H.; Kim, I.; Lee, H.J.; Choi, E.K.; Kim, K.P.; et al. CD226(hi)CD8(+) T Cells Are a Prerequisite for Anti-TIGIT Immunotherapy. Cancer Immunol. Res. 2020, 8, 912–925. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lu, P.H.; Liu, L.; Fang, Z.M.; Duan, W.; Liu, Z.L.; Wang, C.Y.; Zhou, P.; Yu, X.F.; He, W.T. TIGIT negatively regulates inflammation by altering macrophage phenotype. Immunobiology 2016, 221, 48–55. [Google Scholar] [CrossRef]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, C.A.; Yeh, W.I.; Seay, H.R.; Saikumar Lakshmi, P.; Chopra, G.; Zhang, L.; Perry, D.J.; McClymont, S.A.; Yadav, M.; Lopez, M.C.; et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J. Immunol. 2015, 195, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Chauvin, J.M.; Ka, M.; Davar, D.; Pagliano, O.; Wang, H.; Saada, S.; Menna, C.; Amin, R.; et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Z.; Kim, H.J.; Wu, H.; Jalali, S.; Tang, X.; Krull, J.; Ding, W.; Novak, A.J.; Ansell, S.M. TIGIT expression is associated with T-cell suppression and exhaustion and predicts clinical outcome and anti-PD-1 response in follicular lymphoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhang, H.; Han, F.; Chen, X.; Lin, R.; Wang, W.; Qiu, H.; Zhuang, Z.; Liao, Q.; Zhang, W.; et al. CD155T/TIGIT Signaling Regulates CD8(+) T-cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [Green Version]
- Lucca, L.E.; Lerner, B.A.; Park, C.; DeBartolo, D.; Harnett, B.; Kumar, V.P.; Ponath, G.; Raddassi, K.; Huttner, A.; Hafler, D.A.; et al. Differential expression of the T-cell inhibitor TIGIT in glioblastoma and MS. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostroumov, D.; Duong, S.; Wingerath, J.; Woller, N.; Manns, M.P.; Timrott, K.; Kleine, M.; Ramackers, W.; Roessler, S.; Nahnsen, S.; et al. Transcriptome profiling identifies TIGIT as a marker of T cell exhaustion in liver cancer. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Stalhammar, G.; Seregard, S.; Grossniklaus, H.E. Expression of immune checkpoint receptors Indoleamine 2,3-dioxygenase and T cell Ig and ITIM domain in metastatic versus nonmetastatic choroidal melanoma. Cancer Med. 2019, 8, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mao, L.; Liu, J.F.; Chen, L.; Yu, G.T.; Yang, L.L.; Wu, H.; Bu, L.L.; Kulkarni, A.B.; Zhang, W.F.; et al. Blockade of TIGIT/CD155 Signaling Reverses T-cell Exhaustion and Enhances Antitumor Capability in Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Res. 2019, 7, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhao, E.; Zhu, C.; Zhao, W.; Wang, C.; Zhang, Z.; Zhao, G. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology 2020, 225, 151915. [Google Scholar] [CrossRef]
- Duan, X.; Liu, J.; Cui, J.; Ma, B.; Zhou, Q.; Yang, X.; Lu, Z.; Du, Y.; Su, C. Expression of TIGIT/CD155 and correlations with clinical pathological features in human hepatocellular carcinoma. Mol. Med. Rep. 2019, 20, 3773–3781. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Li, L.; Lu, F.; Yue, J.; Liu, Z.; Zhang, W.; Fu, R. Overexpression of TIGIT in NK and T Cells Contributes to Tumor Immune Escape in Myelodysplastic Syndromes. Front. Oncol. 2020, 10, 1595. [Google Scholar] [CrossRef]
- Westergaard, M.C.W.; Milne, K.; Pedersen, M.; Hasselager, T.; Olsen, L.R.; Anglesio, M.S.; Borch, T.H.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Changes in the Tumor Immune Microenvironment during Disease Progression in Patients with Ovarian Cancer. Cancers 2020, 12, 3828. [Google Scholar] [CrossRef]
- Tang, W.; Pan, X.; Han, D.; Rong, D.; Zhang, M.; Yang, L.; Ying, J.; Guan, H.; Chen, Z.; Wang, X. Clinical significance of CD8(+) T cell immunoreceptor with Ig and ITIM domains(+) in locally advanced gastric cancer treated with SOX regimen after D2 gastrectomy. Oncoimmunology 2019, 8, e1593807. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, A.W.; Yeung, H.M.; Alpaugh, R.K.; Dulaimi, E.; Engstrom, P.F.; Dasari, A.; Campbell, K.S.; Vijayvergia, N. Impacts of pembrolizumab therapy on immune phenotype in patients with high-grade neuroendocrine neoplasms. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef]
- Dixon, K.O.; Schorer, M.; Nevin, J.; Etminan, Y.; Amoozgar, Z.; Kondo, T.; Kurtulus, S.; Kassam, N.; Sobel, R.A.; Fukumura, D.; et al. Functional Anti-TIGIT Antibodies Regulate Development of Autoimmunity and Antitumor Immunity. J. Immunol. 2018, 200, 3000–3007. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Hall, B.; Sun, Z.J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.K.; Yuen, V.W.; Wing-Sum Cheu, J.; Wei, L.L.; Ting, V.; Fehlings, M.; Sumatoh, H.; Nardin, A.; Newell, E.W.; Oi-Lin Ng, I.; et al. Hepatocellular Carcinoma Cells Up-regulate PVRL1, Stabilizing Poliovirus Receptor and Inhibiting the Cytotoxic T-Cell Response via TIGIT to Mediate Tumor Resistance to PD1 Inhibitors in Mice. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Lee, B.R.; Chae, S.; Moon, J.; Kim, M.J.; Lee, H.; Ko, H.W.; Cho, B.C.; Shim, H.S.; Hwang, D.; Kim, H.R.; et al. Combination of PD-L1 and PVR determines sensitivity to PD-1 blockade. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Jin, H.S.; Choi, D.S.; Ko, M.; Kim, D.; Lee, D.H.; Lee, S.; Lee, A.Y.; Kang, S.G.; Kim, S.H.; Jung, Y.; et al. Extracellular pH modulating injectable gel for enhancing immune checkpoint inhibitor therapy. J. Control Release 2019, 315, 65–75. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Ka, M.; Pagliano, O.; Menna, C.; Ding, Q.; DeBlasio, R.; Sanders, C.; Hou, J.; Li, X.Y.; Ferrone, S.; et al. IL15 Stimulation with TIGIT Blockade Reverses CD155-mediated NK-Cell Dysfunction in Melanoma. Clin. Cancer Res. 2020, 26, 5520–5533. [Google Scholar] [CrossRef]
- Beers, S.A.; Glennie, M.J.; White, A.L. Influence of immunoglobulin isotype on therapeutic antibody function. Blood 2016, 127, 1097–1101. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Li, K.; Zhang, T. FcgammaR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front. Immunol. 2019, 10, 292. [Google Scholar] [CrossRef]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef]
- Waight, J.D.; Chand, D.; Dietrich, S.; Gombos, R.; Horn, T.; Gonzalez, A.M.; Manrique, M.; Swiech, L.; Morin, B.; Brittsan, C.; et al. Selective FcgammaR Co-engagement on APCs Modulates the Activity of Therapeutic Antibodies Targeting T Cell Antigens. Cancer Cell 2018, 33, 1033–1047.e5. [Google Scholar] [CrossRef] [Green Version]
- Preillon, J.; Cuende, J.; Rabolli, V.; Garnero, L.; Mercier, M.; Wald, N.; Pappalardo, A.; Denies, S.; Jamart, D.; Michaux, A.C.; et al. Restoration of T-cell Effector Function, Depletion of Tregs, and Direct Killing of Tumor Cells: The Multiple Mechanisms of Action of a-TIGIT Antagonist Antibodies. Mol. Cancer Ther. 2021, 20, 121–131. [Google Scholar] [CrossRef]
- Anonymous. Tiragolumab Impresses in Multiple Trials. Cancer Discov. 2020, 10, 1086–1087. [Google Scholar] [CrossRef]
- Huang, Z.; Qi, G.; Miller, J.S.; Zheng, S.G. CD226: An Emerging Role in Immunologic Diseases. Front. Cell Dev. Biol. 2020, 8, 564. [Google Scholar] [CrossRef]
- Wang, H.; Qi, J.; Zhang, S.; Li, Y.; Tan, S.; Gao, G.F. Binding mode of the side-by-side two-IgV molecule CD226/DNAM-1 to its ligand CD155/Necl-5. Proc. Natl. Acad. Sci. USA 2019, 116, 988–996. [Google Scholar] [CrossRef] [Green Version]
- Deuss, F.A.; Watson, G.M.; Fu, Z.; Rossjohn, J.; Berry, R. Structural Basis for CD96 Immune Receptor Recognition of Nectin-like Protein-5, CD155. Structure 2019, 27, 219–228.e3. [Google Scholar] [CrossRef] [Green Version]
- Tahara-Hanaoka, S.; Shibuya, K.; Onoda, Y.; Zhang, H.; Yamazaki, S.; Miyamoto, A.; Honda, S.; Lanier, L.L.; Shibuya, A. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int. Immunol. 2004, 16, 533–538. [Google Scholar] [CrossRef]
- Kearney, C.J.; Ramsbottom, K.M.; Voskoboinik, I.; Darcy, P.K.; Oliaro, J. Loss of DNAM-1 ligand expression by acute myeloid leukemia cells renders them resistant to NK cell killing. Oncoimmunology 2016, 5, e1196308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi-Manaka, A.; Okumura, G.; Kojima, H.; Cho, Y.; Hirochika, R.; Bando, H.; Sato, T.; Yoshikawa, H.; Hara, H.; Shibuya, A.; et al. Increased Soluble CD155 in the Serum of Cancer Patients. PLoS ONE 2016, 11, e0152982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baury, B.; Masson, D.; McDermott, B.M., Jr.; Jarry, A.; Blottiere, H.M.; Blanchardie, P.; Laboisse, C.L.; Lustenberger, P.; Racaniello, V.R.; Denis, M.G. Identification of secreted CD155 isoforms. Biochem. Biophys. Res. Commun. 2003, 309, 175–182. [Google Scholar] [CrossRef]
- Okumura, G.; Iguchi-Manaka, A.; Murata, R.; Yamashita-Kanemaru, Y.; Shibuya, A.; Shibuya, K. Tumor-derived soluble CD155 inhibits DNAM-1-mediated antitumor activity of natural killer cells. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [Green Version]
- Ralston, K.J.; Hird, S.L.; Zhang, X.; Scott, J.L.; Jin, B.; Thorne, R.F.; Berndt, M.C.; Boyd, A.W.; Burns, G.F. The LFA-1-associated molecule PTA-1 (CD226) on T cells forms a dynamic molecular complex with protein 4.1G and human discs large. J. Biol. Chem. 2004, 279, 33816–33828. [Google Scholar] [CrossRef] [Green Version]
- Hara, H.; Wada, T.; Bakal, C.; Kozieradzki, I.; Suzuki, S.; Suzuki, N.; Nghiem, M.; Griffiths, E.K.; Krawczyk, C.; Bauer, B.; et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity 2003, 18, 763–775. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, K.; Lanier, L.L.; Phillips, J.H.; Ochs, H.D.; Shimizu, K.; Nakayama, E.; Nakauchi, H.; Shibuya, A. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity 1999, 11, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wu, N.; Lu, Y.; Davidson, D.; Colonna, M.; Veillette, A. DNAM-1 controls NK cell activation via an ITT-like motif. J. Exp. Med. 2015, 212, 2165–2182. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Long, E.O. Complementary phosphorylation sites in the adaptor protein SLP-76 promote synergistic activation of natural killer cells. Sci. Signal. 2012, 5, ra49. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; de Almeida, P.; Manieri, N.; de Almeida Nagata, D.; Wu, T.D.; Harden Bowles, K.; Arumugam, V.; Schartner, J.; Cubas, R.; Mittman, S.; et al. CD226 regulates natural killer cell antitumor responses via phosphorylation-mediated inactivation of transcription factor FOXO1. Proc. Natl. Acad. Sci. USA 2018, 115, E11731–E11740. [Google Scholar] [CrossRef] [Green Version]
- Iguchi-Manaka, A.; Kai, H.; Yamashita, Y.; Shibata, K.; Tahara-Hanaoka, S.; Honda, S.; Yasui, T.; Kikutani, H.; Shibuya, K.; Shibuya, A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J. Exp. Med. 2008, 205, 2959–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilfillan, S.; Chan, C.J.; Cella, M.; Haynes, N.M.; Rapaport, A.S.; Boles, K.S.; Andrews, D.M.; Smyth, M.J.; Colonna, M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J. Exp. Med. 2008, 205, 2965–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Zhang, W.; Jankovic, V.; Golubov, J.; Poon, P.; Oswald, E.M.; Gurer, C.; Wei, J.; Ramos, I.; Wu, Q.; et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory phenotype. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2012, 90, 109–115. [Google Scholar] [CrossRef]
- Cella, M.; Presti, R.; Vermi, W.; Lavender, K.; Turnbull, E.; Ochsenbauer-Jambor, C.; Kappes, J.C.; Ferrari, G.; Kessels, L.; Williams, I.; et al. Loss of DNAM-1 contributes to CD8+ T-cell exhaustion in chronic HIV-1 infection. Eur. J. Immunol. 2010, 40, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Tauriainen, J.; Scharf, L.; Frederiksen, J.; Naji, A.; Ljunggren, H.G.; Sonnerborg, A.; Lund, O.; Reyes-Teran, G.; Hecht, F.M.; Deeks, S.G.; et al. Perturbed CD8(+) T cell TIGIT/CD226/PVR axis despite early initiation of antiretroviral treatment in HIV infected individuals. Sci. Rep. 2017, 7, 40354. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.; Aguilera, A.R.; Sundarrajan, A.; Corvino, D.; Stannard, K.; Krumeich, S.; Das, I.; Lima, L.G.; Meza Guzman, L.G.; Li, K.; et al. CD155 on Tumor Cells Drives Resistance to Immunotherapy by Inducing the Degradation of the Activating Receptor CD226 in CD8(+) T Cells. Immunity 2020, 53, 805–823.e15. [Google Scholar] [CrossRef]
- Weulersse, M.; Asrir, A.; Pichler, A.C.; Lemaitre, L.; Braun, M.; Carrie, N.; Joubert, M.V.; Le Moine, M.; Do Souto, L.; Gaud, G.; et al. Eomes-Dependent Loss of the Co-activating Receptor CD226 Restrains CD8(+) T Cell Anti-tumor Functions and Limits the Efficacy of Cancer Immunotherapy. Immunity 2020, 53, 824–839.e10. [Google Scholar] [CrossRef]
- Jin, Z.; Lan, T.; Zhao, Y.; Du, J.; Chen, J.; Lai, J.; Xu, L.; Chen, S.; Zhong, X.; Wu, X.; et al. Higher TIGIT(+)CD226(-) gammadelta T cells in Patients with Acute Myeloid Leukemia. Immunol. Invest. 2020, 1–11. [Google Scholar] [CrossRef]
- Minnie, S.A.; Kuns, R.D.; Gartlan, K.H.; Zhang, P.; Wilkinson, A.N.; Samson, L.; Guillerey, C.; Engwerda, C.; MacDonald, K.P.A.; Smyth, M.J.; et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 2018, 132, 1675–1688. [Google Scholar] [CrossRef]
- Song, Y.; Wang, B.; Song, R.; Hao, Y.; Wang, D.; Li, Y.; Jiang, Y.; Xu, L.; Ma, Y.; Zheng, H.; et al. T-cell Immunoglobulin and ITIM Domain Contributes to CD8(+) T-cell Immunosenescence. Aging Cell 2018, 17. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TIGIT Inhibitor | Sponsor | Isotype | Identifiers | Cancer Type | Combination | Phase | Recruitment Status | Start Date |
|---|---|---|---|---|---|---|---|---|
| ASP-8374 | Astellas Pharma Inc. | IgG4 | NCT03260322 | Advanced solid tumor | ASP-8374 alone; Pembrolizumab (anti-PD-1) | Phase 1b | No longer recruiting | 8 September 2017 |
| NCT03945253 | Advanced solid tumor | ASP-8374 alone | Phase 1 | No longer recruiting | 5 August 2019 | |||
| BGB-A1217 | BeiGene Co Ltd. | IgG1 | NCT04047862 | Advanced solid tumor | Tislelizumab (anti-PD-1) | Phase 1 | Recruiting | 26 August 2019 |
| BMS-986207 | Bristol-Myers Squibb Co. | IgG1 (Fc receptor disabled) | NCT02913313 | Advanced solid tumor | BMS-986207 alone; Nivolumab (anti-PD-1) | Phase 1/2 | No longer recruiting | 29 November 2016 |
| NCT04150965 | Multiple myeloma | BMS-986207 alone; Dexamethasone+Pomalidomide | Phase 1/2 | Recruiting | 16 April 2018 | |||
| NCT04570839 | Advanced solid tumor | COM-701 (PVRIG inhibitor) + Nivolumab (anti-PD-1) | Phase 1/2 | Recruiting | 31 August 2020 | |||
| NCT04065425 | Multiple myeloma | Dexamethasone + Pomalidomide | Phase 1/2 | Not yet recruiting | 1 October 2019 | |||
| COM-902 | Compugen Ltd. | IgG4 | NCT04354246 | Advanced solid tumor | COM-902 alone | Phase 1 | Recruiting | 31 March 2020 |
| AB154 (Domvanalimab) | Arcus Biosciences Inc. | IgG1 (Fc receptor disabled) | NCT03628677 | Advanced malignancy | AB154 alone; Zimberelimab (anti-PD-1) | Phase 1 | Recruiting | 12 September 2018 |
| NCT04656535 | Recurrent Glioblastoma | Zimberelimab (anti-PD-1) | Phase 1 | Not yet recruiting | 31 January 2021 | |||
| NCT04262856 | PD-L1 positive lung cancer | Zimberelimab (anti-PD-1); Zimberelimab + etrumadenant (A2aR and A2bR antagonist) | Phase 2 | Recruiting | 28 May 2020 | |||
| EOS-884448 | iTeos Therapeutics | IgG1 | NCT04335253 | Advanced tumor | EOS-884448 alone | Phase 1/2 | Recruiting | 18 February 2020 |
| Etigilimab (OMP-313M32) | OncoMed | IgG1 | NCT03119428 | Advanced solid tumor | Etigilimab alone; Nivolumab (anti-PD-1) | Phase 1 | Terminated | 2 May 2017 |
| IBI-939 | Innovent Biologics Inc. | Not disclosed | NCT04353830 | Advanced tumor | IBI-939 alone; Sintilimab (anti-PD-1) | Phase 1a | Recruiting | 22 May 2020 |
| NCT04672356 | Advanced lung cancer | Sintilimab (anti-PD-1) | Phase 1a | Not yet recruiting | 28 January 2021 | |||
| NCT04672369 | Advanced NSCLC | Sintilimab (anti-PD-1) | Phase 1b | Not yet recruiting | 6 June 2021 | |||
| M-6223 | Serono Research Institute Inc, Merck KGaA | Not disclosed | NCT04457778 | Advanced solid tumor | M-6223 alone; Bintrafusp alfa (TGF beta ligand inhibitor) | Phase 1 | Recruiting | 10 July 2020 |
| Vibostolimab (MK-7684) | Merck Sharp & Dohme Corp. | IgG1 | NCT02964013 | Advanced solid tumor | Vibostolimab alone; Pembrolizumab (anti-PD-1); Pembrolizumab + Pemetrexed + Carboplatin; Pembrolizumab + Carboplatin or Cisplatin + Etoposide | Phase 1 | Recruiting | 13 December 2016 |
| NCT04305054 | Advanced melanoma | Pembrolizumab (anti-PD-1); | Phase 1/2 | Recruiting | 1 July 2020 | |||
| NCT04303169 | Melanoma | Pembrolizumab (anti-PD-1) | Phase 1/2 | Recruiting | 26 June 2020 | |||
| NCT04305041 | Refractory melanoma | Pembrolizumab + Quavonlimab (anti-CTLA4) | Phase 1/2 | Recruiting | 26 June 2020 | |||
| NCT04165070 | Advanced NSCLC | Pembrolizumab + Carboplatin + Paclitaxel; Pembrolizumab + Pemetrexed | Phase 2 | Recruiting | 19 December 2019 | |||
| NCT02861573 | Prostate cancer | Pembrolizumab (anti-PD-1) | Phase 1/2 | Recruiting | 17 November 2016 | |||
| Tiragolumab (MTIG7192A) | Genentech Inc., Chugai Pharmaceutical Co. Ltd., Roche Holding AG | IgG1 | NCT04045028 | Relapse/Refractory Multiple myeloma and B-cell Non-Hodgkin lymphoma | Tiragolumab alone; Daratumumab (anti-CD38); Rituximab (anti-CD20) | Phase 1 | Recruiting | 22 July 2019 |
| NCT02794571 | Metastatic solid tumor | Tiragolumab alone; Atezolizumab (anti-PD-L1); Chemotherapy (Carboplatin, Cisplatin, Etoposide, Paclitaxel, Pemetrexed) | Phase 1 | Recruiting | 23 May 2016 | |||
| NCT03281369 | Metastatic esophageal cancer | Atezolizumab (anti-PD-L1); Atezolizumab + Cisplatin+5FU | Phase 1/2 | Recruiting | 13 October 2017 | |||
| NCT04513925 | NSCLC | Atezolizumab (anti-PD-L1) | Phase 3 | Recruiting | 24 August 2020 | |||
| NCT04294810 | Metastatic NSCLC, PD-L1 selected | Atezolizumab (anti-PD-L1) | Phase 3 | Recruiting | 04 March 2020 | |||
| NCT04665843 | Metastatic head and neck cancer, PD-L1 positive | Atezolizumab (anti-PD-L1) | Phase 2 | Not yet recruiting | 21 January 2021 | |||
| NCT04543617 | Esophagus squamous cell carcinoma | Atezolizumab (anti-PD-L1) | Phase 3 | Recruiting | 28 September 2020 | |||
| NCT04300647 | Metastasis/Recurrent uterine cervix tumor, PD-L1 positive | Atezolizumab (anti-PD-L1) | Phase 2 | Recruiting | 30 June2020 | |||
| NCT03563716 | NSCLC, chemotherapy-naïve | Atezolizumab (anti-PD-L1) | Phase 2 | No longer recruiting | 10 August 2018 | |||
| NCT04665856 | Small-cell lung cancer | Atezolizumab + Carboplatin + Etoposide | Phase 3 | Recruiting | 4 January 2021 | |||
| NCT04619797 | Metastatic NSCLC | Atezolizumab + Pemetrexed + Carboplatin or Cisplatin | Phase 2 | Recruiting | 11 December 2020 | |||
| NCT04584112 | Triple-negative breast cancer | Atezolizumab + Nab-paclitaxel; Atezolizumab + Nab-pac-carbo-AC; Atezolizumab+Nab-pac-AC; | Phase 1b | Recruiting | 28 September 2020 | |||
| NCT04256421 | Metastatic small-cell lung cancer | Atezolizumab + Carboplatin + Etoposide | Phase 3 | Recruiting | 4 February 2020 | |||
| NCT04540211 | Metastatic esophageal cancer | Atezolizumab + Paclitaxel + Cisplatin | Phase 3 | Recruiting | 4 November 2020 | |||
| NCT04524871 | Metastatic hepatocellular carcinoma | Atezolizumab + Bevacizumab (anti-VEGF) | Phase 1/2 | Recruiting | 2 November 2020 | |||
| NCT03869190 | Advanced urothelial carcinoma | Atezolizumab (anti-PD-L1) | Phase 1/2 | Recruiting | 1 June 2019 | |||
| NCT03193190 | Metastatic pancreatic ductal adenocarcinoma | Atezolizumab + Nab-Paclitaxe l+ Gemcitabine | Phase 1/2 | Recruiting | 5 July 2017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, J.; Ko, M.; Lee, D.-H.; Park, Y.; Jin, H.-s. TIGIT/CD226 Axis Regulates Anti-Tumor Immunity. Pharmaceuticals 2021, 14, 200. https://doi.org/10.3390/ph14030200
Yeo J, Ko M, Lee D-H, Park Y, Jin H-s. TIGIT/CD226 Axis Regulates Anti-Tumor Immunity. Pharmaceuticals. 2021; 14(3):200. https://doi.org/10.3390/ph14030200
Chicago/Turabian StyleYeo, Jinah, Minkyung Ko, Dong-Hee Lee, Yoon Park, and Hyung-seung Jin. 2021. "TIGIT/CD226 Axis Regulates Anti-Tumor Immunity" Pharmaceuticals 14, no. 3: 200. https://doi.org/10.3390/ph14030200
APA StyleYeo, J., Ko, M., Lee, D. -H., Park, Y., & Jin, H. -s. (2021). TIGIT/CD226 Axis Regulates Anti-Tumor Immunity. Pharmaceuticals, 14(3), 200. https://doi.org/10.3390/ph14030200