
A Comprehensive Approach to Compatibility Testing Using Chromatographic, Thermal and Spectroscopic Techniques: Evaluation of Potential for a Monolayer Fixed-Dose Combination of 6-Mercaptopurine and Folic Acid

 ,
,  ,
,  , and
, and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Differential Scanning Calorimetry (DSC)
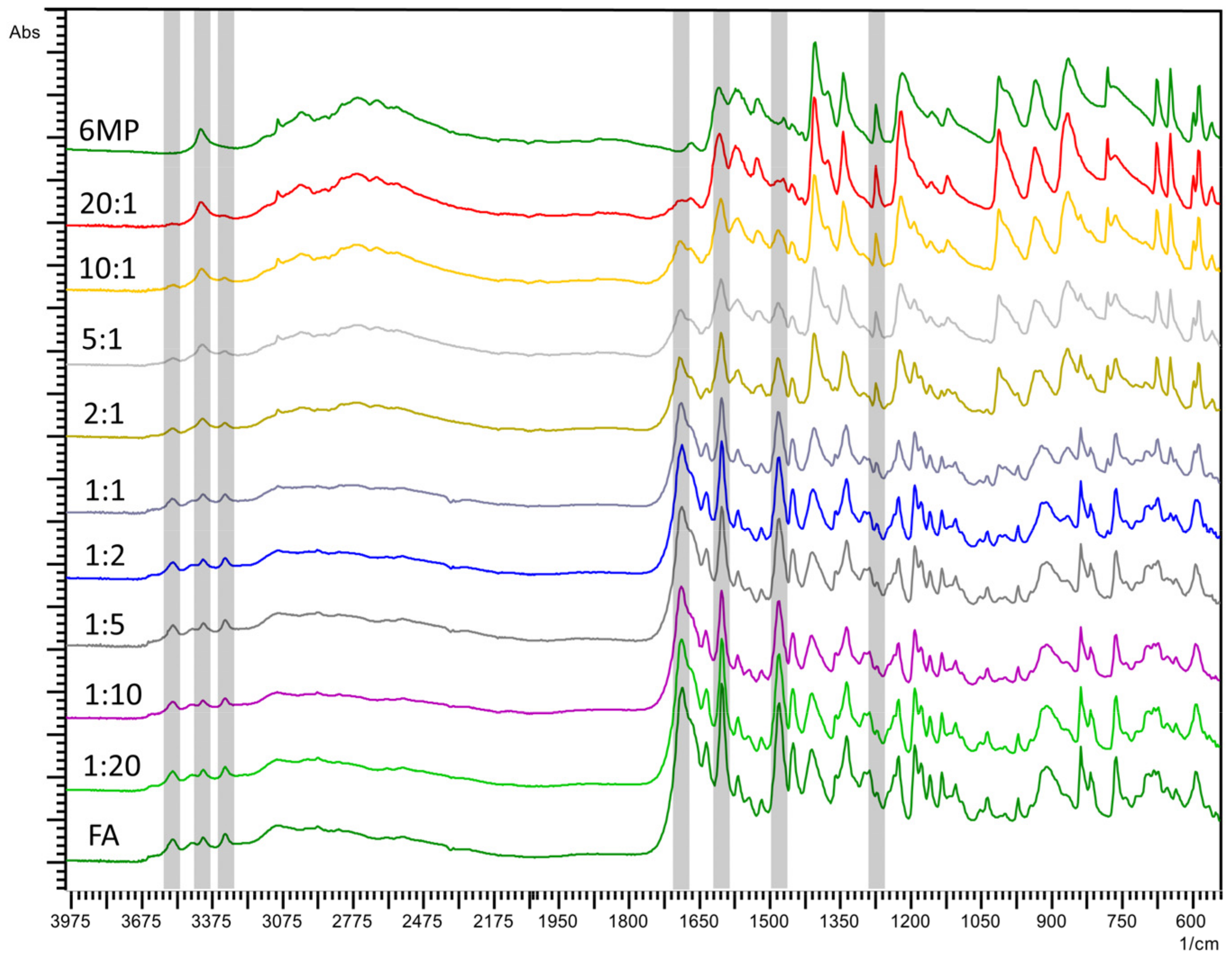
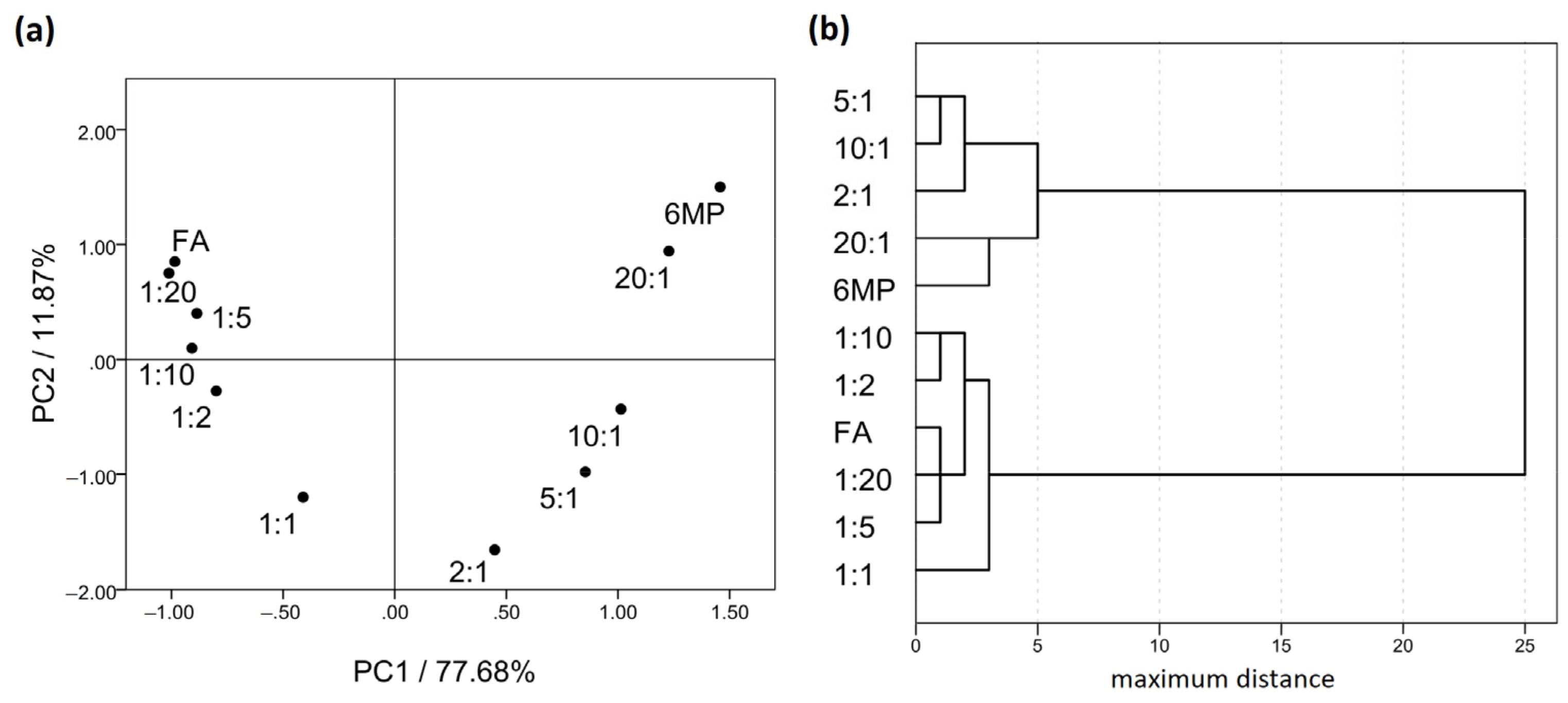
2.2. Fourier-Transform Infrared Spectroscopy (FTIR)
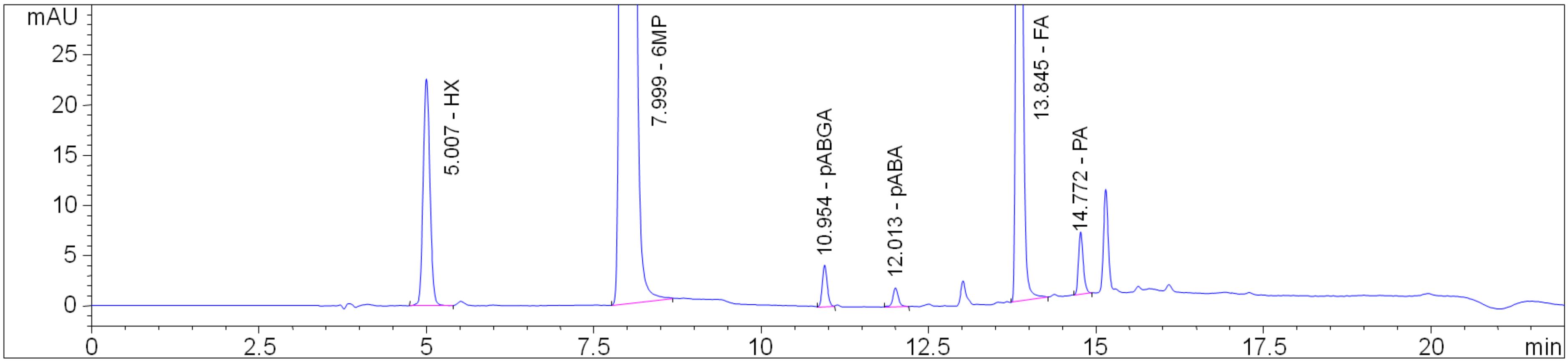
2.3. HPLC Method Development
2.4. Chromatographic Method Validation
2.4.1. Selectivity
2.4.2. Linearity and Limits of Detection and Quantitation
2.4.3. Accuracy and Precision
2.4.4. Robustness
2.5. Isothermal Stress Studies
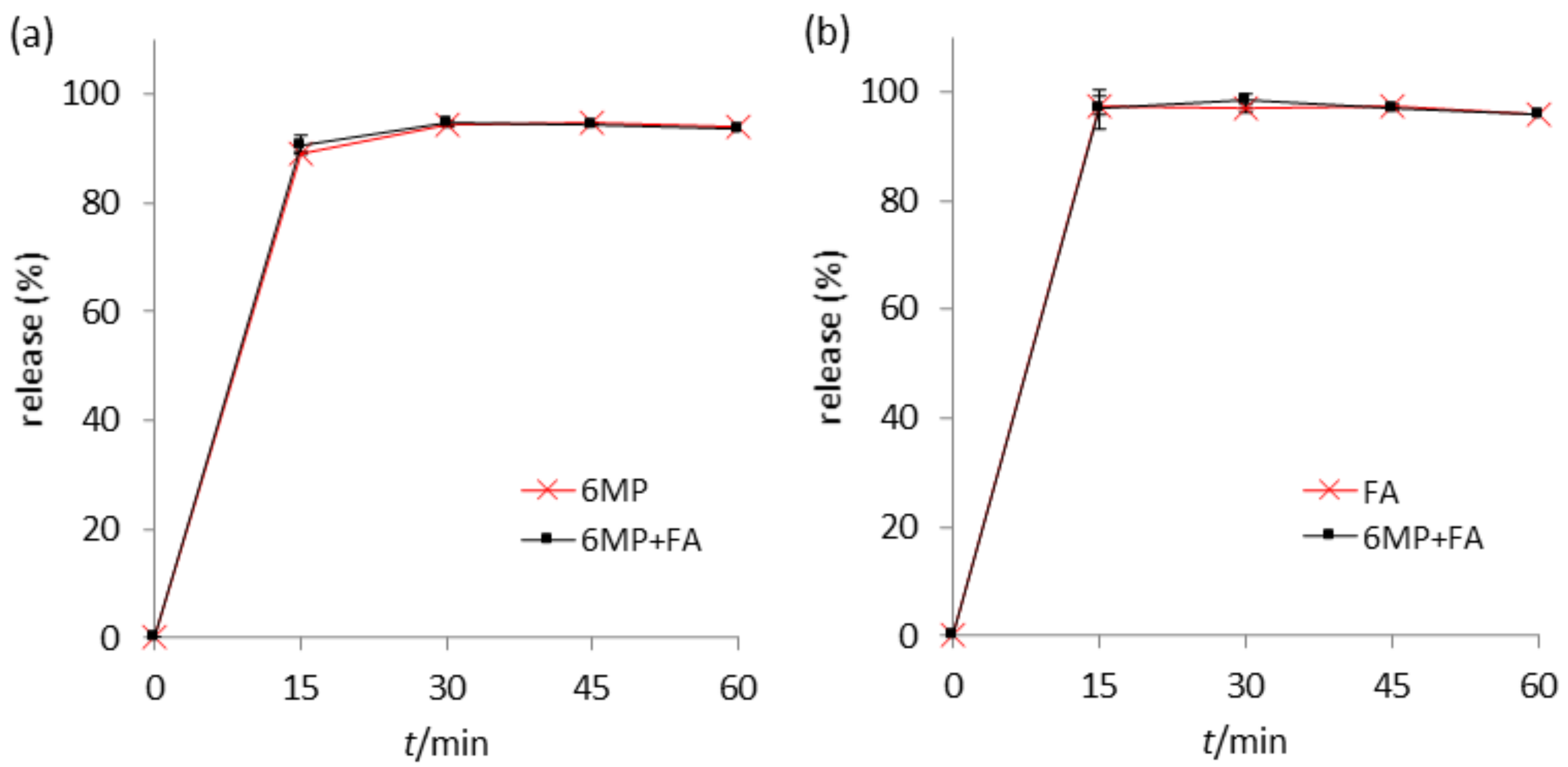
2.6. Stability in Fasted-State Simulated Intestinal Fluid (FaSSIF)
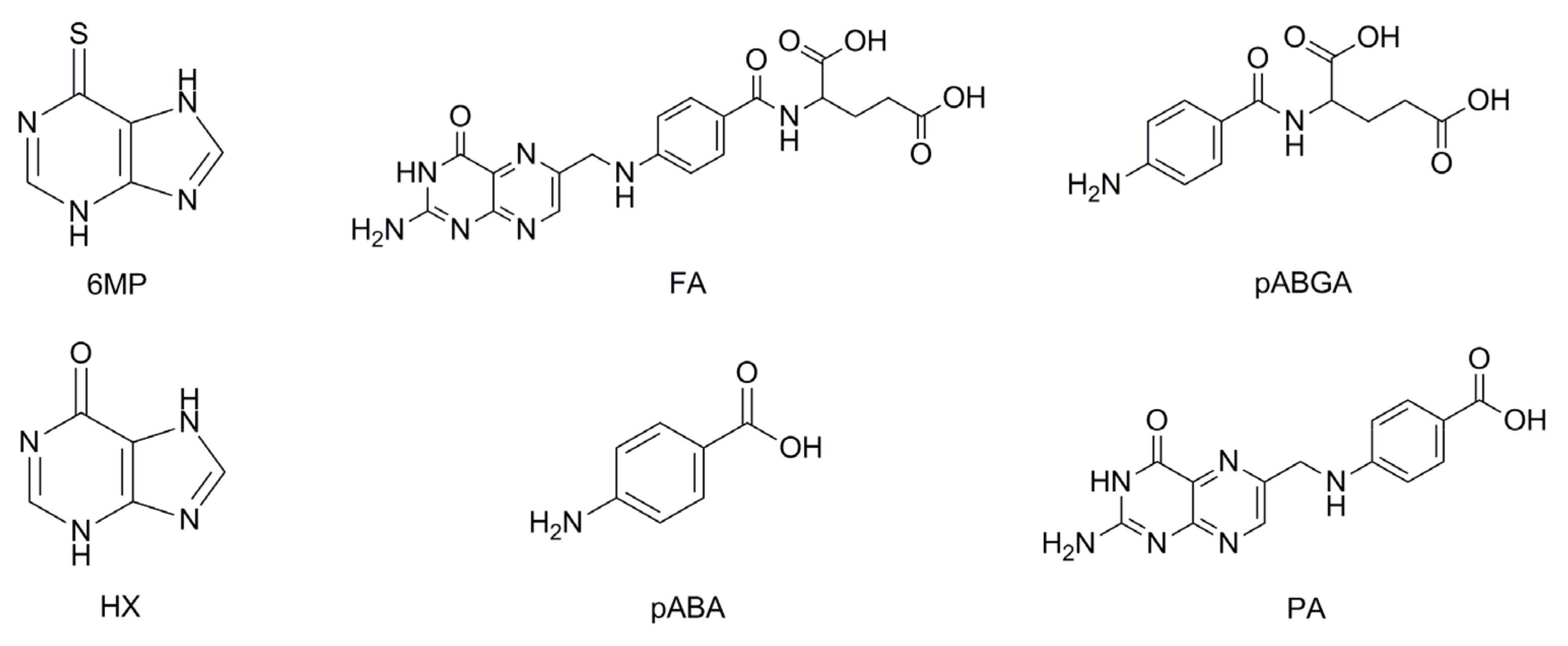
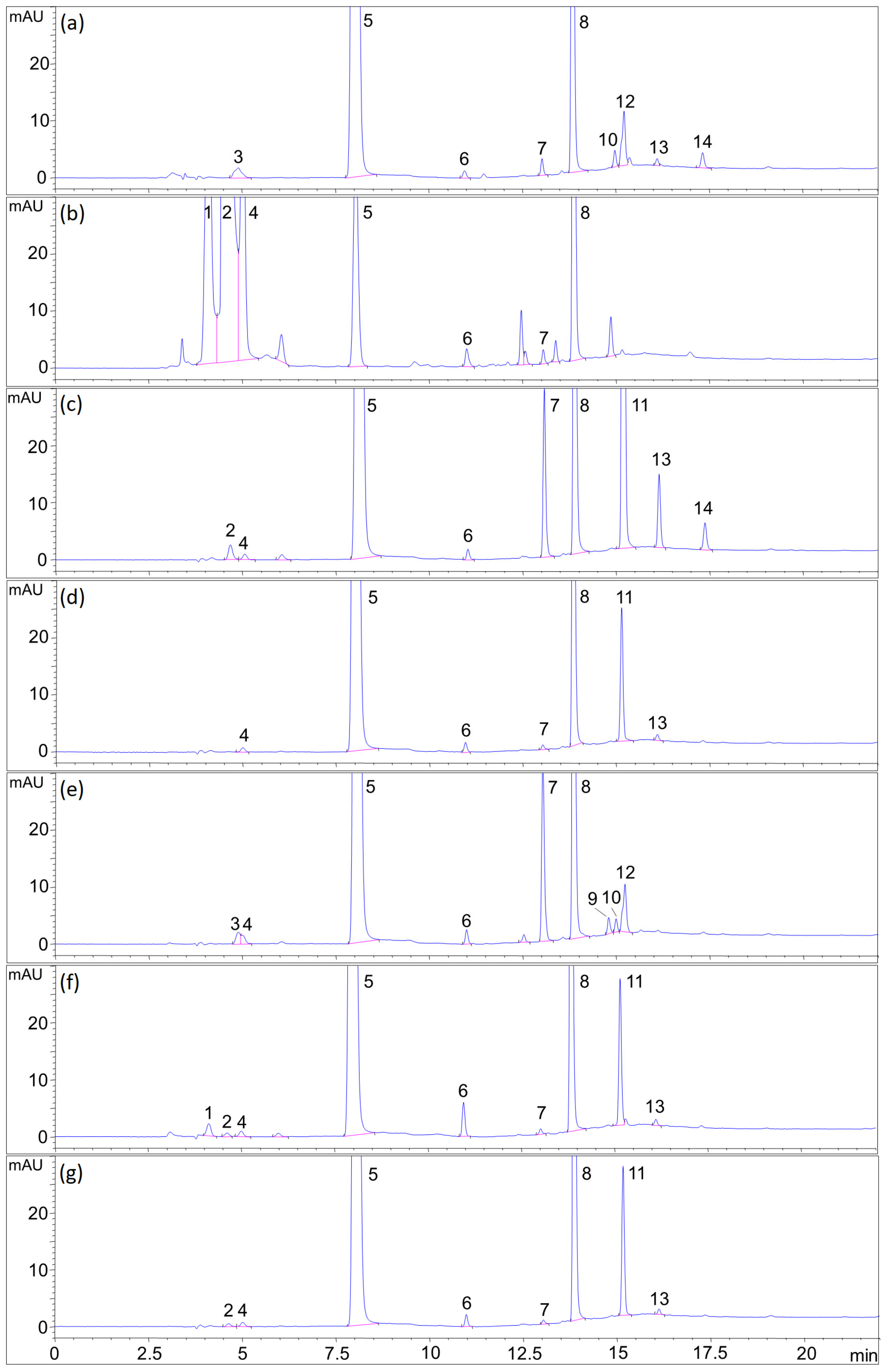
2.7. Forced Degradation
2.7.1. Acidic Hydrolysis
2.7.2. Basic Hydrolysis
2.7.3. Oxidative Stress
2.7.4. Thermal Stress
2.7.5. Photolysis
2.7.6. Summarized Results of Forced Degradation
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Sample Preparation
3.2.1. Preparation of Standard Blends, Formulation Blends and Placebo
3.2.2. Preparation of Standard, Placebo and Blend Solutions
3.3. Differential Scanning Calorimetry
3.4. Isothermal Stress Testing
3.5. Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy
3.5.1. Acquisition of FTIR Spectra
3.5.2. Multivariate Analysis of FTIR Spectra
3.6. Chromatographic Conditions
3.7. Dissolution Medium Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernstein, C.N.; Blanchard, J.F.; Rawsthorne, P.; Yu, N. The prevalence of extraintestinal diseases in inflammatory bowel disease: A population-based study. Am. J. Gastroenterol. 2001, 96, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Danese, S.; Sans, M.; Fiocchi, C. Inflammatory bowel disease: The role of environmental factors. Autoimmun. Rev. 2004, 3, 394–400. [Google Scholar] [CrossRef]
- Ravaglia, G.; Forti, P.; Maioli, F.; Martelli, M.; Servadei, L.; Brunetti, N.; Porcellini, E.; Licastro, F. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am. J. Clin. Nutr. 2005, 82, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhai, J.-X.; Liu, D.-W. Serum folate levels in schizophrenia: A meta-analysis. Psychiatry Res. 2016, 235, 83–89. [Google Scholar] [CrossRef]
- Lee, T.-Y.; Chiang, E.-P.; Shih, Y.-T.; Lane, H.-Y.; Lin, J.-T.; Wu, C.-Y. Lower serum folate is associated with development and invasiveness of gastric cancer. World J. Gastroenterol. 2014, 20, 11313–11320. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, Y.; Guo, H.; Jabir, M.S.; Liu, X.; Cui, W.; Li, D. Associations between folate and vitamin B12 levels and inflammatory bowel disease: A meta-analysis. Nutrients 2017, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Pan, F.; Chernew, M.E.; Fendrick, A.M. Impact of fixed-dose combination drugs on adherence to prescription medications. J. Gen. Intern. Med. 2008, 23, 611–614. [Google Scholar] [CrossRef]
- Thom, S.; Poulter, N.; Field, J.; Patel, A.; Prabhakaran, D.; Stanton, A.; Grobbee, D.E.; Bots, M.L.; Reddy, K.S.; Cidambi, R.; et al. Effects of a fixed-dose combination strategy on adherence and risk factors in patients with or at high risk of CVD: The UMPIRE randomized clinical trial. JAMA J. Am. Med. Assoc. 2013, 310, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Castellano, J.M.; Sanz, G.; Peñalvo, J.L.; Bansilal, S.; Fernández-Ortiz, A.; Alvarez, L.; Guzmán, L.; Linares, J.C.; García, F.; D’Aniello, F.; et al. A polypill strategy to improve adherence: Results from FOCUS (Fixed-dose Combination Drug for Secondary Cardiovascular Prevention) Project. J. Am. Coll. Cardiol. 2014, 64, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Ramjan, R.; Calmy, A.; Vitoria, M.; Mills, E.J.; Hill, A.; Cooke, G.; Ford, N. Systematic review and meta-analysis: Patient and programme impact of fixed-dose combination antiretroviral therapy. Trop. Med. Int. Health 2014, 19, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, C.N.; Fried, M.; Krabshuis, J.H.; Cohen, H.; Eliakim, R.; Fedail, S.; Gearry, R.; Goh, K.L.; Hamid, S.; Khan, A.G.; et al. World Gastroenterology Organization practice guidelines for the diagnosis and management of IBD in 2010. Inflamm. Bowel Dis. 2010, 16, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Chadha, R.; Bhandari, S. Drug-excipient compatibility screening-role of thermoanalytical and spectroscopic techniques. J. Pharm. Biomed. Anal. 2014, 87, 82–97. [Google Scholar] [CrossRef]
- De Gomes, E.C.L.; Mussel, W.N.; Resende, J.M.; Fialho, S.L.; Barbosa, J.; Yoshida, M.I. Chemical interactions study of antiretroviral drugs efavirenz and lamivudine concerning the development of stable fixed-dose combination formulations for AIDS treatment. J. Braz. Chem. Soc. 2013, 24, 573–579. [Google Scholar] [CrossRef]
- Jeličić, M.-L.; Brusač, E.; Amidžić Klarić, D.; Nigović, B.; Keser, S.; Mornar, A. Physicochemical Compatibility Investigation of Mesalazine and Folic Acid Using Chromatographic and Thermoanalytical Techniques. Pharmaceuticals 2020, 13, 187. [Google Scholar] [CrossRef]
- Petruševska, V.; Lasić, K.; Mornar, A. Compatibility investigation for a new antituberculotic fixed dose combination with an adequate drug delivery. Drug Dev. Ind. Pharm. 2020, 46, 1298–1307. [Google Scholar] [CrossRef]
- Rosasco, M.A.; Bonafede, S.L.; Faudone, S.N.; Segall, A.I. Compatibility study of tobramycin and pharmaceutical excipients using differential scanning calorimetry, FTIR, DRX, and HPLC. J. Therm. Anal. Calorim. 2018, 134, 1929–1941. [Google Scholar] [CrossRef]
- Franklin, S.J.; Younis, U.S.; Myrdal, P.B. Estimating the Aqueous Solubility of Pharmaceutical Hydrates. J. Pharm. Sci. 2016, 105, 1914–1919. [Google Scholar] [CrossRef] [Green Version]
- Kersten, K.M.; Matzger, A.J. Improved Pharmacokinetics of Mercaptopurine Afforded by a Thermally Robust Hemihydrate. Chem. Commun. 2016, 52, 5281–5284. [Google Scholar] [CrossRef] [Green Version]
- Varuna Kumara, J.B.; Ravikumara, N.R.; Madhusudhan, B. Evaluation of Surfactants-Assisted Folic Acid-Loaded Pectin Submicrospheres: Characterization and Hemocompatibility Assay. Ind. J. Clin. Biochem. 2016, 31, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Luo, Y.-H.; Zhao, J.; Sun, B.-W. Selection of excipients for dispersible tablets of itraconazole through the application of thermal techniques and Raman spectroscopy. J. Therm. Anal. Calorim. 2014, 115, 2391–2400. [Google Scholar] [CrossRef]
- Rojek, B.; Wesolowski, M.; Suchacz, B. Detection of compatibility between baclofen and excipients with aid of infrared spectroscopy and chemometry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 116, 532–538. [Google Scholar] [CrossRef]
- British Pharmacopoeia Commision. British Pharmacopoeia 2019; The Stationery Office: London, UK, 2019. [Google Scholar]
- ICH Secretariat. Validation of Analytical Procedures: Text and Methodology, Q2 (R1); ICH Secretariat: Geneva, Switzerland, 2005. [Google Scholar]
- Grimm, M.; Koziolek, M.; Kühn, J.P.; Weitschies, W. Interindividual and intraindividual variability of fasted state gastric fluid volume and gastric emptying of water. Eur. J. Pharm. Biopharm. 2018, 127, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Impact from the Recent Issuance of ANVISA Resolution RDC-53/2015 on Pharmaceutical Small Molecule Forced Degradation Study Requirements. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/184364-Impact-from-the-Recent-Issuance-of-ANVISA-Resolution-RDC-53-2015-on-Pharmaceutical-Small-Molecule-Forced-Degradation-Study-Requirements (accessed on 8 March 2021).
- Thomas, A.H.; Suárez, G.; Cabrerizo, F.M.; Martino, R.; Capparelli, A.L. Study of the photolysis of folic acid and 6-formylpterin in acid aqueous solutions. J. Photochem. Photobiol. A Chem. 2000, 135, 147–154. [Google Scholar] [CrossRef]
- Gazzali, A.M.; Lobry, M.; Colombeau, L.; Acherar, S.; Azaïs, H.; Mordon, S.; Arnoux, P.; Baros, F.; Vanderesse, R.; Frochot, C. Stability of folic acid under several parameters. Eur. J. Pharm. Sci. 2016, 93, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Jamil Akhtar, M.; Ataullah Khan, M.; Ahmad, I. Identification of photoproducts of folic acid and its degradation pathways in aqueous solution. J. Pharm. Biomed. Anal. 2003, 31, 579–588. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Rojek, B.; Wesolowski, M. FTIR and TG analyses coupled with factor analysis in a compatibility study of acetazolamide with excipients. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 208, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusač, E.; Jeličić, M.-L.; Klarić, D.A.; Nigović, B.; Turk, N.; Klarić, I.; Mornar, A. Pharmacokinetic profiling and simultaneous determination of thiopurine immunosuppressants and folic acid by chromatographic methods. Molecules 2019, 24, 3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Range (µg/mL) | Regression Equation | Correlation Coefficient (r) | Relative Retention Time 1 | Relative Response Factor 1 | LOD (µg/mL) | LOQ (µg/mL) |
|---|---|---|---|---|---|---|---|
| 6MP | 400–600 | y = 8.48 x − 10.37 | 0.9999 | / | / | n/a 2 | n/a |
| FA | 20–30 | y = 34.10 x + 5.12 | 0.9999 | / | / | n/a | n/a |
| 6MP 3 | 1.00–15.00 | y = 8.57 x − 1.63 | 0.9997 | / | 1.00 | n/a | n/a |
| FA 3 | 0.05–0.75 | y = 33.45 x + 0.57 | 0.9995 | / | 1.00 | n/a | n/a |
| HX | 0.15–15.00 | y = 12.54 x − 0.97 | 0.9999 | 0.63 | 1.46 | 0.06 | 0.15 |
| pABGA | 0.05–0.75 | y = 24.95 x + 0.04 | 0.9999 | 0.79 | 0.75 | 0.02 | 0.05 |
| pABA | 0.05–0.20 | y = 57.51 x + 0.25 | 0.9995 | 0.87 | 1.72 | 0.02 | 0.05 |
| PA | 0.05–0.75 | y = 40.40 x − 0.29 | 0.9992 | 1.07 | 1.21 | 0.02 | 0.05 |
| Analyte | Low/Medium/High Concentrations (μg/mL) | Accuracy (Recovery, Mean (%) ± RSD, n = 3) | Precision (RSD, %) | |||
|---|---|---|---|---|---|---|
| Low | Medium | High | Intra-Day Precision (n = 6) | Inter-Day Precision (n = 9) | ||
| 6MP | 400/500/600 | 99.64 ± 1.73 | 99.78 ± 0.38 | 100.09 ± 0.11 | 0.23 | 0.28 |
| FA | 20/25/30 | 99.98 ± 1.74 | 100.13 ± 0.77 | 100.93 ± 1.06 | 0.49 | 0.70 |
| HX | 1.50/7.50/12.50 | 103.82 ± 1.51 | 100.21 ± 1.72 | 97.40 ± 0.77 | 1.64 | 2.06 |
| pABGA | 0.15/0.50/0.75 | 102.83 ± 4.16 | 102.15 ± 1.04 | 101.68 ± 1.03 | 1.18 | 3.24 |
| pABA | 0.07/0.15/0.20 | 98.38 ± 3.53 | 104.63 ± 3.19 | 105.85 ± 0.48 | 4.34 | 4.72 |
| PA | 0.15/0.50/0.75 | 97.09 ± 3.45 | 104.45 ± 1.23 | 105.53 ± 1.42 | 2.23 | 2.96 |
| Sample | Physical Change (In Comparison To Nonstressed Sample) | Recovery (In Comparison To Nonstressed Sample) Mean (%) ± RSD, n = 3 | |
|---|---|---|---|
| 6MP drug substance | no significant changes | 100.89 ± 0.44 | |
| FA drug substance | 99.86 ± 0.75 | ||
| drug substance blend | 6MP | 100.37 ± 0.68 | |
| FA | 98.97 ± 0.49 | ||
| formulation blend | 6MP | 101.09 ± 0.43 | |
| FA | 100.94 ± 0.21 | ||
| Stress Type | Degradation Condition | Degradation, % | Degradation Profile Remarks (Peaks Numbered as in Figure 7) | |||||
|---|---|---|---|---|---|---|---|---|
| 6MP | FA | Drug Substance Blends | Formulation Blends | |||||
| 6MP | FA | 6MP | FA | |||||
| acid hydrolytic | 0.1 M HCl, 4 h | 3.1 | 8.9 | n.d. 1 | 10.9 | n.d. | 9.2 | FA degraded to pABGA and impurities 10 and 12 |
| base hydrolytic | 0.1 M NaOH, 5 days | 7.2 | 1.8 | 2.6 | 1.9 | 90.0 | 1.8 | 6MP markedly degraded in formulation blend |
| oxidative | 0.1% H2O2, 16 h | 4.4 | n.d. | 6.1 | 1.5 | 5.1 | 1.7 | possible dimerization and oxidation of 6MP to degradants 7, 11 and 13 |
| thermal (solid) | 60 °C, 7 days | 0.7 | n.d. | n.d. | n.d. | 1.2 | n.d. | / |
| thermal (solution) | 60 °C, 5 days | 4.5 | 7.7 | 1.2 | 6.2 | 2.7 | 4.4 | formation of PA |
| photolytic (solid) | indirect sunlight, 7 days | n.d. | 2.1 | n.d. | 4.4 | n.d. | 6.9 | pABGA as principal FA degradant |
| photolytic (solution) | indirect sunlight, 15 min | 0.5 | 5.1 | n.d. | n.d. | 0.2 | 1.4 | pABGA as principal FA degradant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brusač, E.; Jeličić, M.-L.; Cvetnić, M.; Amidžić Klarić, D.; Nigović, B.; Mornar, A. A Comprehensive Approach to Compatibility Testing Using Chromatographic, Thermal and Spectroscopic Techniques: Evaluation of Potential for a Monolayer Fixed-Dose Combination of 6-Mercaptopurine and Folic Acid. Pharmaceuticals 2021, 14, 274. https://doi.org/10.3390/ph14030274
Brusač E, Jeličić M-L, Cvetnić M, Amidžić Klarić D, Nigović B, Mornar A. A Comprehensive Approach to Compatibility Testing Using Chromatographic, Thermal and Spectroscopic Techniques: Evaluation of Potential for a Monolayer Fixed-Dose Combination of 6-Mercaptopurine and Folic Acid. Pharmaceuticals. 2021; 14(3):274. https://doi.org/10.3390/ph14030274
Chicago/Turabian StyleBrusač, Edvin, Mario-Livio Jeličić, Matija Cvetnić, Daniela Amidžić Klarić, Biljana Nigović, and Ana Mornar. 2021. "A Comprehensive Approach to Compatibility Testing Using Chromatographic, Thermal and Spectroscopic Techniques: Evaluation of Potential for a Monolayer Fixed-Dose Combination of 6-Mercaptopurine and Folic Acid" Pharmaceuticals 14, no. 3: 274. https://doi.org/10.3390/ph14030274
APA StyleBrusač, E., Jeličić, M.-L., Cvetnić, M., Amidžić Klarić, D., Nigović, B., & Mornar, A. (2021). A Comprehensive Approach to Compatibility Testing Using Chromatographic, Thermal and Spectroscopic Techniques: Evaluation of Potential for a Monolayer Fixed-Dose Combination of 6-Mercaptopurine and Folic Acid. Pharmaceuticals, 14(3), 274. https://doi.org/10.3390/ph14030274