2.1. Antidiabetic Activity of Pyrrolo[3,4-c]pyridine Derivatives
Knutsen et al. [
8] described 4-substituted 6-methyl-pyrrolo[3,4-
c]pyridine-1,3(
2H)-dione derivatives
6 (
Figure 3) that effectively reduce blood glucose levels without affecting the concentration of circulating insulin. The present compounds reduce blood glucose levels by stimulating glucose uptake into muscle and fat cells.
The ability of pyrrolo[3,4-
c]pyridine-1,3(
2H)-dione derivatives
6 to stimulate the incorporation of glucose into lipids was tested. The maximum increase in insulin sensitivity achieved in the dose range of 0.3–100 µM of the tested compounds, normalized to the full insulin dose-response (100%), was determined. The phenoxy substituent in the 4-position turned out to significantly influence the activity of the derivatives. While 4-phenoxy-6-methyl-pyrrolo[3,4-
c]pyridine-1,3(
2H)-dione derivatives increased the insulin sensitivity of mouse adipocytes by 7.4–37.4% (
Figure 3). The substituent in the para position of the phenyl ring increased the activity. Compounds with 4-phenoxy-(
6a), 4-(4-iodophenoxy)-(
6b), 4-(3,4-dichlorophenoxy)-(
6c) and 4-(4-methylphenoxy)-(
6d) derivatives increased the insulin sensitivity by more than 30% [
8].
Due to the efficacy of the present compounds 6 to reduce the blood glucose, they may find application in the prevention and treatment of disorders involving elevated plasma blood glucose, such as hyperglycemia and ailments in which such a reduction of blood glucose is beneficial: type 1 diabetes, diabetes as a consequence of obesity, diabetic dyslipidemia, hypertriglyceridemia, insulin resistance, impaired glucose tolerance, hyperlipidemia, cardiovascular diseases, and hypertension.
Aldose reductase (AR) is an enzyme present in the lens and brain that eliminates excess glucose by catalyzing glucose reduction to sorbitol. The accumulation of sorbitol may cause neuropathy in the peripheral nerves and cataracts in the lens. AR inhibitors reduce secondary complications caused by diabetes mellitus, especially in tissues where glucose uptake is not insulin-dependent.
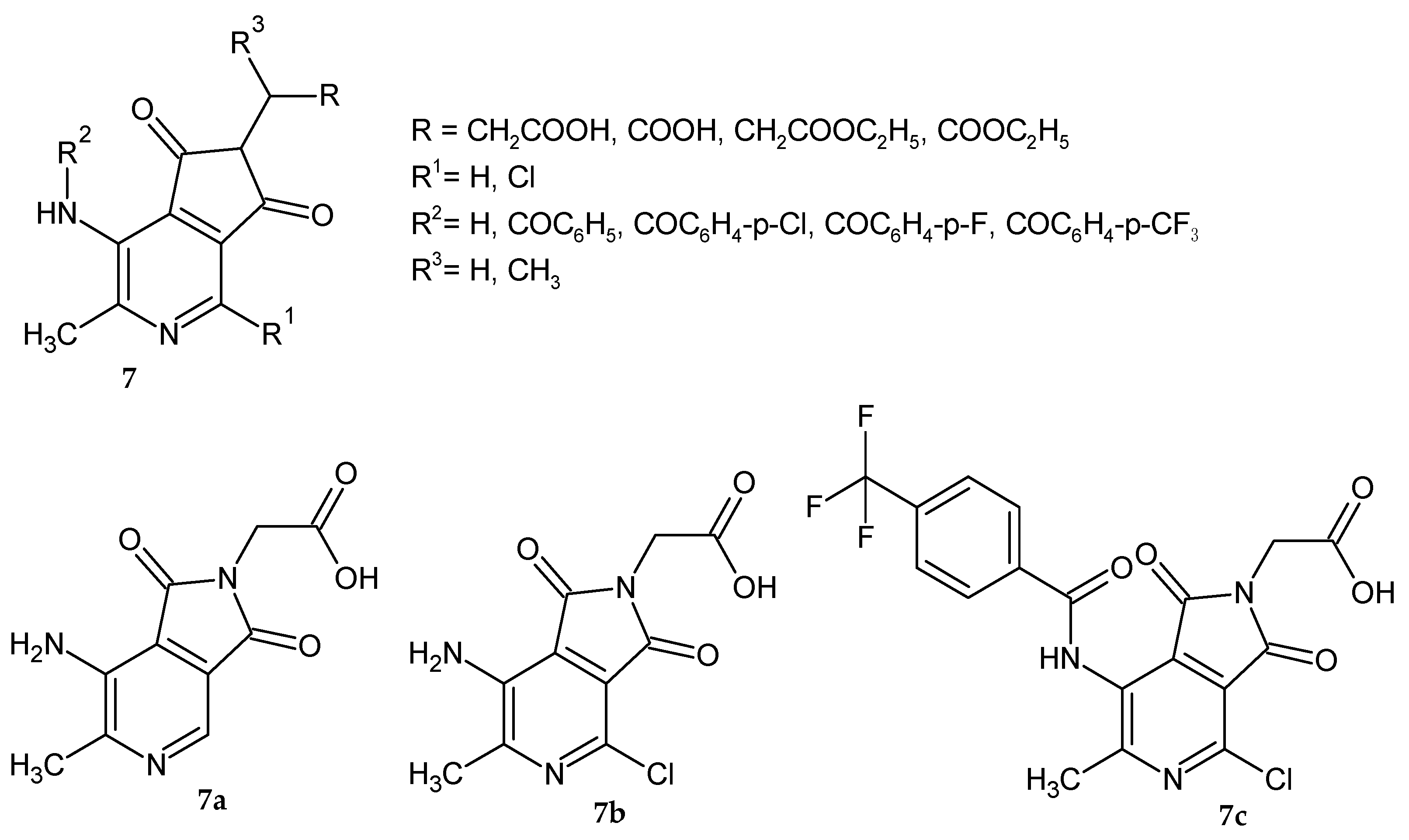
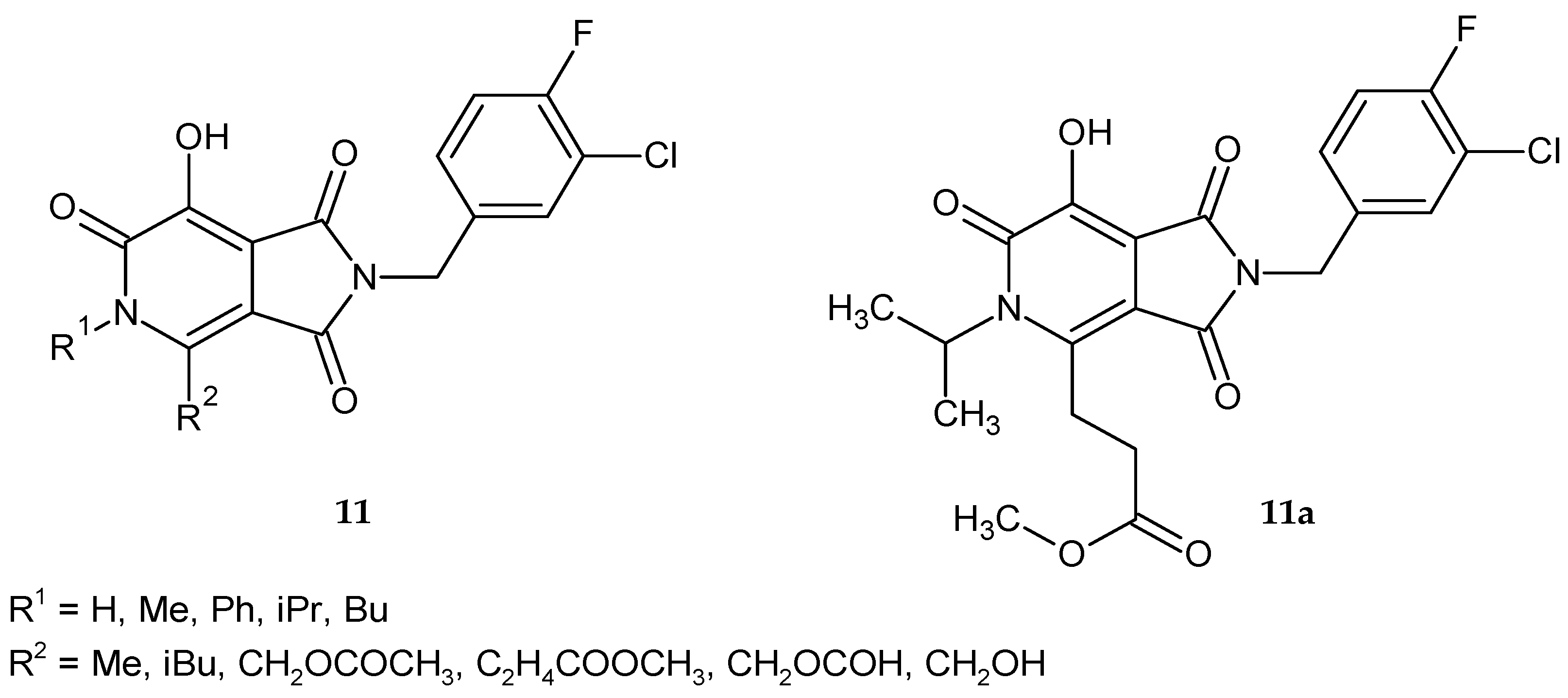
Da Settimo et al. [
9] synthesized a series of 6-methylpyrrolo[3,4-
c]pyridine-1,3-dione alkanoic acid derivatives
7 with potential AR inhibitory activity (
Figure 4). The obtained compounds were evaluated for their ability to inhibit AR in vitro in the extract of rat lenses. The biological data showed that the acidic derivatives proved to be new AR inhibitors. The presence of the carboxylic group at an adequate distance from the pyrrolopyridine scaffold is important for their activity. Compounds with a small chain length of carboxylic acid proved to be much more potent inhibitors than propionic or iso-propionic acid derivatives. Its corresponding ethyl ester analogs were inactive even at a higher concentration range. Among amide analogs most potent was 4-trifluoromethyl-benzylamide derivative
7c, but no more active than
7a. The inhibition potency of compounds
7a–
c was similar to the reference Alrestatin and Sorbinil at concentrations: 10
−5 M (70–90%) and 10
−6 M (33–43%). IC
50 (the concentration at which a substance exerts half of its maximal inhibitory effect) values for compounds
7a–
c were 1.4 µM, 2.5 µM, and 1.7 µM, respectively. The most active derivatives
7a–
b were evaluated as inhibitors of glutathione lens depletion in galactosemic rats. Glutathione depletion in the lens occurs rapidly after induction of galactosemia in rats (following the galactose diet). None of the tested compounds were active in maintaining glutathione levels in the rats’ lenses, which may be due to problems with metabolism or ocular bioavailability.
GPR119 is a cannabinoid receptor expressed predominantly in the incretin-releasing intestinal cells and pancreatic islet β-cells. GPR119 regulates the secretion of incretin and insulin. Therefore, new drugs acting on the GPR119 could find application in treating obesity and type 2 diabetes.
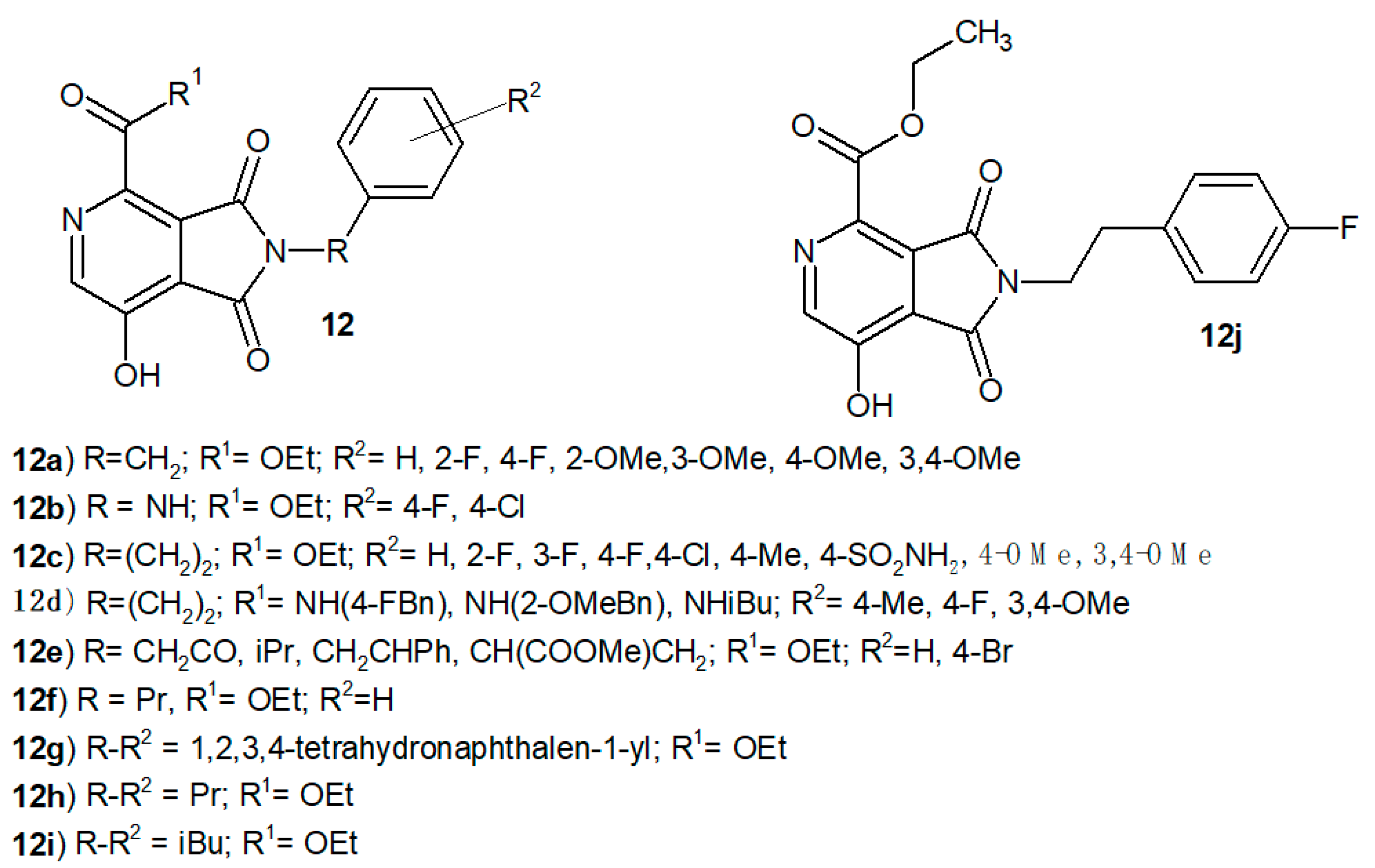
Yu et al. [
10] synthesized a series of N-[3-(1,3-dioxo-2,3-dihydro-
1H-pyrrolo[3,4-
c]pyridin-4-yloxy)phenyl]benzenesulfonamide derivatives
8 as novel GPR119 agonists. They replaced the -O- linker with the -S- or -NH-, as exemplified in
9, but both modifications did not bring any benefit, only an efficacy reduction of about 20% (
Figure 5).
Scientists investigated the effect of pyrrolo[3,4-
c]pyridine ring substituents on potency [
10]. The methyl group for R
1 was selected for its potential to improve metabolic stability. Biological studies have shown that the potency of derivatives 8 also depends on the R
2 substituent. Activity increased for the ethyl and propyl chains as R
2 compared to the methyl substituent or lack thereof, but large substituents (e.g., phenyl, cyclopentyl) lead to a significant loss of potency. Pharmacokinetic studies showed different metabolic decomposition of the tested compounds in both rat liver microsomes (RLM) depending on the substitution of the middle phenyl ring. The most active 6-F-phenyl derivative 8a exhibited an EC
50 of 0.016 µM in the human GPR119 cAMP assay and good RLM stability with clearance intrinsic (CL- the liver ability to remove (metabolize) a drug without the restrictions placed on it being delivered to the liver cell by blood flow or protein binding) = 84 µL/min/mg. Compound
8a was also evaluated during in vivo experiments and exhibited 0.073 µM in the cynomolgus monkey GPR119 cAMP assay with 100% efficacy, and in human liver microsomes, the CL was 29 µL/min/mg. Derivative
8a showed good bioavailability in rats (95%) and 29% in cynomolgus monkeys following a 2.0 mg/kg PO dose. It did not bind to BSEP (bile salt export pump), did not show hPXR (the human pregnane X receptor) activation and cytochrome P450 inhibition. Although the pharmacological properties were excellent, compound
8a showed high plasma protein binding, which precluded the assessment of its antidiabetic efficacy in vivo [
10].
2.3. Anticancer Activity of Pyrrolo[3,4-c]pyridine Derivatives
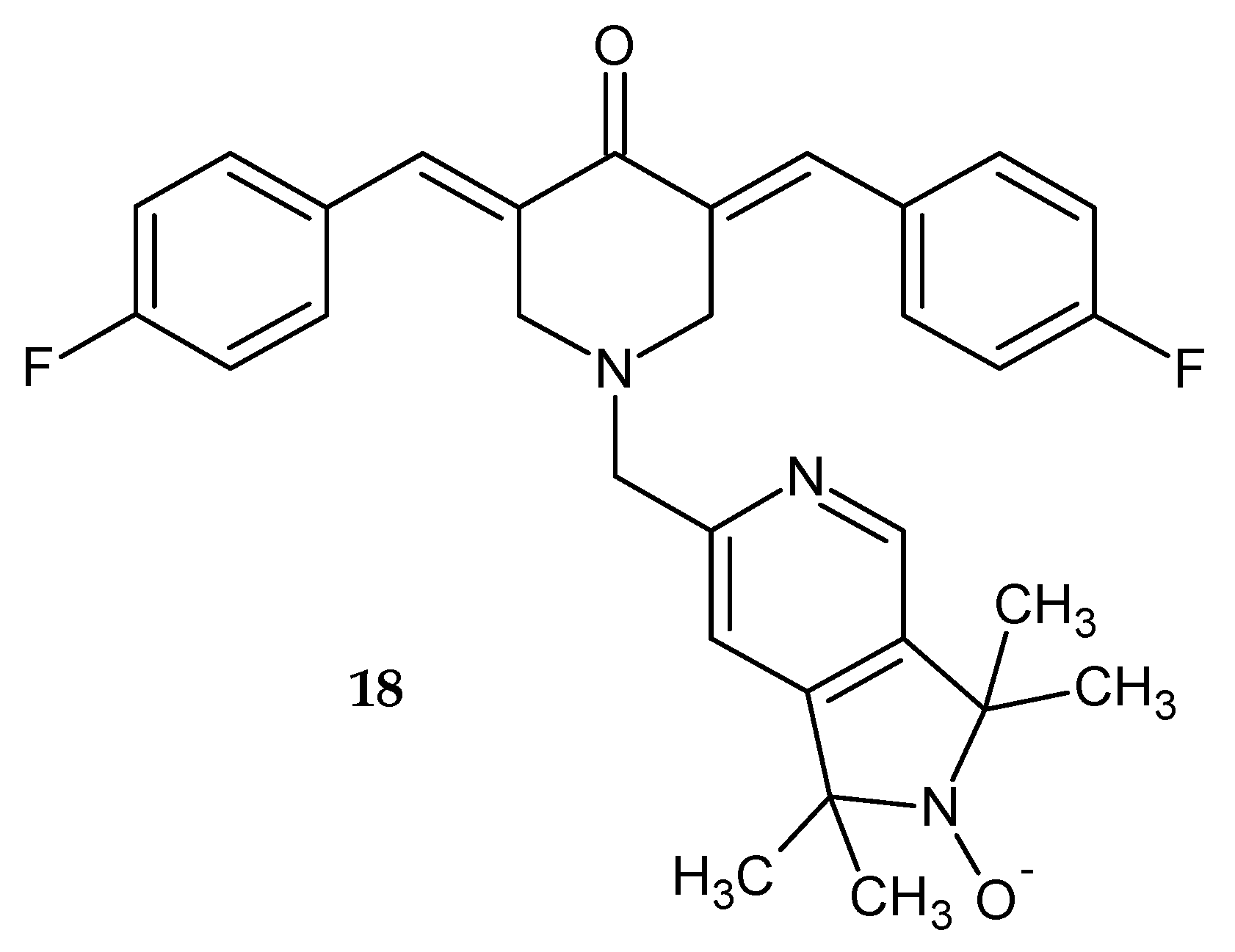
In a study by Kalai et al. [
21], 3,5-bis(4-fluorobenzylidene)-1-(1,1,3,3-tetramethyl-1,2-dihydropyrrolo[3,4-
c]pyridin-6-yl)piperidin-4-on N-oxide
18 was synthesized (
Figure 13). The antitumor activity of the obtained compound was assessed by measuring its cytotoxicity against ovarian and breast cancer cell lines and noncancerous cardiac cell lines. Tested compound
18 exhibited moderate cytotoxicity against ovarian cancer cells and limited toxicity toward breast cancer and healthy cardiac cell lines [
21].
Nicotinamide phosphoribosyltransferase (NAMPT) catalyzes the condensation of nicotinamide (NAM) with 5-phosphoribosyl-1-pyrophosphate to nicotinamide mononucleotide (NMN), which is the first step in NAD+ biosynthesis. Blocking NAMPT activity may impair cell growth. Thus, NAMPT inhibition is one of the anti-cancer therapy strategies.
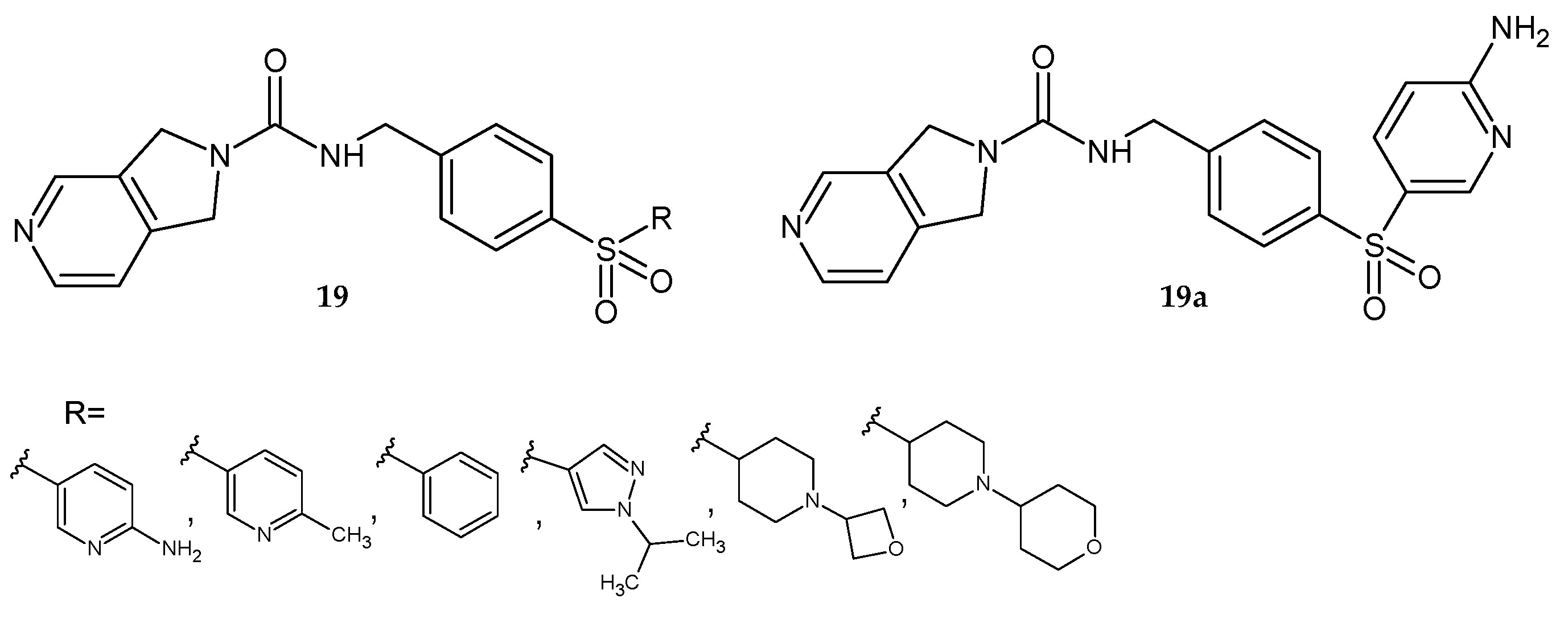
Dragovich et al. [
22] synthesized 4-sulfonylobenzyl- derivatives of pyrrolo[3,4-
c]pyridine-2-carboxamide
19 (
Figure 14) and evaluated them as potent NAMPT inhibitors. Derivatives that showed strong activity against NAMPT, good in vitro stability against human liver microsomes, and sufficient water solubility were referred for further studies. Many of these compounds exhibited minimal inhibition of the cytochrome P450 3A4 isoform and several CYP isoforms (2D6, 1A2, 2C19), but were strong CYP2C9 inhibitors. The binding of these derivatives to mouse plasma protein was typically moderate (approximately 90% bound). Molecule
19a also showed antiproliferative effects against several human tumor cell lines. Based on that data, compound
19a was accepted for in vivo mouse PK and xenograft efficacy studies. The
19a derivative showed satisfactory PK properties in mice and was effective in the PC-3 mouse xenograft model. Derivative
19a exhibited good bioavailability and plasma exposure after oral administration to female NCR mice [
22].
Wojcicka et al. [
20] synthesized a series of 4-methyl-6-phenylpyrrolo[3,4-
c]pyridine-1,3-dione derivatives by modifying of the substituent at position 2 of the pyrrolo[3,4
c]pyridine scaffold (
Figure 15).
N-alkil-4-methyl-6-phenyl-1
H-pyrrolo[3,4-
c]pyridine-1,3-diones
20a–
s were obtained and screened for their antitumor activity in vitro. Mannich bases
20g–
s with an IC
50 value in the range of 19–29 µg/mL were the most active.
Phosphoinositide 3-kinases (PI3Ks) are a family of intracellular lipid signaling enzymes that catalyze the phosphorylation of the hydroxyl group in the 3-position of the phosphatidylinositol ring. PI3Ks can control many vital cellular processes such as growth, proliferation, and survival. Therefore, the inhibition of PI3Ks can be used in anticancer therapy.
Collier et al. [
23] synthesized 4-methyl-2-[1-(2,2,2,-trifluoroethyl)-1
H-pyrazol-4-yl]-1
H-pyrrolo[3,4-
c]pyridine-3,6-dione derivatives
21 (
Figure 16). The substitution of the carbonyl group in the 6-position with a 5,6-dimethoxypyridin-3-yl ring led to derivative
21a, which showed high PI3Kγ affinity and inhibited the monocyte chemoattractant protein-1 MCP-1 induced chemotaxis of THP-1 cells (IC
50 = 270 nM).
Spleen tyrosine kinase (SYK) is a non-receptor cytoplasmic kinase that is a key mediator for various inflammatory cells. Therefore its inhibition is an essential therapeutic target in both neoplastic and inflammatory diseases. FLT3 (fms like tyrosine kinase 3) is a cytokine receptor expressed on the surface of many hematopoietic progenitor cells.
Lam et al. [
24] prepared 6-[(2-aminocyclohexyl)amino]-1,2-dihydro-3
H-pyrrolo[3,4-
c]pyridin-3-one derivatives
22 as SYK inhibitors (
Figure 17). The 4-(3-methylanilino)-substituted compounds exhibited good enzymatic and cellular potency. It turned out that the 7-fluoro substitution had a significant effect on the activity. Replacing methylaniline with a methylpyrazole ring increased the activity of the 4-fluoro derivative. The methylpyrazole derivative
22a showed the best in vitro profile and was selective for kinases (it was potent toward both SYK and FLT3). Compound
22a showed inhibition toward an SYK-dependent and FLT3-dependent cell line in a cell proliferation assay. It demonstrated strong tumor growth inhibition (TGI) after 20 days of treatment. This derivative blocked anti-IgD stimulated expression of CD86 in murine peripheral B cells in vivo. Compound
22a named TAK-659 has been used in clinical trials to treat advanced solid tumors and lymphoma malignancies [
24].
2.5. Sedative Activity of Pyrrolo[3,4-c]pyridine Derivatives
While conducting studies on pyrrolo[3,4-
c]pyridines to obtain new anxiolytic derivatives of the buspirone type, Śladowska’s team noticed that these compounds are often not active in this regard. Instead, they show other pharmacological effects, in particular, sedative [
27] and analgesic activity [
27,
28,
29]. The analgesic activity of the new pyrrolo[3,4-
c]pyridines is described in the following section.
Taking into account the results of previous work [
28,
29], in 1994, the above researchers obtained a series of nine new N-substituted piperazinalkyl derivatives of 6-methyl-2-(1-piperidine)-pyridine-1,3-dione (
Figure 20) [
27].
Several pharmacological studies were performed with regard to the newly obtained structures, including testing for:
acute toxicity in mice;
spontaneous locomotor activity in mice;
pantetrazol-induced seizures in mice;
anxiolytic properties in a four-plate test in mice;
amphetamine-induced motor hyperactivity in mice;
pain reactivity in a writhing syndrome test in mice.
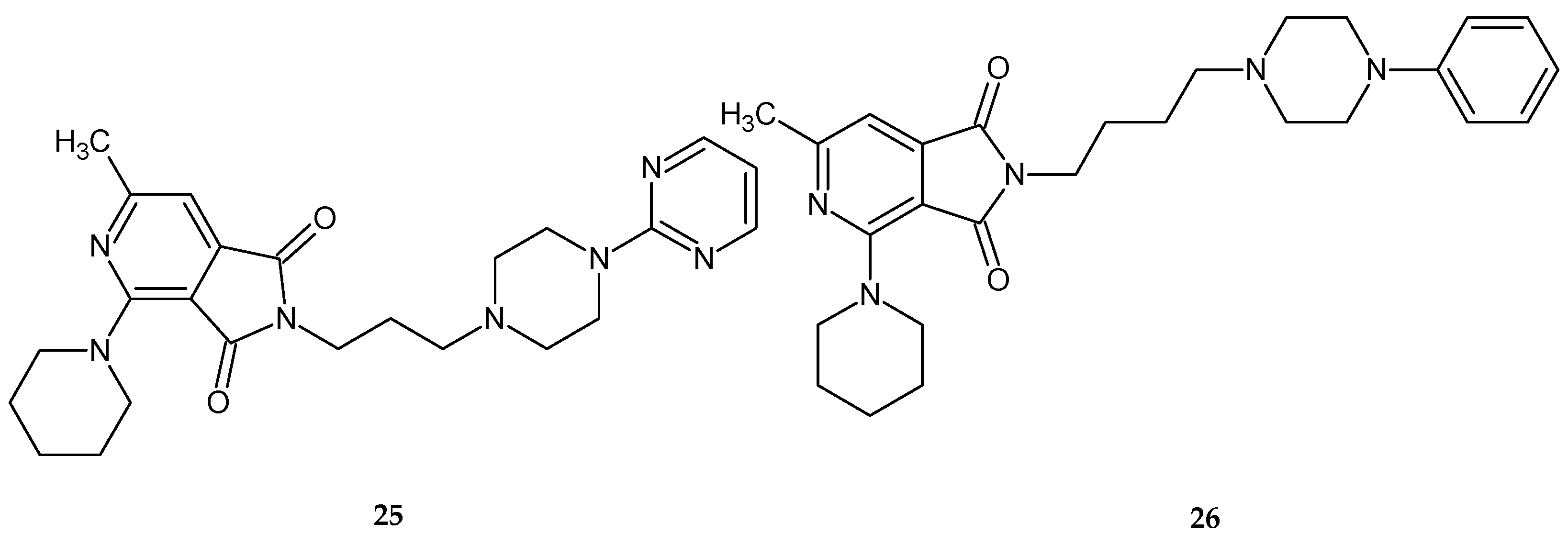
The test results showed that out of the tested series of compounds, derivatives
25 and
26 (
Figure 21) significantly decreased locomotor activity during a one-hour observation.
Compound 25 showed this effect at doses of 1/10 and 1/20 LD
50, 26 at doses of 1/10, 1/20, 1/40, and even 1/80 of LD
50 [
27]. In the other tests mentioned above, the activity of compounds was not as promising. None of the compounds tested at doses of 1/10 LD
50 showed antinociceptive activity in the writhing test [
27].
A year later, the same scientists obtained another series of compounds derived from pyrrolo[3,4-
c]pyridines [
30]. This time, they studied the influence of other amino- and alkyl-(aryl-)aminoalkyl(hydroxyalkyl) substituents on the activity of 3,4-pyridinedicarboximide. They synthesized fifteen differently substituted derivatives, of which only compound
27 (
Figure 22) deserved attention. Only compound 27 significantly depressed locomotor activity at doses of 1/10 and 1/20 LD
min. [
30].
The strong pharmacological activity of compound
27 encouraged scientists to introduce further modifications. They obtained a series of compounds in which the piperidine group present at position 2 was replaced with pyrrolidine or morpholine. Phenylpiperazine or pyrimidinylpiperazine was retained as an amine residue of the pharmacophore. The link between the core and the pharmacophore fragment was a propyl, butyl, or 2-hydroxypropyl chain [
31].
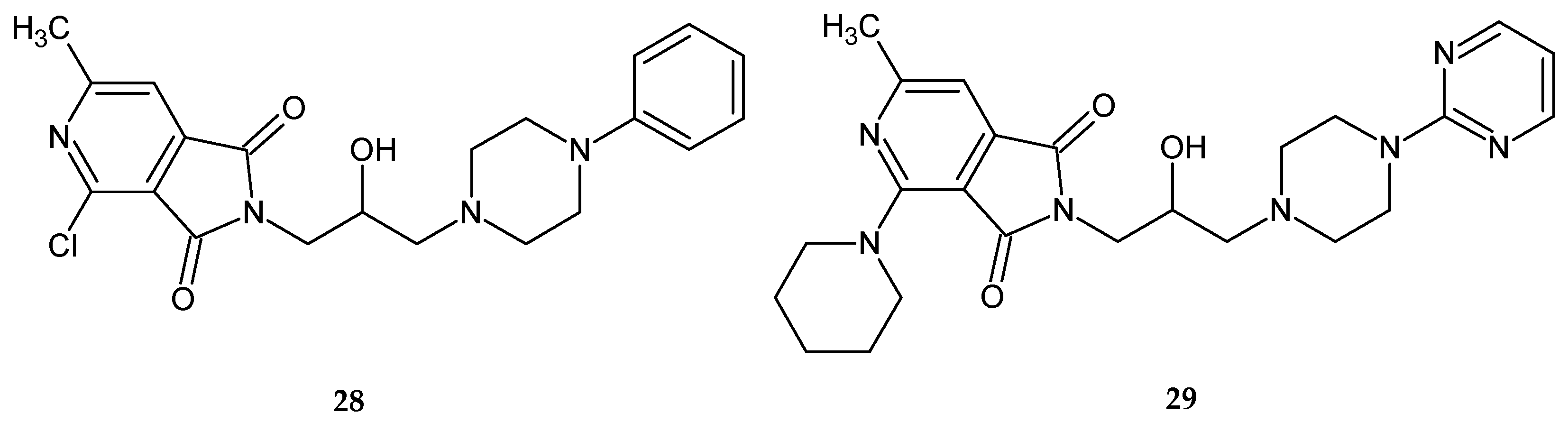
This time, all the tested substances significantly inhibited the spontaneous locomotor activity of mice during a one-hour observation. Compounds
28 and
29 showed the most potent sedative effects (
Figure 23). These compounds were active up to doses of 1/160 LD
50 (
28) and 1/80 LD
50 (
29).
The results indicate that replacing the piperidine ring at position 2 of the pyrrolo[3,4-
c]pyridine scaffold with other pharmacophore substituents does not substantially alter the pharmacological action profile [
31].
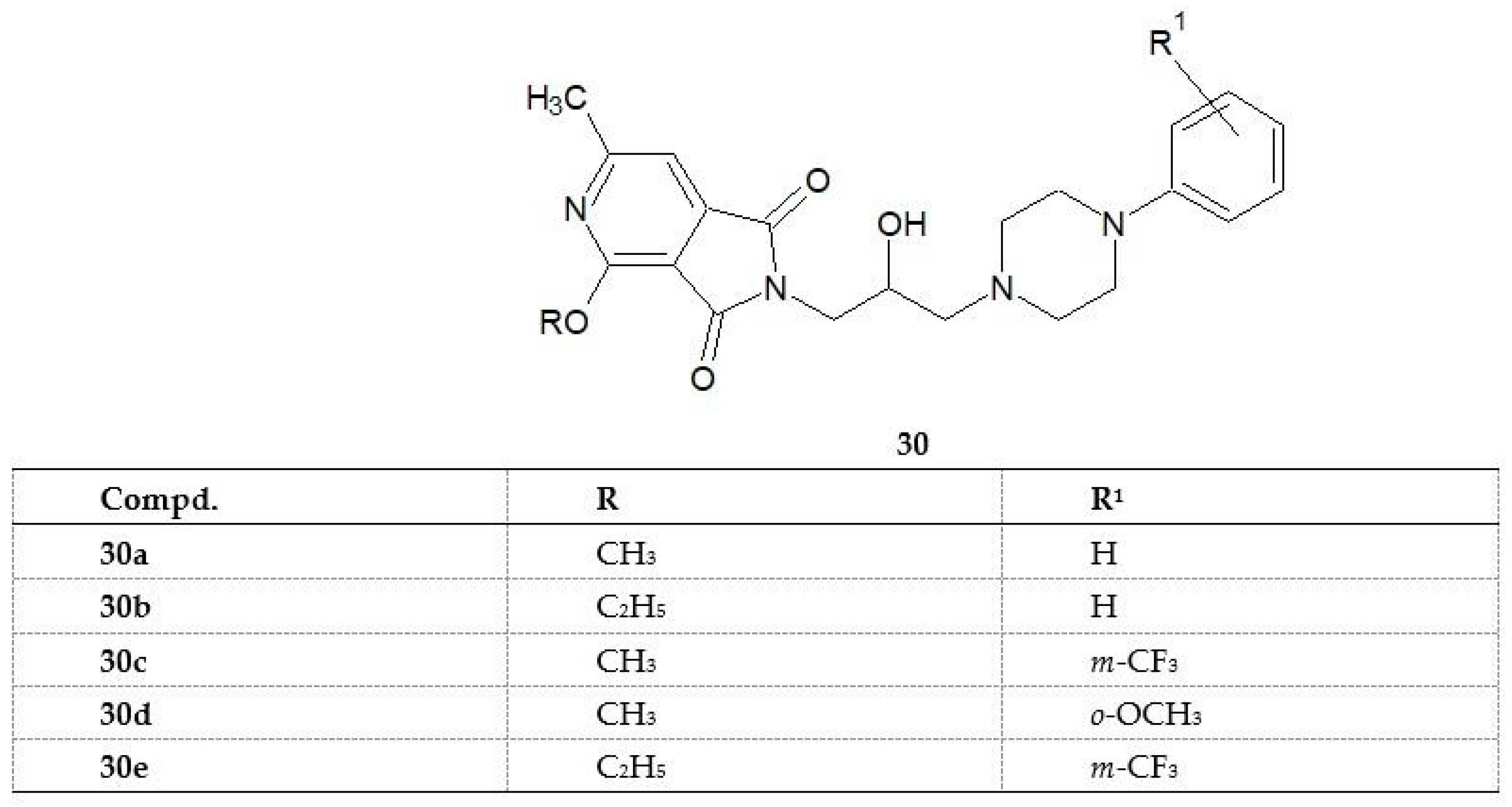
Continuing the research, the scientists synthesized the next series of compounds in which the piperidine group was replaced by an alkoxy group [
32]. The structures of compounds
30–
34 are shown in
Figure 24.
Compounds
30c,
30d, and
30e significantly suppressed the spontaneous locomotor activity of mice during a 30 min observation up to a dose of 12.5mg/kg. Imide 31 was active at a dose of 200 mg/kg. All of the studied compounds (
30a–
e) exhibited analgesic activity [
32]. It is described in the following section.
In the next publication [
33], Śladowska’s team described the synthesis and properties of 2-(4-substituted)butyl derivatives of some 2,3-dihydro-1,3-dioxo-1
H-pyrrolo[3,4-
c]pyridines. New derivatives have an alkoxy group at position 2, butyl linker, and an amine residue (arylpiperazines, or 1,2,3,4-tetrahydroisoquinoline substituent) instead of a piperazinyl group. All tested compounds significantly suppressed the spontaneous locomotor activity of mice during a 30-min observation. While 2-[(4-phenyl-1-piperazinyl)butyl]-4-methoxy-6-methyl-1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-dione 31 (
Figure 25), given in doses of 1/20, 1/40 and 1/80 LD
50, inhibited spontaneous locomotor activity in mice by 86 (
p < 0.02), 67 (
p < 0.05), and 47%, respectively.
The other compounds in this series significantly reduced locomotor activity in mice, by 87–82% when administered at 1/20 LD
50. However, when administered at doses of 1/40 and 1/80LD
50, they decreased spontaneous locomotor activity by 59–47% and 26–13%, respectively [
33].
In 2009, researchers reported [
34] the synthesis and pharmacological results of novel 1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-diones. These compounds were obtained by modifying previously described structures [
32,
35,
36]. The modifications consisted of the following:
replacement of the phenyl ring at the N-4 position of piperazine with benzyl or benzhydryl groups;
replacement of N-substituted piperazines by other cyclic amines (morpholine, piperidine, or pyrrolidine).
The authors aimed was to investigate if and how the performed structural changes would affect the toxicity, analgesic, and sedative effects of the tested compounds [
34]. The results indicated that all tested pyrrolo[3,4-
c]pyridine-1,3(2
H)-diones significantly suppressed the spontaneous locomotor activity of mice during a 30-min observation.
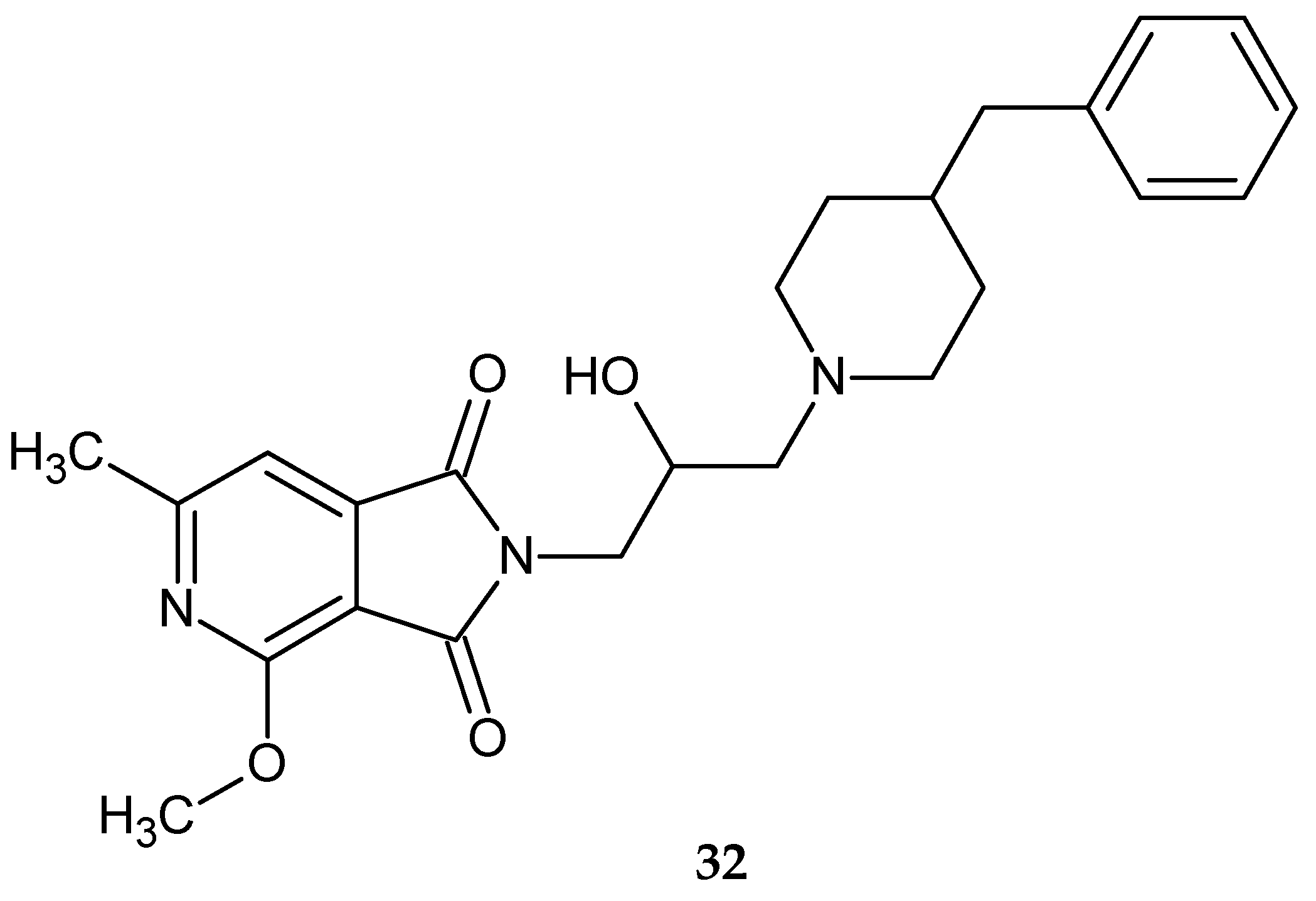
The most potent effect (ED
50 = 12.03 mg/kg) was produced by compound
32 (
Figure 26). Moreover, compound
32 was not toxic (LD
50 >2000 mg/kg) [
34]. The antinociceptive activity of the pyrrolo[3,4-
c]pyridines derivatives described in this publication is reported in the following section.
In 2020, Szkatuła et al. [
37] published a paper reporting the synthesis and pharmacological results of novel 1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-dione derivatives with potential sedative and analgesic activity. The structures of new derivatives
33a–
33h are shown in
Figure 27 [
37].
All new compounds
33a–
h inhibited the locomotor activity in mice, and the two most active (
33b,
33d) also extended the duration of thiopental anesthesia. Biological studies concerning imides
33b and
33d were supplemented with the determination of the effect of intraperitoneal administration of tested compounds on the duration of thiopental-induced sleep. The mechanism and extent of blood-brain barrier crossing were not defined. However, the observed sedative effect may indicate good penetration of the compounds in question into the central nervous system, which has not been proven either [
37]. As mentioned earlier, the authors also described the analgesic activity of the new derivatives. Their results are presented in the section Analgesic activity.
2.6. Analgesic Activity of Pyrrolo[3,4-c]pyridine Derivatives
As mentioned above, Śladowska’s team initiated work on pyrrolo[3,4-
c]pyridine derivatives to synthesize buspirone-inspired compounds. As a result of pharmacological studies, these compounds were found not to have the intended anxiolytic effect but to exhibit other pharmacological effects. The first structure that inspired the authors to work on analgesic pyrrolo[3,4-
c]pyridine derivatives was the non-toxic compound
27 [
27,
30,
31]. The researchers decided to modify the structure of this compound to obtain new pyrrolo[3,4-
c]pyridine derivatives with potential analgesic activity. These modifications consisted mainly of replacing the pyridine present in the structure of
27 with another pharmacophore group. At position 2, the pyridine ring was replaced by morpholine, pyrrolidine, 4-methylpiperazine, or a chlorine atom. The analgesic effect of compounds was determined in two behaviorally different tests: phenylbenzoquinone-induced writhing syndrome test and hot-plate (thermal analgesic stimulus) test in mice. Unfortunately, most of the imides tested did not show any analgesic effect during testing [
31].
Continuing their study, researchers observed that replacement of the piperidine group by an alkoxy one gave non-toxic substances with powerful analgesic properties [
32]. The structures of compounds
30a–
30e are presented in
Figure 24.
The analgesic action of the new 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-dione derivatives 30a–30e was investigated on mice using the hot-plate and writhing tests.
In both tests (writhing and hot-plate tests), all structures (
30a–
30e) tested showed analgesic activity superior to that of the reference acetylsalicylic acid (ASA). In the writhing test, imide
30a, containing a methoxy group at position 2 of the pyridine ring and an unsubstituted phenyl at the N-4 position of piperazine, proved to be the most active compound. Replacement of the methoxy group with an ethoxy group (imide
30b) contributed to an almost fourfold decrease in analgesic activity. Similarly, the activity of imide
30c with a methoxy group in the pyridine ring was higher than that of its ethoxy analog
30e. This result indicates that the potency of the analgesic effect in the writhing test is influenced by the type of alkoxy group at position 2 of the pyridine ring. The authors also observed that the introduction of trifluoromethyl or methoxy groups to the phenyl substituent caused a significant inhibition of spontaneous locomotor activity in mice. As it is known, analgesics can act centrally, peripherally, or both centrally and peripherally. Opioid analgesics, like morphine, base their analgesic effects mainly on a central mechanism, whereas non-steroidal anti-inflammatory drugs, like ASA, on a peripheral mechanism [
38]. However, this division is not unambiguous as salicylates exert their analgesic effects also partly through the central mechanism [
39]. Since the structures studied were active in both pharmacological tests, they may show analgesic effects both in the central and peripheral mechanism. However, as suggested by the authors, elucidation of the exact mechanism requires further pharmacological studies [
32].
Continuing the research in 2005, Śladowska’s team designed another series of compounds with potential analgesic activity [
35]. To this end, the researchers further modified the structure of 1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-diones
30a and
30b by:
elimination of the hydroxyl group from the alkyl chain;
introduction of a methoxy group at position 2 of the phenyl ring on N-4-piperazine;
replacement of the phenyl ring with a 2-pyrimidine ring;
replacement of the N-aryl(heteroaryl) piperazine group with tetrahydroisoquinoline and morpholine substituents;
shortening of the alkyl chain at the nitrogen atom to C-1.
Considering the above rationale, they synthesized the corresponding 1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-diones (
Figure 28) to obtain compounds with potent analgesic activity [
35].
The analgesic activity of compounds 34 and 35 was tested in two assays: hot-plate and writhing tests. In the writhing syndrome test, all tested substances, i.e.,
34 and
35, showed strong analgesic activity. The strongest effect was produced by compounds 34a and
34c, which were effective up to a dose of 0.78 mg/kg. The most active compound in this test was imide
34c, containing an o-methoxyphenyl substituent at the N-4 position of piperazine and a methoxy group at position 2 of the pyridine ring. None of the structures obtained was more active than the leading structure 30a in this test. This indicates that the most preferable modification involves removing a hydroxyl group at the β propyl position and introducing of a methoxy group at the ortho position of the phenyl ring. Only compound
35a (the most toxic imide) was more active in the hot-plate test than compound
30a. Compound
35a showed significant activity up to a dose of 9 mg/kg in this test. In the case of 4-ethoxy derivatives, no increase in analgesic activities was observed concerning imide
30b. The pharmacological results obtained indicate that most of the structures studied show very interesting analgesic properties. The results of preliminary radioligand binding studies suggest that these compounds show a weak affinity for µ-opioid receptors, which probably play a role in their mechanism of action. However, the authors do not explain the mechanism of action of the obtained pyrrolo[3,4-
c]pyridine derivatives [
35].
In 2006, the researchers obtained another series of compounds. This time, modifications to model structure
30a involved introducing chlorine and fluorine atoms to the phenyl ring at the N-4 position of piperazine. Additionally, in several cases, the OH group was removed from the linker [
36]. The analgesic activity of the compounds obtained in this study was measured using the writhing syndrome and hot-plate tests. All tested imides showed significant antinociceptive activity in phenylbenzoquinone-induced writhing test, and the ED
50 values ranged from 3.51 to 16.04 mg/kg [
36].
Importantly, these compounds were non-toxic. However, the analgesic activity (tested in both the hot-plate and writhing syndrome tests) of the compounds in this series was lower than the model structure
30a previously adopted by the authors [
34].
In 2020, Dziubina et al. [
40] published a paper that aimed to investigate the potential analgesic, antiedematous (anti-inflammatory) and antiallodynic activities using two 1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-dione derivatives
36 and
37 (
Figure 29) in various experimental models of pain.
The authors performed a number of pharmacological tests, including the hot-plate test, formalin test, capsaicin test, and oxaliplatin-induced allodynia test. The hot-plate test is commonly used to evaluate centrally acting analgesics, and nociceptive responses in this test are of supraspinal origin. In the experiment conducted, only
36 at a dose of 20 mg/kg significantly increased the latency time of the pain response. However, as the authors suggest, its effect seems to be due to sedation (compound
36 at the highest analgesic dose significantly decreased locomotor activity) rather than antinociceptive action [
40].
The formalin test is used to identify tonic inflammatory pain. It assumes two phases of stimulus perception – the first, lasting about 5 min after administration of an irritant (formalin), manifested by licking, shaking, biting of the paw by the animal, and the second, following several minutes, by tonic pain. The first phase results from the conduction of the pain stimulus along the C-fiber, whereas the second phase is the effect of increasing inflammation and central sensitization in response to a peripheral stimulus. In this test compounds
36 and
37 (5–20 mg/kg) showed antinociceptive activity in both phases, but it was more pronounced in the second phase of the test. Formalin-induced pain involves numerous channels, receptors, and signaling pathways; therefore, the authors verified the involvement of opioidergic, adenosinergic and nitrergic systems in the analgesic effects of compounds
36 and
37 in the model of tonic pain. The antinociceptive effect of both compounds was not reversed by systemic injection of naloxone, suggesting an opioid-independent analgesic effect of the test substances. Next, the effects of the compounds on the adenosinergic system were tested. In this test, caffeine (10 mg/kg), which by itself has no effect, reversed the effect of compound
37 on formalin-induced pain responses in both phases. Moreover, the antinociceptive effects of
37 were blocked by DPCPX (1,3-dipropyl-8-cyclopentyl-xanthine), an adenosine A1 antagonist, confirming that they were mediated by adenosine A1 receptors. As for the effects of these compounds on the nitrergic system (nitric oxide is involved in the analgesic effect of many drugs with different analgesia mechanisms), L-NAME (nω-nitro-L-arginine methyl ester hydrochloride) administration reduced the antinociceptive effect of compound
36 only in behaviors associated with the second phase. L-NAME administration did not affect the antinociceptive effect of compound
55. The possible analgesic effect of compounds
36 and
37 on neurogenic pain was investigated using a capsaicin-induced pain model in mice. Capsaicin induces an immediate response at the application site, called neurogenic inflammation, which results from the activation of transient receptor potential vanilloid-1 and the release of mediators substance P and glutamate. Both compounds
36 and
37 reduced capsaicin-induced pain behaviors in a dose-dependent manner. In the next step of the study, the authors tested the potential anti-allodynic efficacy of the tested compounds
36 and
37 in a mouse model of chemotherapy-induced peripheral neuropathy (CIPN) pain induced by oxaliplatin, an anticancer drug commonly used in the model of neuropathy in humans. A single administration of oxaliplatin in animals (mice and rats) induces a painful peripheral neuropathy with associated mechanical allodynia. It is characterized by two phases: an early phase that develops soon after cytostatic administration (several hours after administration), and a late phase that occurs after several days [
41]. The results of compounds
36 and
37 showed that as little as one dose of oxaliplatin decreased the threshold of pain sensitivity to mechanical stimuli, and tactile allodynia was observed as early as three hours after oxaliplatin injection. The observed effect of oxaliplatin was durable, as it was also found seven days after its administration. Test compounds
36 and
37 significantly reduced mechanical hypersensitivity at doses of 5 and 10 mg/kg. In the late phase of allodynia, both compounds
36 and
37 were significantly but slightly less effective than in the early phase of the test. The researchers also conducted in vitro studies. In this study, a biochemical assay was performed to determine the effect of the tested compounds on COX-2 levels. The COX-2 enzyme plays an essential role in the inflammatory response and its expression is induced after exposure of macrophages to LPS (lipopolysaccharide) or other pro-inflammatory stimuli. The RAW 264.7 cell line was used for this study. Compounds
36 and
37 significantly reduced COX-2 levels in LPS-stimulated cells. This result suggests at least partial anti-inflammatory properties of the tested compounds.
The obtained pharmacological results showed that compounds
36 and
37 exhibits broad-spectrum activity in several pain models (neurogenic pain, inflammatory pain, and chemotherapy-induced peripheral neuropathic pain) [
40].
Krzyżak et al. [
42] synthesized a novel N-substituted 1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-diones derivatives
38–
39. The compounds were tested for their inhibitory activity against COX-1/ COX-2 and BSA (bovine serum albumin) interaction. The researchers obtained two series of compounds (
Figure 30). Series I—Mannich bases with an arylpiperazine moiety
38a–
38c. Series II—structures with a two-carbon linker and a cyclic amine
39a,
39b (
Figure 30) [
42].
In vitro, COX-1 and COX-2 inhibition assays were performed, and pharmacological results showed that all compounds
38–
39 have the potential to inhibit both enzymes. It should be noted that COX-1 is involved in the synthesis of prostaglandins responsible for maintaining normal body function in the kidneys, intestines, and other organs, while COX-2 is an isoform that plays a major role in inflammation and associated pain [
43,
44]. In the study presented by the researchers, the COX-1 enzyme is inhibited more effectively (than the reference meloxicam) by compounds
38a and
39a, while COX-2 is inhibited more strongly than the reference drug by all compounds (except
38c). The highest selectivity towards the COX-2 enzyme was shown by structure
39a, for which the COX selectivity ratio (IC
50(COX-2)/IC
50(COX-1)) is 0.55 (for meloxicam—0.71) [
42].
The interaction with BSA was investigated by fluorescence spectroscopy and circular dichroism measurement. To understand the binding interaction of compounds
38–
39 in the active site of COX and BSA, a molecular docking study was performed [
42].
The obtained experimental and molecular docking results confirmed that the main interaction forces between the studied
38–
39 structures and BSA are hydrogen bonding and van der Waals forces [
42].
As mentioned in the previous section, compounds
33a–
33h (
Figure 27) presented interesting antinociceptive activity in addition to the strong sedative activity that has already been described [
37]. Compounds 33a–33h (except
33c) were not toxic (LD
50 >2000mg/kg). The analgesic activity of new compounds was studied using the hot-plate test and the writhing test. All tested 3,4-pyridinedicarboximide
33a–
33h were active in the writhing test (ED
50 = 3.25–19.2 mg/kg), and their analgesic activities in this study exceeded the effect of the reference ASA (ED
50 = 39.15 mg/kg). In addition, the pharmacological activity of the two imides
33b (ED
50 = 3.25 mg/kg and
33d (ED
50 = 3.67mg/kg) was comparable to that of morphine used as a second reference drug (ED
50 = 2.44 mg/kg). However, in the case of the hot-plate test, the analgesic effects observed for the tested structures did not reach statistical significance [
37].
Based on the obtained results and taking into account the results obtained in previous studies [
31,
33,
34,
35], the authors aimed to determine the relationship between the structure and biological activities in the group of N-substituted 1
H-pyrrolo[3,4-
c]pyridine-1,3(2
H)-diones. In their results, the researchers considered the effect of the following modifications on pharmacological activity (
Figure 31):
- (I)
an alkoxy substituent in the pyridine ring;
- (II)
the length of the alkyl link;
- (III)
the type of amino residue.
The potency of the analgesic effect of the studied compounds is decisively influenced by the type of alkoxy substituent in the pyridine ring (I). Compounds substituted with methoxy group in this position are more active than their ethoxy analogues.
In most cases, a phenylpiperazine moiety was introduced as the amine residue (III). The most active compounds had an unsubstituted phenyl ring in this grouping. Introduction of alkyl substituents at the ortho or meta position (
o-OCH
3,
m-CF
3) or halogen atoms was not preferred. Replacement of the phenylpiperazine moieties with another cyclic amine (morpholine, tetrahydroisoquinoline) also led to attenuation of the analgesic activity. The length of the alkyl bond between the pyrrole ring (II) and the amine residue (III) also affects the analgesic potency. Removal of the hydroxyl group weakens the pharmacological effect. Shortening of this linker to 2 or 1 carbon atom also causes a decrease in activity, whereas lengthening the linker to 4 carbon atoms (butyl derivatives) did not increase the potency [
37].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






























