
Structure-Based Virtual Screening and De Novo Design of PIM1 Inhibitors with Anticancer Activity from Natural Products


Abstract
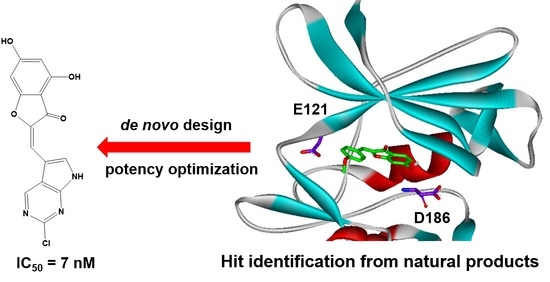
:
1. Introduction
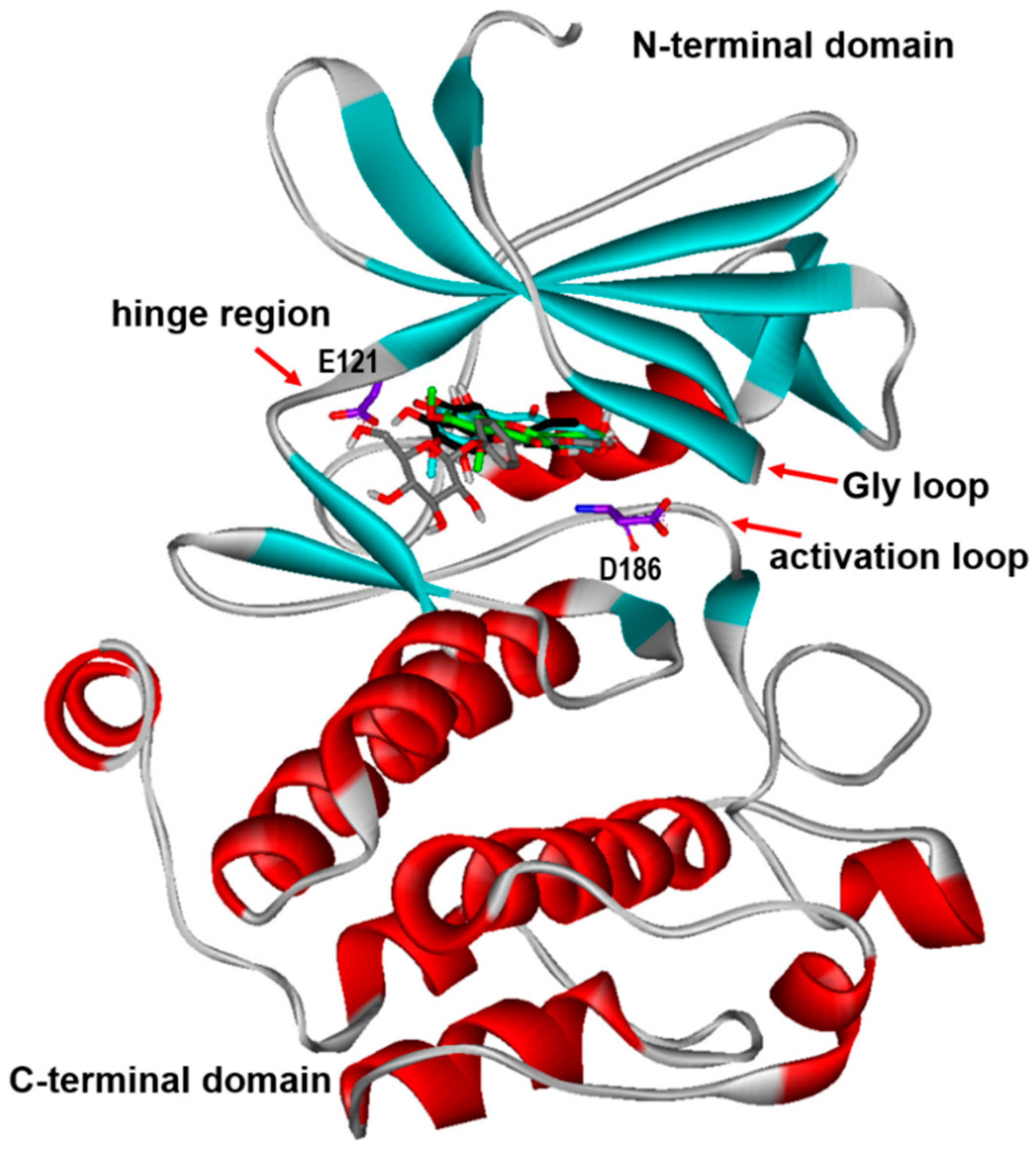
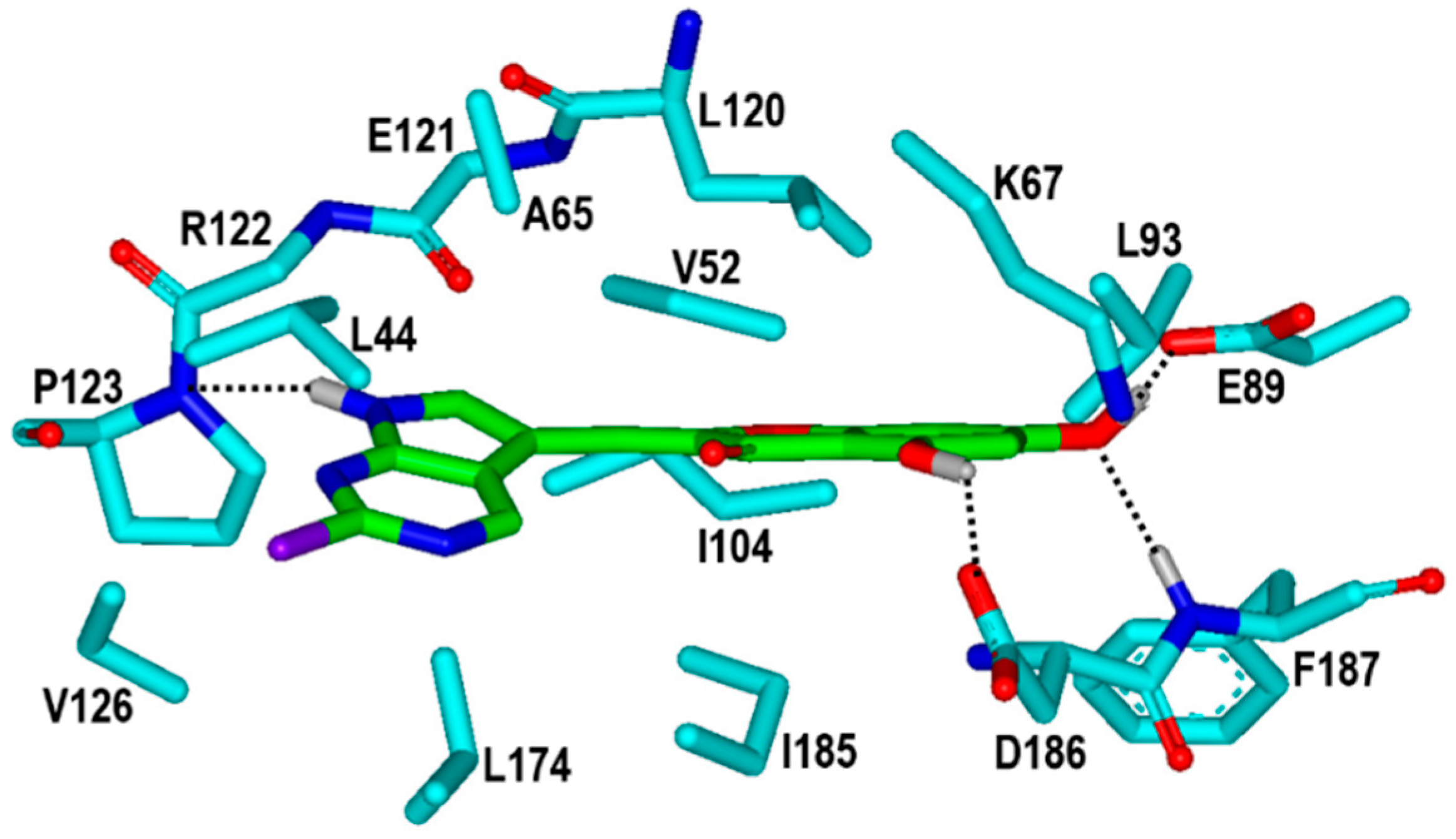
2. Results and Discussion
3. Materials and Methods
3.1. Preparation of the Atomic Coordinates of PIM1 and the Natural Product Library
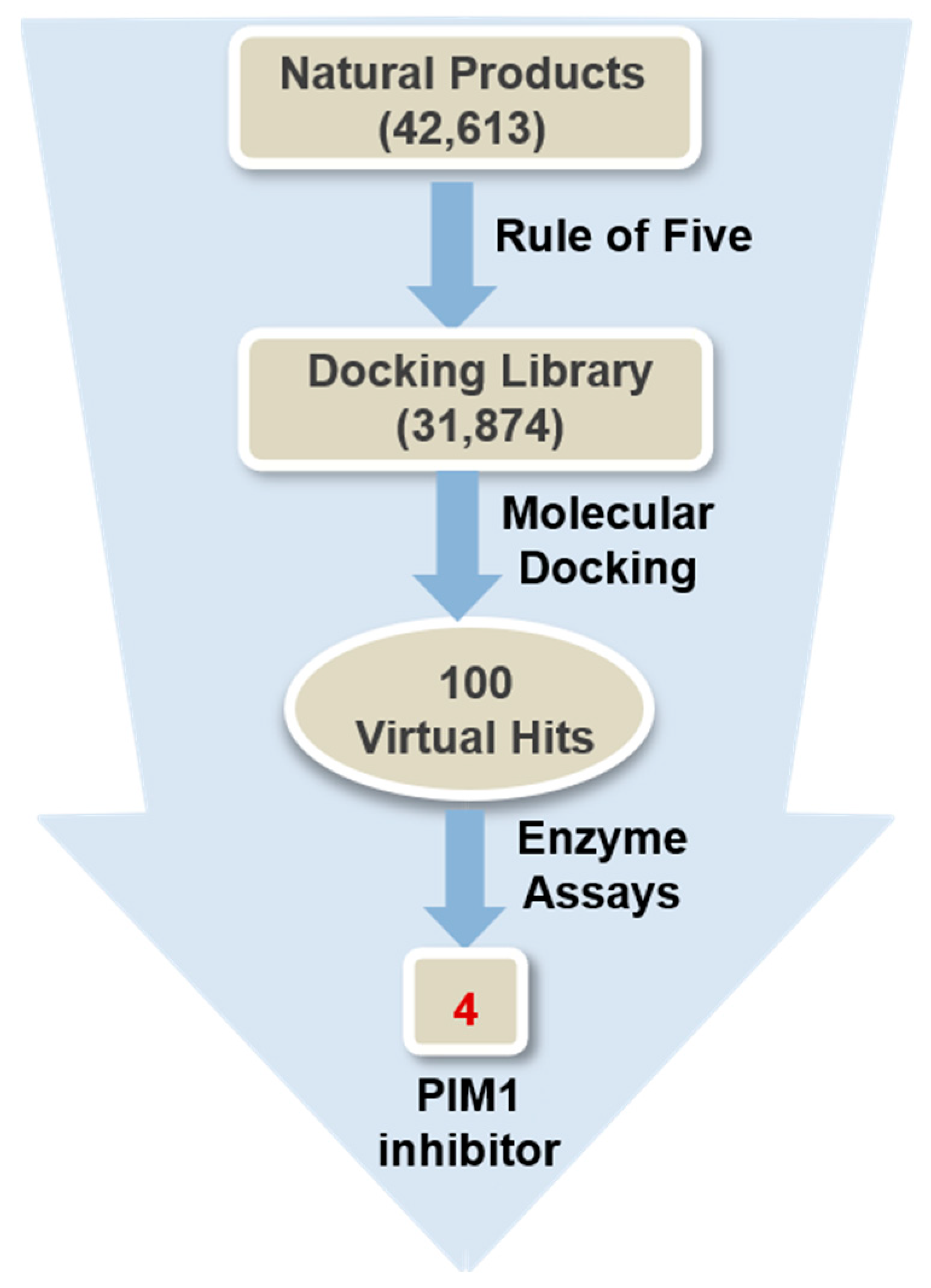
3.2. Virtual Screening to Identify the PIM1 Inhibitors of Natural Origin
3.3. De Novo Design
3.4. Chemical Synthesis
3.4.1. General Methods
3.4.2. General Procedure I (GPI) (Compound 5–14)
3.4.3. General Procedure II (GPII) (Compound 15 and 16)
3.5. Enzyme Inhibition Assays
3.6. Cell Proliferation Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brault, L.; Gasser, C.; Bracher, F.; Huber, K.; Knapp, S.; Schwaller, J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 2010, 95, 1004–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawijn, M.C.; Alendar, A.; Berns, A. For better or for worse: The role of PIM oncogenes in tumorigenesis. Nat. Rev. Cancer 2011, 11, 23–34. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Verbeek, S.; Krimpenfort, P.; Domen, J.; Saris, C.; Radaszkiewicz, T.; Berns, A. Predisposition to lymphomagenesis in PIM-1 transgenic mice: Cooperation with C-myc and N-myc in murine leukemia virus-induced tumors. Cell 1989, 56, 673–682. [Google Scholar] [CrossRef]
- Cen, B.; Xiong, Y.; Song, J.H.; Mahajan, S.; DuPont, R.; McEachern, K.; DeAngelo, D.J.; Cortes, J.E.; Minden, M.D.; Ebens, A.; et al. The Pim-1 protein kinase is an important regulator of MET receptor tyrosine kinase levels and signaling. Mol. Cell. Biol. 2014, 34, 2517–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.W.; Chan, D.C.; Donald, C.; Lilly, M.B.; Kraft, A.S. PIM family kinases enhance tumor growth of prostate cancer cells. Mol. Cancer Res. 2005, 3, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, C.; Nakamoto, Y.; Lu, P.; Tsuneyama, K.; Popivanova, B.K.; Kaneko, S.; Mukaida, N. Aberrant expression of serine/threonine kinase PIM-3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int. J. Cancer 2005, 114, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Popivanova, B.K.; Li, Y.-Y.; Zheng, H.; Omura, K.; Fujii, C.; Tsuneyama, K.; Mukaida, N. Proto-oncogene, PIM-3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent bad-mediated apoptosis. Cancer Sci. 2007, 98, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-C.; Tsuneyama, K.; Takahashi, H.; Miwa, S.; Sugiyama, T.; Popivanova, B.K.; Fujii, C.; Nomoto, K.; Mukaida, N.; Takano, Y. Aberrant PIM-3 expression is involved in gastric adenoma-adenocarcinoma sequence and cancer progression. J. Cancer Res. Clin. Oncol. 2008, 134, 481–488. [Google Scholar] [CrossRef]
- Laird, P.W.; van der Lugt, N.M.T.; Clarke, A.; Domen, J.; Linders, K.; McWhir, J.; Berns, A.; Hooper, M. In vivo analysis of PIM-1 deficiency. Nucleic Acids Res. 1993, 21, 4750–4755. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, K.; Linn, D.E.; Yang, X.; Guo, Z.; Shimelis, H.; Nakanishi, T.; Ross, D.D.; Chen, H.; Fazli, L.; et al. The 44-kDa PIM-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J. Biol. Chem. 2008, 283, 3349–3356. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Burcu, M.; Linn, D.E.; Qiu, Y.; Baer, M.R. PIM-1 kinase protects p-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol. Pharmacol. 2010, 78, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.D.; Black, J.; Futer, O.; Swenson, L.; Hare, B.; Fleming, M.; Saxena, K. PIM-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J. Biol. Chem. 2005, 280, 13728–13734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Muller, S.; Bullock, A.N.; Schwaller, J.; Sundstrom, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef] [Green Version]
- Kutchukian, P.S.; Wassermann, A.M.; Lindvall, M.K.; Wright, S.K.; Ottl, J.; Jacob, J.; Scheufler, C.; Marzinzik, A.; Brooijmans, N.; Glick, M. Large scale meta-analysis of fragment-based screening campaigns: Privileged fragments and complementary technologies. J. Biomol. Screen. 2015, 20, 588–596. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Zhang, W.; Li, L.; Xie, Y.; Li, W.; Huang, N. A new target for an old drug: Identifying mitoxantrone as a nanomolar inhibitor of PIM1 kinase via kinome-wide selectivity modeling. J. Med. Chem. 2013, 56, 2619–2629. [Google Scholar] [CrossRef]
- Lee, S.J.; Han, B.G.; Cho, J.W.; Choi, J.S.; Lee, J.K.; Song, H.J.; Koh, J.S.; Lee, B.I. Crystal structure of PIM1 kinase in complex with a pyrido[4,3-D]pyrimidine derivative suggests a unique binding mode. PLoS ONE 2013, 8, e70358. [Google Scholar] [CrossRef] [Green Version]
- Bogusz, J.; Zrubek, K.; Rembacz, K.P.; Grudnik, P.; Golik, P.; Romanowska, M.; Wladyka, B.; Dubin, G. Structural analysis of PIM1 kinase complexes with ATP-competitive inhibitors. Sci. Rep. 2017, 7, 13399. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Debreczeni, J.E.; Fedorov, O.Y.; Nelson, A.; Marsden, B.D.; Knapp, S. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in Moloney murine leukemia virus (PIM-1) kinase. J. Med. Chem. 2005, 48, 7604–7614. [Google Scholar] [CrossRef]
- Pogacic, V.; Bullock, A.N.; Fedorov, O.; Filippakopoulos, P.; Gasser, C.; Biondi, A.; Meyer-Monard, S.; Knapp, S.; Schwaller, J. Structural analysis identifies imidazo[1,2-b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res. 2007, 67, 6916–6924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, A.C.; Jacobs, M.; Stuver-Moody, C. Docking study yields four novel inhibitors of the protooncogene Pim-1 kinase. J. Med. Chem. 2008, 51, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Knaak, C.; Ma, J.; Beharry, Z.M.; McInnes, C.; Wang, W.; Kraft, A.S.; Smith, C.D. Synthesis and evaluation of novel inhibitors of Pim-1 and Pim-2 protein kinases. J. Med. Chem. 2009, 52, 74–86. [Google Scholar] [CrossRef] [Green Version]
- Qian, K.; Wang, L.; Cywin, C.L.; Farmer, B.T., II; Hickey, E.; Homon, C.; Jakes, S.; Kashem, M.A.; Lee, G.; Leonard, S.; et al. Hit to lead account of the discovery of a new class of inhibitors of PIM kinases and crystallographic studies revealing an unusual kinase binding mode. J. Med. Chem. 2009, 52, 1814–1827. [Google Scholar] [CrossRef]
- Nakano, H.; Saito, N.; Parker, L.; Tada, Y.; Abe, M.; Tsuganezawa, K.; Yokoyama, S.; Tanaka, A.; Kojima, H.; Okabe, T.; et al. Rational evolution of a novel type of potent and selective proviral integration site in Moloney murine leukemia virus kinase 1 (PIM1) inhibitor from a screening-hit compound. J. Med. Chem. 2012, 55, 5151–5164. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Hasegawa, T.; Kojima, H.; Okabe, T.; Nagano, T. Design and synthesis of potent and selective PIM kinase inhibitors by targeting unique structure of ATP-binding pocket. ACS Med. Chem. Lett. 2017, 8, 504–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, M.T.; Nishiguchi, G.; Han, W.; Lan, J.; Simmons, R.; Atallah, G.; Ding, Y.; Tamez, V.; Zhang, Y.; Mathur, M.; et al. Identification of N-(4-((1R,3S,5S)-3-amino-5-methylcyclohexyl)pyridin-3-yl)-6-(2,6-difluorophenyl)-5-fluoropicolinamide (PIM447), a potent and selective proviral insertion site of Moloney murine leukemia (PIM) 1, 2, and 3 kinase inhibitor in clinical trials for hematological malignancies. J. Med. Chem. 2015, 58, 8373–8386. [Google Scholar]
- Andreoli, M.; Persico, M.; Kumar, A.; Orteca, N.; Kumar, V.; Pepe, A.; Mahalingam, S.; Alegria, A.E.; Petrella, L.; Sevciunaite, L.; et al. Identification of the first inhibitor of the GBP1:PIM1 interaction. Implications for the development of a new class of anticancer agents against paclitaxel resistant cancer cells. J. Med. Chem. 2014, 57, 7916–7932. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, R.L.; Goyal, N.; Bratton, M.; Townley, I.; Pham, N.A.; Tram, P.; Stone, T.; Geathers, J.; Nguyen, K.; Sridhar, J. Identification of quinones as novel PIM1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3187–3191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barberis, C.; Moorcroft, N.; Arendt, C.; Levit, M.; Moreno-Mazza, S.; Batchelor, J.; Mechin, I.; Majid, T. Discovery of N-substituted 7-azaindoles as PIM1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4730–4734. [Google Scholar] [CrossRef]
- Holder, S.; Lilly, M.; Brown, M.L. Comparative molecular field analysis of flavonoid inhibitors of the PIM-1 kinase. Bioorg. Med. Chem. 2007, 15, 6463–6473. [Google Scholar] [CrossRef] [PubMed]
- Cheney, I.W.; Yan, S.; Appleby, T.; Walker, H.; Vo, T.; Yao, N.; Hamatake, R.; Hong, Z.; Wu, J.Z. Identification and structure—activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg. Med. Chem. Lett. 2007, 17, 1679–1683. [Google Scholar] [CrossRef]
- Aouidate, A.; Ghaleb, A.; Ghamali, M.; Ousaa, A.; Choukrad, M.; Sbai, A.; Bouachrine, M.; Lakhlifi, T. 3D QSAR studies, molecular docking and ADMET evaluation, using thiazolidine derivatives as template to obtain new inhibitors of PIM1 kinase. Comput. Biol. Chem. 2018, 74, 201–211. [Google Scholar] [CrossRef]
- Watanabe, C.; Watanabe, H.; Fukuzawa, K.; Parker, L.J.; Okiyama, Y.; Yuki, H.; Yokoyama, S.; Nakano, H.; Tanaka, S.; Honma, T. Theoretical analysis of activity cliffs among benzofuranone-class Pim1 inhibitors using the fragment molecular orbital method with molecular mechanics Poisson–Boltzmann surface area (FMO+MM-PBSA) approach. J. Chem. Inf. Model. 2017, 57, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.J.; Carroll, A.R.; Pham, N.B.; Baron, P.; Palframan, M.E.; Suraweera, L.; Pierens, G.K.; Muresan, S. Developing a drug-like natural product library. J. Nat. Prod. 2008, 71, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, W.; Sun, Z.; Zhang, J.Z.H.; Ji, C. Protein-ligand empirical interaction components for virtual wcreening. J. Chem. Inf. Model. 2017, 57, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Su, M.; Han, L.; Liu, J.; Yang, Q.; Li, Y.; Wang, R. Forging the basis for developing protein–ligand interaction scoring functions. Acc. Chem. Res. 2017, 50, 302–309. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Leach, A.R.; Kuntz, I.D. Ligand solvation in molecular docking. Proteins 1999, 34, 4–16. [Google Scholar] [CrossRef]
- Park, H.; Jung, H.-Y.; Mah, S.; Kim, K.; Hong, S. Kinase and GPCR polypharmacological approach for the identification of efficient anticancer medicines. Org. Biomol. Chem. 2020, 18, 8402–8413. [Google Scholar] [CrossRef]
- Park, H.; Jung, H.-Y.; Kim, K.; Kim, M.; Hong, S. Rational computational design of fourth-generation EGFR inhibitors to combat drug-resistant non-small cell lung cancer. Int. J. Mol. Sci. 2020, 21, 9323. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Tsuganezawa, K.; Watanabe, H.; Parker, L.; Yuki, H.; Taruya, S.; Nakagawa, Y.; Kamei, D.; Mori, M.; Ogawa, N.; Tomabechi, Y.; et al. A novel Pim-1 kinase inhibitor targeting residues that bind the substrate peptide. J. Mol. Biol. 2012, 417, 240–252. [Google Scholar] [CrossRef]
- Schneider, P.; Welin, M.; Svensson, B.; Walse, B.; Schneider, G. Virtual screening and design with machine intelligence applied to Pim-1 kinase inhibitors. Mol. Inform. 2020, 39, e2000109. [Google Scholar] [CrossRef] [PubMed]
- Casuscelli, F.; Ardini, E.; Avanzi, N.; Casale, E.; Cervi, G.; D’Anello, M.; Donati, D.; Faiardi, D.; Ferguson, R.D.; Fogliatto, G.; et al. Discovery and optimization of pyrrolo[1,2-a]pyrazinones leads to novel and selective inhibitors of Pim kinases. Bioorg. Med. Chem. 2013, 21, 7364. [Google Scholar] [CrossRef]
- Henley, Z.A.; Bax, B.D.; Inglesby, L.M.; Champigny, A.; Gaines, S.; Faulder, P.; Le, J.; Thomas, D.A.; Washio, Y.; Baldwin, I.R. From PIM1 to PI3K delta via GSK3 beta: Target hopping through the kinome. ACS Med. Chem. Lett. 2017, 8, 1093–1098. [Google Scholar] [CrossRef]
- Wang, H.L.; Cee, V.J.; Chavez, F.; Lanman, B.A.; Reed, A.B.; Wu, B.; Guerrero, N.; Lipford, J.R.; Sastri, C.; Winston, J.; et al. The discovery of novel 3-(pyrazin-2-yl)-1H-indazoles as potent pan-Pim kinase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 834–840. [Google Scholar] [CrossRef]
- Muley, L.; Baum, B.; Smolinski, M.; Freindorf, M.; Heine, A.; Klebe, G.; Hangauer, D.G. Enhancement of hydrophobic interactions and hydrogen bond strength by cooperativity: Synthesis, modeling, and molecular dynamics simulations of a congeneric series of thrombin inhibitors. J. Med. Chem. 2010, 53, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-H.; Ueng, S.-H.; Tseng, C.-T.; Hung, M.-S.; Song, J.-S.; Wu, J.-S.; Liao, F.-Y.; Fan, Y.-S.; Wu, M.-H.; Hsiao, W.-C.; et al. Important hydrogen bond networks in indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor design revealed by crystal structures of imidazoleisoindole derivatives with IDO1. J. Med. Chem. 2016, 59, 282–293. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Chew, E.-H.; Go, M.-L. Functionalized aurones as inducers of NAD(P)H:quinone oxidoreductase 1 that activate AhR/XRE and Nrf2/ARE signaling pathways: Synthesis, evaluation and SAR. Eur. J. Med. Chem. 2010, 45, 2957–2971. [Google Scholar] [CrossRef]
- Gao, X.; Liu, X.; Lu, Y.; Wang, Y.; Cao, W.; Liu, X.; Hu, H.; Wang, H. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer 2019, 26, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerns, E.H.; Di, L. Pharmaceutical profiling in drug discovery. Drug Discov. Today 2003, 8, 316–323. [Google Scholar] [CrossRef]
- Innocenti, P.; Cheung, K.-M.J.; Solanki, S.; Mas-Droux, C.; Rowan, F.; Yeoh, S.; Boxall, K.; Westlake, M.; Pickard, L.; Hardy, T.; et al. Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: Structure–activity relationship, structural biology, and cellular activity. J. Med. Chem. 2012, 55, 3228–3241. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, J.; Gasteiger, J.; Klebe, G. Comparison of automatic three-dimensional model builders using 639 X-ray structures. J. Chem. Inf. Model. 1994, 34, 1000–1008. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Mehler, E.L.; Solmajer, T. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. 1991, 4, 903–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stouten, P.F.W.; Frömmel, C.; Nakamura, H.; Sander, C. An effective solvation term based on atomic occupancies for use in protein simulations. Mol. Simul. 1993, 10, 97–120. [Google Scholar] [CrossRef]
- Chung, K.-C.; Park, H. Accuracy enhancement in the estimation of molecular hydration free energies by implementing the intramolecular hydrogen bond effects. J. Cheminform. 2015, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Gao, Y.; Lai, L. LigBuilder: A multi-purpose program for structure-based drug design. J. Mol. Model. 2000, 6, 498–516. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
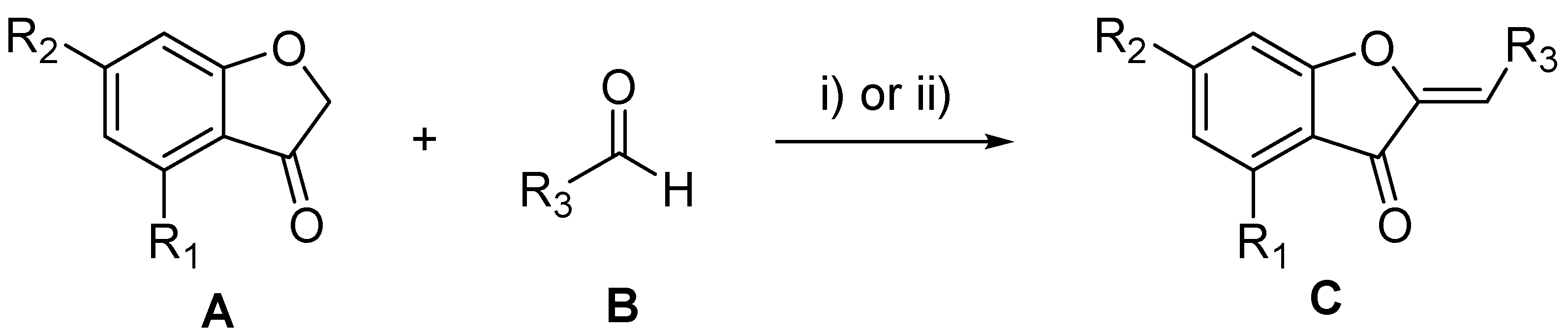{kind=link}
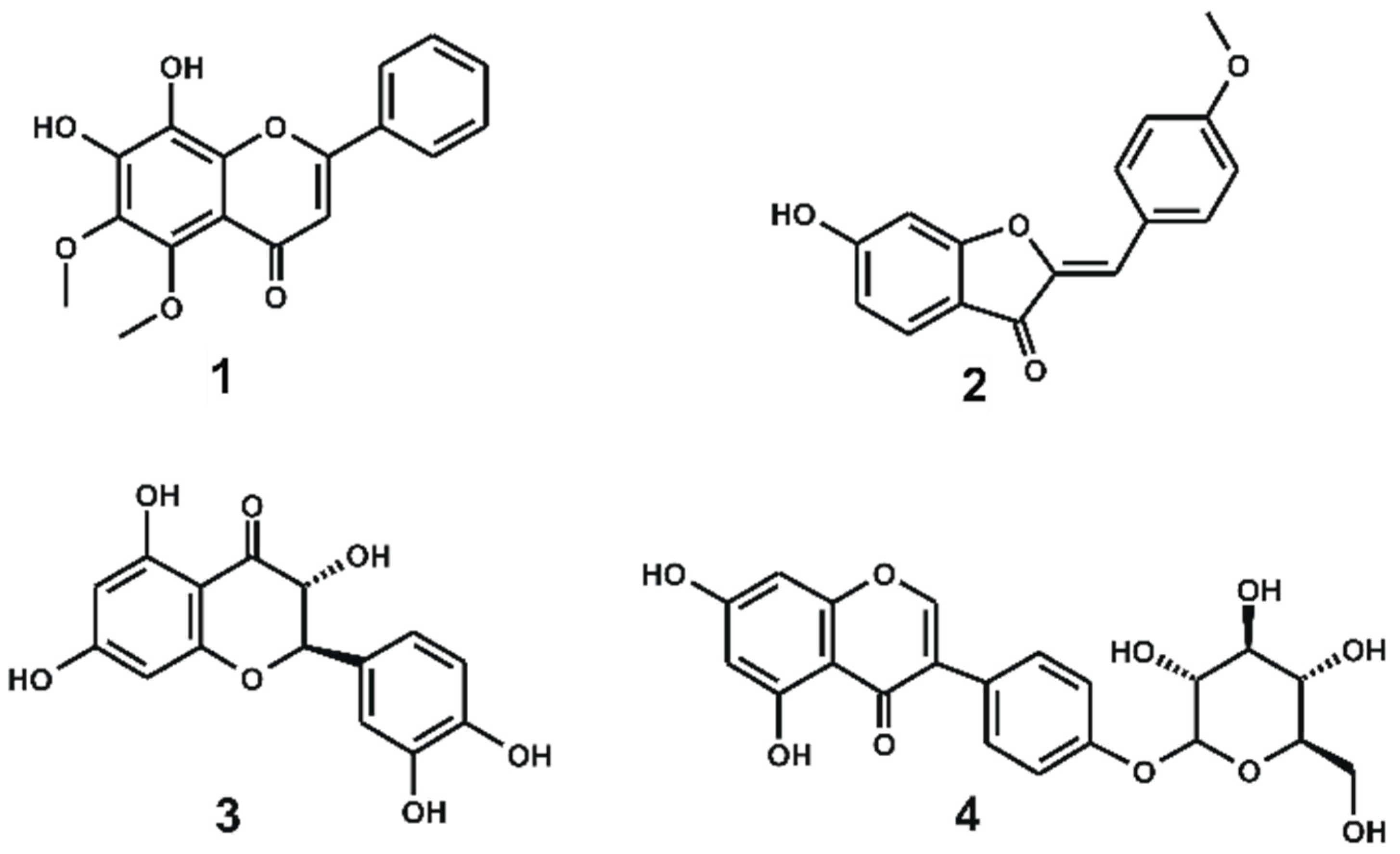
| Inhibitor | Purity (%) | IC50 (μM) a | Plant Source |
|---|---|---|---|
| 1 | 92% | 0.58 ± 0.2 | Scutellaria ramosissima |
| 2 | 95% | 1.2 ± 0.1 | Soja and Lygos spp. |
| 3 | 95% | 4.0 ± 0.6 | Xanthoceras sorbifolia |
| 4 | 95% | 20.2 ± 2 | Sophora japonica and Piptanthus nepalensis |
| staurosporine | 98% | 0.0056 ± 0.0008 | Not applicable |
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | IC50 (μM) b | Synthetic Procedure c |
| 2 | H | OH |  | 1.19 ± 0.07 | GPI |
| 5 | H | OH |  | 0.26 ± 0.05 | GPI |
| 6 | OH | OH |  | 0.31 ± 0.02 | GPI |
| 7 | OH | OH |  | 1.45 ± 0.1 | GPI |
| 8 | H | OH |  | 0.20 ± 0.03 | GPI |
| 9 | OH | OH |  | 0.15 ± 0.01 | GPI |
| 10 | OH | OH |  | 0.68 ± 0.08 | GPI |
| 11 | H | OH |  | 5.23 ± 0.4 | GPI |
| 12 | OH | OH |  | 5.86 ± 1 | GPI |
| 13 | OH | OH |  | 1.73 ± 0.3 | GPI |
| 14 | OH | OCH3 |  | 0.018 ± 0.002 | GPI |
| 15 | OH | OCH3 |  | 0.0088 ± 0.002 | GPII |
| 16 | OH | OH |  | 0.0067 ± 0.003 | GPII |
| staurosporine | 0.0056 ± 0.0008 | ||||
| Inhibitor | IC50 (μM) a |
|---|---|
| 5 | 15.1 ± 1 |
| 8 | 11.2 ± 2 |
| 14 | 5.9 ± 1 |
| 15 | 3.2 ± 0.6 |
| 16 | 4.5 ± 0.8 |
| wortmannin | 3.4 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Jeon, J.; Kim, K.; Choi, S.; Hong, S. Structure-Based Virtual Screening and De Novo Design of PIM1 Inhibitors with Anticancer Activity from Natural Products. Pharmaceuticals 2021, 14, 275. https://doi.org/10.3390/ph14030275
Park H, Jeon J, Kim K, Choi S, Hong S. Structure-Based Virtual Screening and De Novo Design of PIM1 Inhibitors with Anticancer Activity from Natural Products. Pharmaceuticals. 2021; 14(3):275. https://doi.org/10.3390/ph14030275
Chicago/Turabian StylePark, Hwangseo, Jinwon Jeon, Kewon Kim, Soyeon Choi, and Sungwoo Hong. 2021. "Structure-Based Virtual Screening and De Novo Design of PIM1 Inhibitors with Anticancer Activity from Natural Products" Pharmaceuticals 14, no. 3: 275. https://doi.org/10.3390/ph14030275
APA StylePark, H., Jeon, J., Kim, K., Choi, S., & Hong, S. (2021). Structure-Based Virtual Screening and De Novo Design of PIM1 Inhibitors with Anticancer Activity from Natural Products. Pharmaceuticals, 14(3), 275. https://doi.org/10.3390/ph14030275