
Physicochemical Features and Peculiarities of Interaction of AMP with the Membrane

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Interaction with Envelope
3. Physicochemical Properties
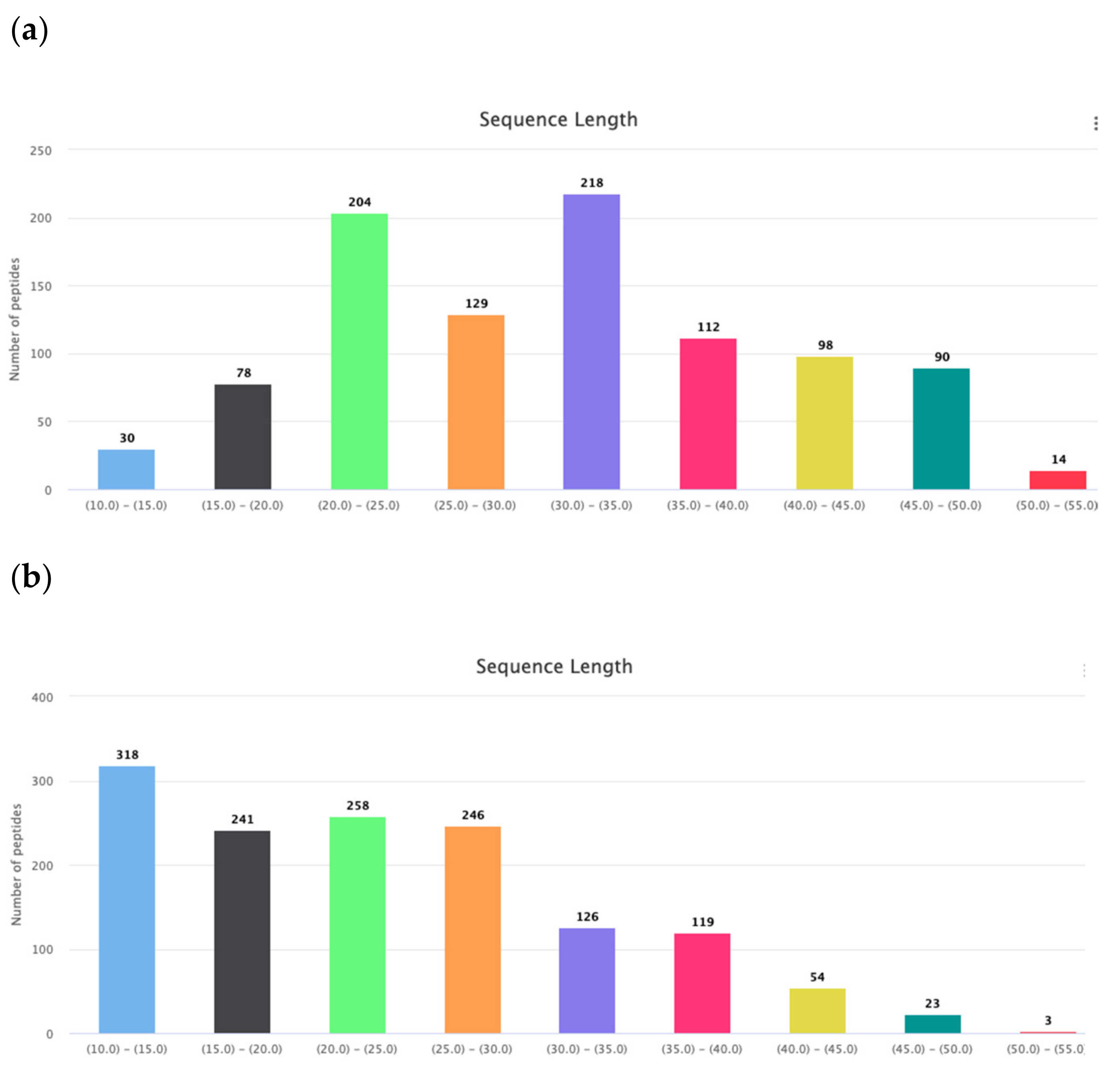
3.1. Amino Acids Composition and Distribution
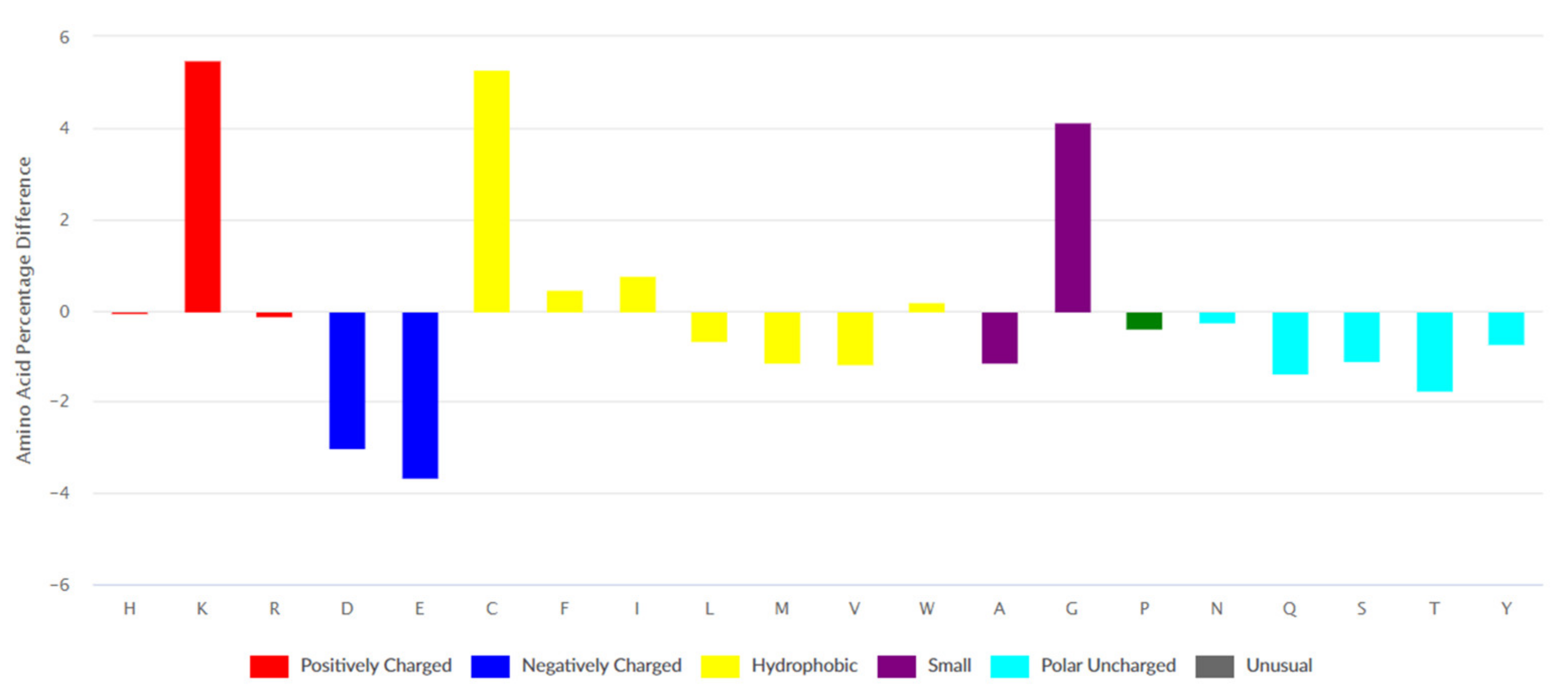
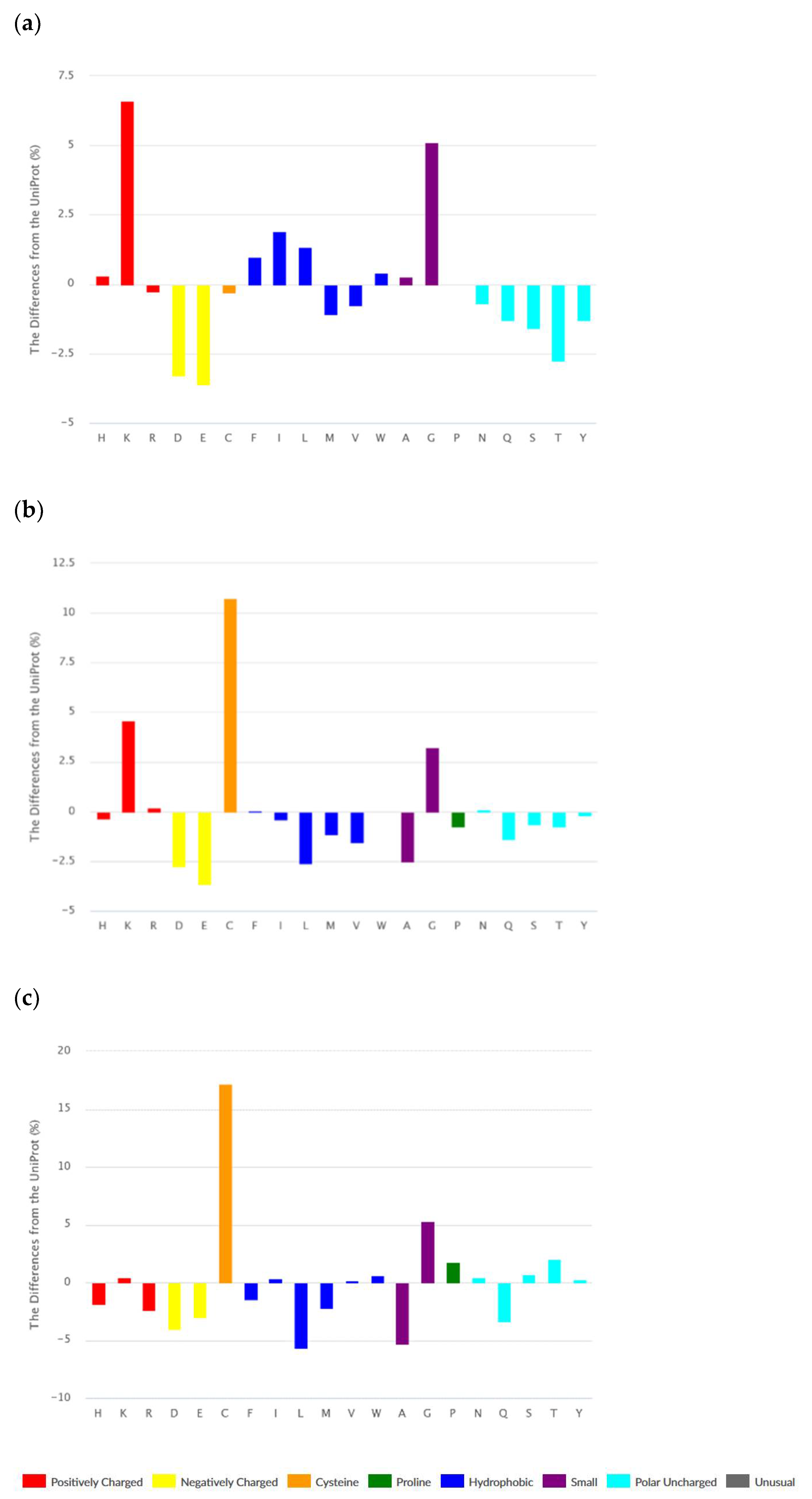
3.1.1. Composition
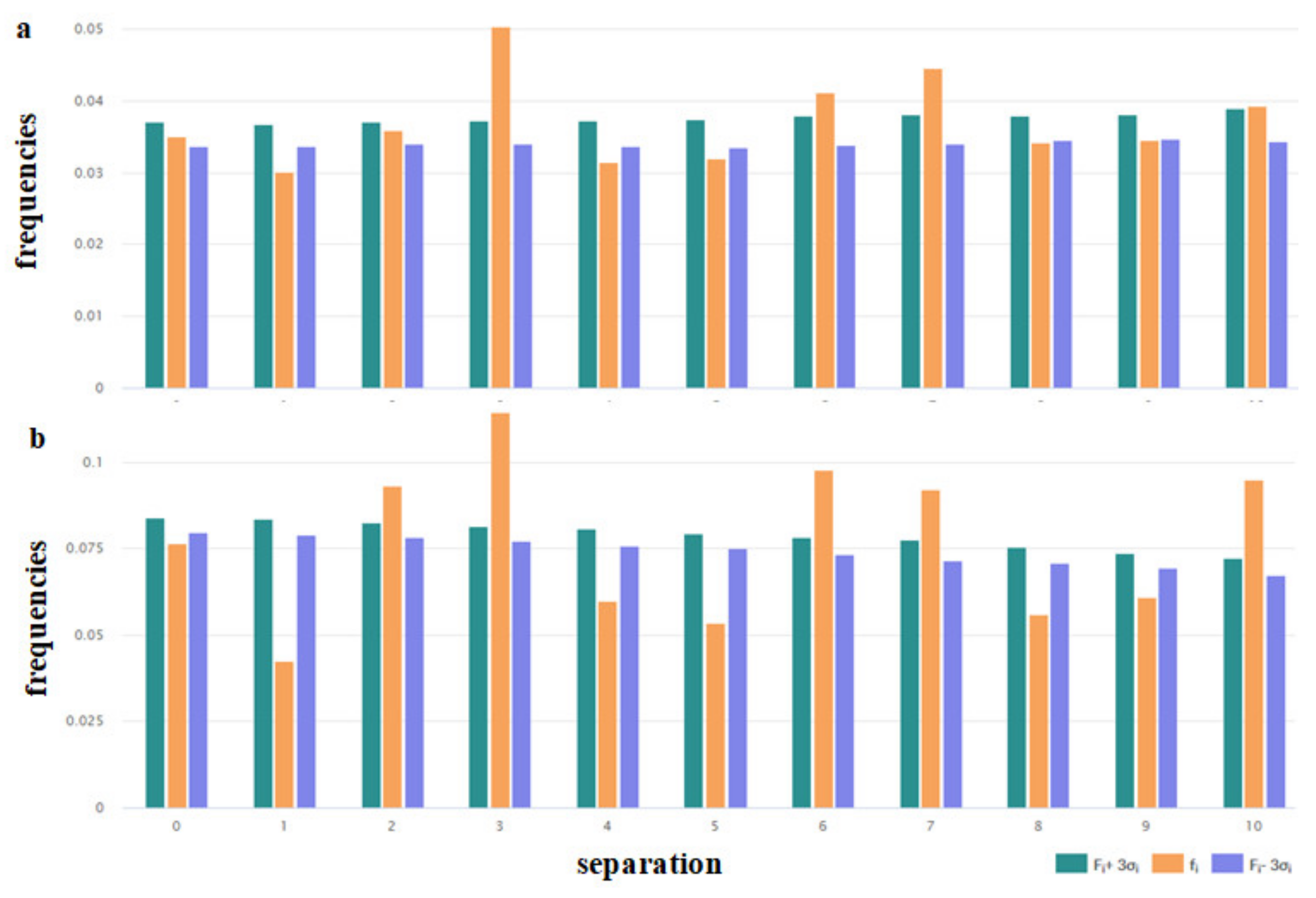
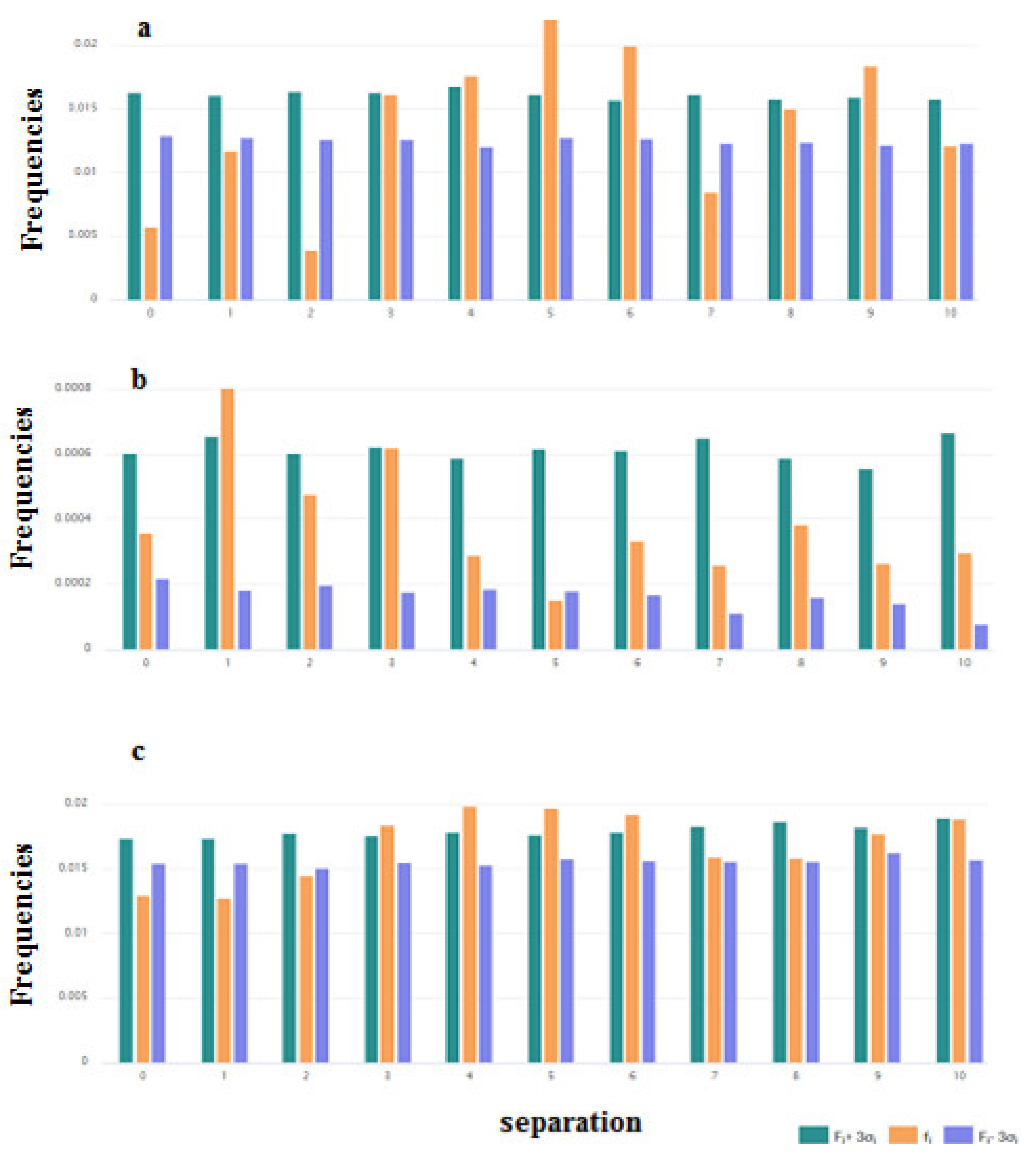
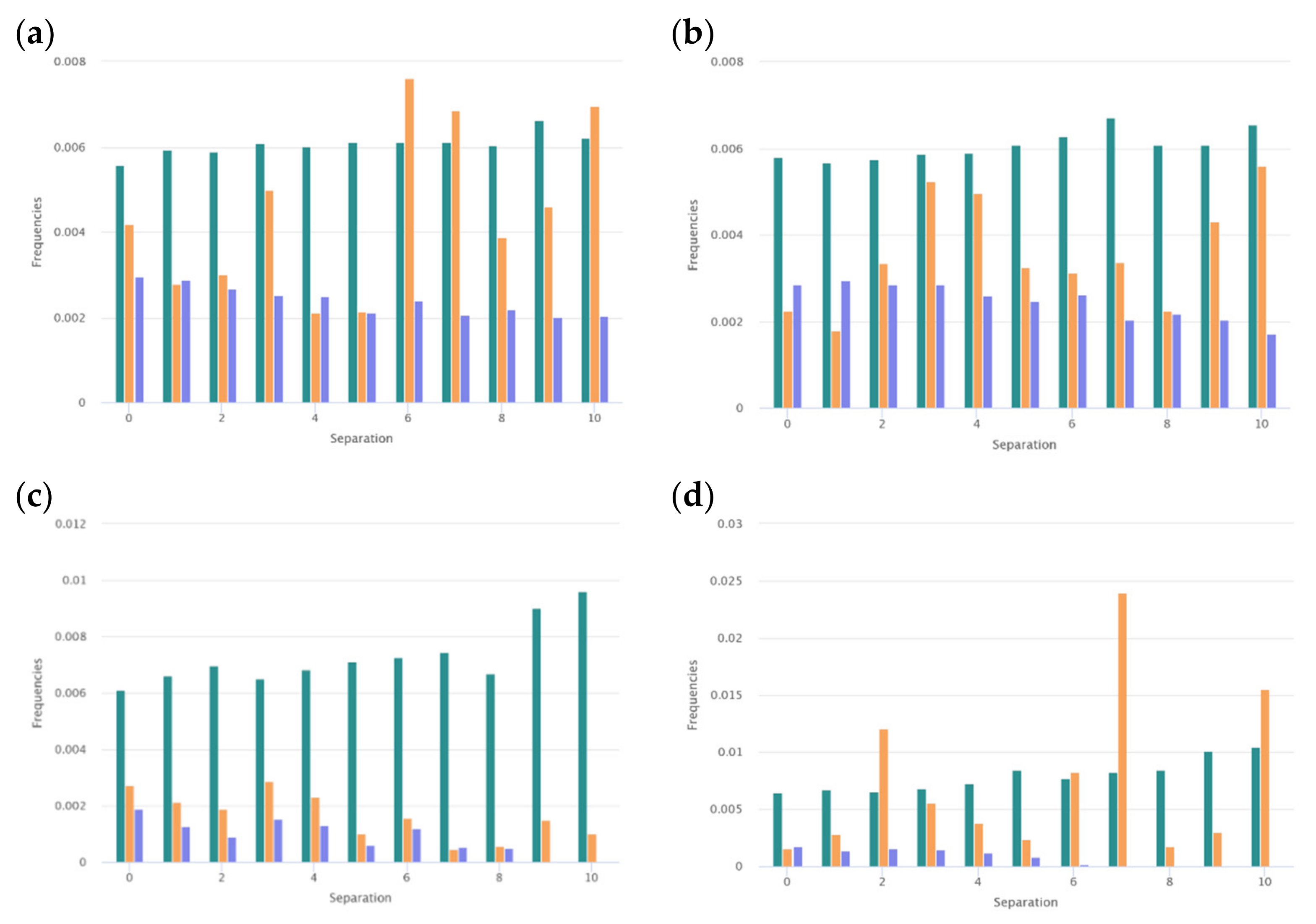
3.1.2. Distribution along the Chain
4. Secondary Structure and Self-Aggregation
4.1. Secondary Structure
4.2. Self-Aggregation
5. Frequently Occurred Amino Acids
5.1. Basic Amino Acids and AMPs
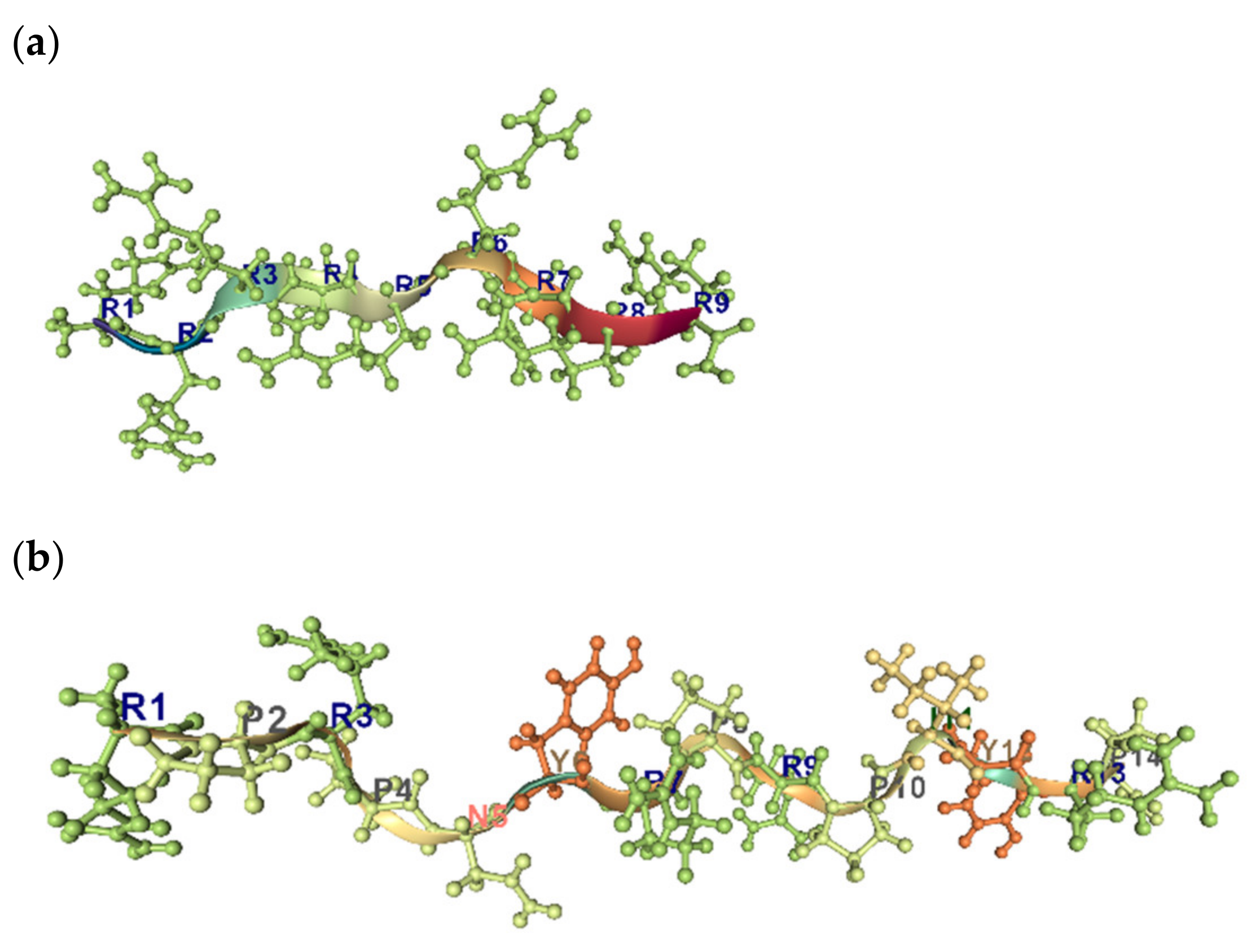
5.1.1. Lysine and Arginine in the AMPs
5.1.2. Histidines in the AMPs
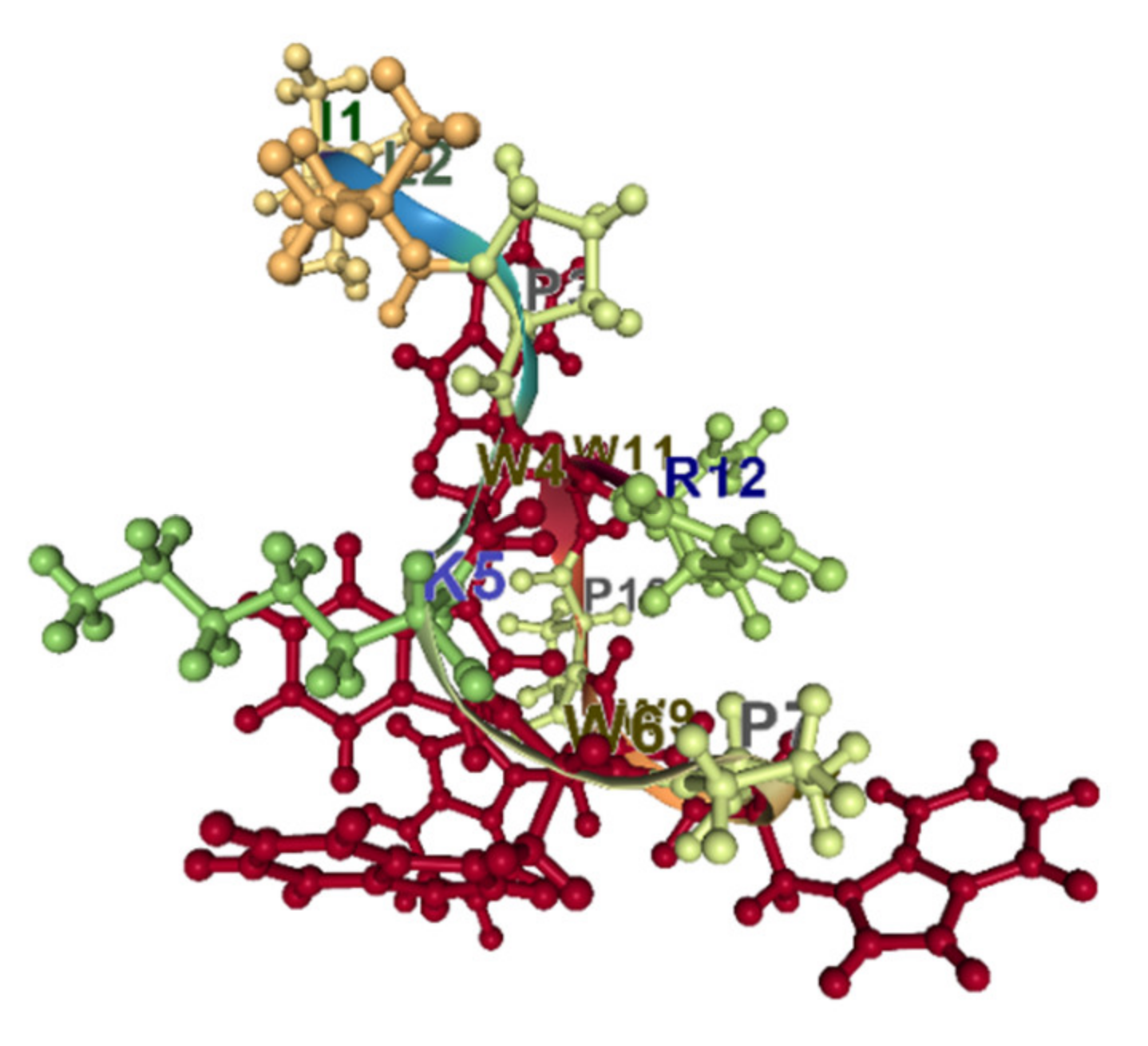
5.2. Aromatic Amino Acids and AMPs. Cation–π Interactions
5.3. Prolines and AMPs. ppII Conformation
5.4. Glycines and AMPs
5.5. Cysteines and AMPs: Intra-Chain Covalent Bonds
6. Modes of AMP Interaction with Biological Membrane
6.1. Permeabilization
6.2. Translocation
6.3. Cooperation among AMPs: Synergism
7. Concluding Remarks on the Grouping of AMP
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cardoso, P.; Glossop, H.; Meikle, T.G.; Aburto-Medina, A.; Conn, C.E.; Sarojini, V.; Valery, C. Molecular engineering of antimicrobial peptides: Microbial targets, peptide motifs and translation opportunities. Biophys. Rev. 2021, 13, 35–69. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 2559. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Pirtskhalava, M. Comment on: ‘Empirical comparison of web-based antimicrobial peptide prediction tools’. Bioinformatics 2019, 35, 2692–2694. [Google Scholar] [CrossRef]
- Espeche, J.C.; Martínez, M.; Maturana, P.; Cutró, A.; Semorile, L.; Maffia, P.C.; Hollmann, A. Unravelling the mechanism of action of “de novo” designed peptide P1 with model membranes and gram-positive and gram-negative bacteria. Arch. Biochem. Biophys. 2020, 693, 108549. [Google Scholar] [CrossRef]
- Savini, F.; Loffredo, M.R.; Troiano, C.; Bobone, S.; Malanovic, N.; Eichmann, T.O.; Caprio, L.; Canale, V.C.; Park, Y.; Mangoni, M.L.; et al. Binding of an antimicrobial peptide to bacterial cells: Interaction with different species, strains and cellular components. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183291. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, T.; Deplazes, E.; Mancera, R.L. Revisiting the Interaction of Melittin with Phospholipid Bilayers: The Effects of Concentration and Ionic Strength. Int. J. Mol. Sci. 2020, 21, 746. [Google Scholar] [CrossRef] [Green Version]
- Palm, C.; Netzereab, S.; Hällbrink, M. Quantitatively determined uptake of cell-penetrating peptides in non-mammalian cells with an evaluation of degradation and antimicrobial effects. Peptides 2006, 27, 1710–1716. [Google Scholar] [CrossRef]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta Biomembr. 2007, 1768, 2500–2509. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, A.; Dupuy, F.G.; Arsov, Z.; Elhady, Y.; Moody, D.; Ernst, R.K.; Deslouches, B.; Montelaro, R.C.; Peter Di, Y.; Tristram-Nagle, S. Elastic behavior of model membranes with antimicrobial peptides depends on lipid specificity and d-enantiomers. Soft Matter 2019, 15, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Remington, J.M.; Liao, C.; Sharafi, M.; Ste Marie, E.J.; Ferrell, J.B.; Hondal, R.J.; Wargo, M.J.; Schneebeli, S.T.; Li, J. Aggregation State of Synergistic Antimicrobial Peptides. J. Phys. Chem. Lett. 2020, 11, 9501–9506. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhu, X.; Acosta, S.; Kumar, D.; Sang, T.; Aparicio, C. Self-assembly dynamics and antimicrobial activity of all l- and d-amino acid enantiomers of a designer peptide. Nanoscale 2019, 11, 266–275. [Google Scholar] [CrossRef]
- Allolio, C.; Magarkar, A.; Jurkiewicz, P.; Baxová, K.; Javanainen, M.; Mason, P.E.; Šachl, R.; Cebecauer, M.; Hof, M.; Horinek, D.; et al. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. USA 2018, 115, 11923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shagaghi, N.; Bhave, M.; Palombo, E.A.; Clayton, A.H.A. Revealing the sequence of interactions of PuroA peptide with Candida albicans cells by live-cell imaging. Sci. Rep. 2017, 7, 43542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechinger, B. The SMART model: Soft Membranes Adapt and Respond, also Transiently, in the presence of antimicrobial peptides. J. Pept. Sci. 2015, 21, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilman, H.R.; Shi, J.; Deane, C.M. Helix kinks are equally prevalent in soluble and membrane proteins. Proteins Struct. Funct. Bioinform. 2014, 82, 1960–1970. [Google Scholar] [CrossRef] [Green Version]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association11Edited by G. von Heijne. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef]
- Bowie, J.U. Membrane protein folding: How important are hydrogen bonds? Curr. Opin. Struct. Biol. 2011, 21, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K.; Toda, H.; Ikeguchi, M. Dependence of α-helical and β-sheet amino acid propensities on the overall protein fold type. BMC Struct. Biol. 2012, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.C.; Deber, C.M. A measure of helical propensity for amino acids in membrane environments. Nat. Struct. Biol. 1994, 1, 558. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Ikonomova, S.P.; Karlsson, A.J. Secondary structure of cell-penetrating peptides during interaction with fungal cells. Protein Sci. 2018, 27, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmouche, N.; Aisenbrey, C.; Porcelli, F.; Xia, Y.; Nelson, S.E.D.; Chen, X.; Raya, J.; Vermeer, L.; Aparicio, C.; Veglia, G.; et al. Solution and Solid-State Nuclear Magnetic Resonance Structural Investigations of the Antimicrobial Designer Peptide GL13K in Membranes. Biochemistry 2017, 56, 4269–4278. [Google Scholar] [CrossRef]
- Shi, Z.; Woody, R.W.; Kallenbach, N.R. Is polyproline II a major backbone conformation in unfolded proteins? Adv. Protein Chem. 2002, 62, 163–240. [Google Scholar]
- Tang, M.; Hong, M. Structure and mechanism of β-hairpinantimicrobialpeptides in lipidbilayers from solid-state NMR spectroscopy. Mol. Biosyst. 2009, 5, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, M.; Resende, J.M.; Moraes, C.M.; Marquette, A.; Chich, J.-F.; Metz-Boutigue, M.-H.; Bechinger, B. Membrane structure and interactions of human catestatin by multidimensional solution and solid-state NMR spectroscopy. FASEB J. 2010, 24, 1737–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean-François, F.; Castano, S.; Desbat, B.; Odaert, B.; Roux, M.; Metz-Boutigue, M.-H.; Dufourc, E.J. Aggregation of Cateslytin β-Sheets on Negatively Charged Lipids Promotes Rigid Membrane Domains. A New Mode of Action for Antimicrobial Peptides? Biochemistry 2008, 47, 6394–6402. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.L.; Moorthy, A.E.; Li, Y.; Tamm, L.K. Fusion Activity of HIV gp41 Fusion Domain Is Related to Its Secondary Structure and Depth of Membrane Insertion in a Cholesterol-Dependent Fashion. J. Mol. Biol. 2012, 418, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, U.H.; Zeng, E.; Pasinetti, G.M. The Use of Antimicrobial and Antiviral Drugs in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 4920. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra372. [Google Scholar] [CrossRef] [Green Version]
- Pagel, K.; Wagner, S.C.; Samedov, K.; von Berlepsch, H.; Böttcher, C.; Koksch, B. Random coils, beta-sheet ribbons, and alpha-helical fibers: One peptide adopting three different secondary structures at will. J. Am. Chem. Soc. 2006, 128, 2196–2197. [Google Scholar] [CrossRef]
- Lan, Y.; Ye, Y.; Kozlowska, J.; Lam, J.K.W.; Drake, A.F.; Mason, A.J. Structural contributions to the intracellular targeting strategies of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarig, H.; Rotem, S.; Ziserman, L.; Danino, D.; Mor, A. Impact of Self-Assembly Properties on Antibacterial Activity of Short Acyl-Lysine Oligomers. Antimicrob. Agents Chemother. 2008, 52, 4308. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, C.J.; Liyanage, W.; Federation, A.J.; Nilsson, B.L. Tuning β-Sheet Peptide Self-Assembly and Hydrogelation Behavior by Modification of Sequence Hydrophobicity and Aromaticity. Biomacromolecules 2011, 12, 2735–2745. [Google Scholar] [CrossRef] [PubMed]
- Feder, R.; Dagan, A.; Mor, A. Structure-activity relationship study of antimicrobial dermaseptin S4 showing the consequences of peptide oligomerization on selective cytotoxicity. J. Biol. Chem. 2000, 275, 4230–4238. [Google Scholar] [CrossRef] [Green Version]
- Torrent, M.; Andreu, D.; Nogués, V.M.; Boix, E. Connecting Peptide Physicochemical and Antimicrobial Properties by a Rational Prediction Model. PLoS ONE 2011, 6, e16968. [Google Scholar] [CrossRef]
- Torrent, M.; Valle, J.; Nogués, M.V.; Boix, E.; Andreu, D. The Generation of Antimicrobial Peptide Activity: A Trade-off between Charge and Aggregation? Angew. Chem. Int. Ed. 2011, 50, 10686–10689. [Google Scholar] [CrossRef]
- Sato, H.; Feix, J.B. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnaider, L.; Brahmachari, S.; Schmidt, N.W.; Mensa, B.; Shaham-Niv, S.; Bychenko, D.; Adler-Abramovich, L.; Shimon, L.J.W.; Kolusheva, S.; DeGrado, W.F.; et al. Self-assembling dipeptide antibacterial nanostructures with membrane disrupting activity. Nat. Commun. 2017, 8, 1365. [Google Scholar] [CrossRef]
- Lombardi, L.; Falanga, A.; Del Genio, V.; Galdiero, S. A New Hope: Self-Assembling Peptides with Antimicrobial Activity. Pharmaceutics 2019, 11, 166. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yang, S.; Li, S.; Lang, J.C.; Mao, C.; Kroll, P.; Tang, L.; Dong, H. Self-Assembled Peptide Nanofibers Display Natural Antimicrobial Peptides to Selectively Kill Bacteria without Compromising Cytocompatibility. ACS Appl. Mater. Interfaces 2019, 11, 28681–28689. [Google Scholar] [CrossRef] [PubMed]
- Glossop, H.D.; De Zoysa, G.H.; Hemar, Y.; Cardoso, P.; Wang, K.; Lu, J.; Valéry, C.; Sarojini, V. Battacin-Inspired Ultrashort Peptides: Nanostructure Analysis and Antimicrobial Activity. Biomacromolecules 2019, 20, 2515–2529. [Google Scholar] [CrossRef]
- Di Pisa, M.; Chassaing, G.; Swiecicki, J.-M. Translocation Mechanism(s) of Cell-Penetrating Peptides: Biophysical Studies Using Artificial Membrane Bilayers. Biochemistry 2015, 54, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Eriksson, L.E.G.; Gräslund, A. Conformational states of the cell-penetrating peptide penetratin when interacting with phospholipid vesicles: Effects of surface charge and peptide concentration. Biochim. Biophys. Acta Biomembr. 2002, 1563, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Eiríksdóttir, E.; Konate, K.; Langel, Ü.; Divita, G.; Deshayes, S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitaku, S.; Hirokawa, T.; Tsuji, T. Amphiphilicity index of polar amino acids as an aid in the characterization of amino acid preference at membrane-water interfaces. Bioinformatics 2002, 18, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Robison, A.D.; Sun, S.; Poyton, M.F.; Johnson, G.A.; Pellois, J.-P.; Jungwirth, P.; Vazdar, M.; Cremer, P.S. Polyarginine Interacts More Strongly and Cooperatively than Polylysine with Phospholipid Bilayers. J. Phys. Chem. B 2016, 120, 9287–9296. [Google Scholar] [CrossRef] [Green Version]
- Schwieger, C.; Blume, A. Interaction of Poly(l-arginine) with Negatively Charged DPPG Membranes: Calorimetric and Monolayer Studies. Biomacromolecules 2009, 10, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Vazdar, M.; Uhlig, F.; Jungwirth, P. Like-Charge Ion Pairing in Water: An Ab Initio Molecular Dynamics Study of Aqueous Guanidinium Cations. J. Phys. Chem. Lett. 2012, 3, 2021–2024. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Hong, S.; Liu, Z.; Yue, T.; Dobnikar, J.; Zhang, X. Membrane potential drives direct translocation of cell-penetrating peptides. Nanoscale 2019, 11, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Lamazière, A.; Wolf, C.; Lambert, O.; Chassaing, G.; Trugnan, G.; Ayala-Sanmartin, J. The Homeodomain Derived Peptide Penetratin Induces Curvature of Fluid Membrane Domains. PLoS ONE 2008, 3, e1938. [Google Scholar] [CrossRef]
- Almeida, C.; Lamazière, A.; Filleau, A.; Corvis, Y.; Espeau, P.; Ayala-Sanmartin, J. Membrane re-arrangements and rippled phase stabilisation by the cell penetrating peptide penetratin. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2584–2591. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Vogel, A.; Correia, I.; Lequin, O.; Olausson, B.E.; Desbat, B.; Sagan, S.; Alves, I.D. Membrane interactions of two arginine-rich peptides with different cell internalization capacities. Biochim. Biophys. Acta 2012, 1818, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Correia, I.; Jiao, C.-Y.; Lequin, O.; Bent, E.H.; Goasdoué, N.; Lacombe, C.; Chassaing, G.; Sagan, S.; Alves, I.D. Different membrane behaviour and cellular uptake of three basic arginine-rich peptides. Biochim. Biophys. Acta Biomembr. 2011, 1808, 382–393. [Google Scholar] [CrossRef]
- Liao, S.-M.; Du, Q.-S.; Meng, J.-Z.; Pang, Z.-W.; Huang, R.-B. The multiple roles of histidine in protein interactions. Chem. Cent. J. 2013, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Kacprzyk, L.; Rydengård, V.; Mörgelin, M.; Davoudi, M.; Pasupuleti, M.; Malmsten, M.; Schmidtchen, A. Antimicrobial activity of histidine-rich peptides is dependent on acidic conditions. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2667–2680. [Google Scholar] [CrossRef] [Green Version]
- Muller, L.; Jackson, S.N.; Woods, A.S. Histidine, the less interactive cousin of arginine. Eur. J. Mass Spectrom. 2019, 25, 212–218. [Google Scholar] [CrossRef]
- Kumar, K.; Woo, S.M.; Siu, T.; Cortopassi, W.A.; Duarte, F.; Paton, R.S. Cation–π interactions in protein–ligand binding: Theory and data-mining reveal different roles for lysine and arginine. Chem. Sci. 2018, 9, 2655–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyda, J.; Mason, P.E.; Jungwirth, P. Attractive Interactions between Side Chains of Histidine-Histidine and Histidine-Arginine-Based Cationic Dipeptides in Water. J. Phys. Chem. B 2010, 114, 8744–8749. [Google Scholar] [CrossRef] [PubMed]
- Sforça, M.L.; Machado, A.; Figureueredo, R.C.R.; Oyama, S.; Silva, F.D.; Miranda, A.; Daffre, S.; Miranda, M.T.M.; Spisni, A.; Pertinhez, T.A. The Micelle-Bound Structure of an Antimicrobial Peptide Derived from the α-Chain of Bovine Hemoglobin Isolated from the Tick Boophilus microplus. Biochemistry 2005, 44, 6440–6451. [Google Scholar] [CrossRef]
- Lee, I.H.; Cho, Y.; Lehrer, R.I. Effects of pH and salinity on the antimicrobial properties of clavanins. Infect. Immun. 1997, 65, 2898–2903. [Google Scholar] [CrossRef] [Green Version]
- Lauth, X.; Shike, H.; Burns, J.C.; Westerman, M.E.; Ostland, V.E.; Carlberg, J.M.; Van Olst, J.C.; Nizet, V.; Taylor, S.W.; Shimizu, C.; et al. Discovery and characterization of two isoforms of moronecidin, a novel antimicrobial peptide from hybrid striped bass. J. Biol. Chem. 2002, 277, 5030–5039. [Google Scholar] [CrossRef] [Green Version]
- van der Spek, J.C.; Wyandt, H.E.; Skare, J.C.; Milunsky, A.; Oppenheim, F.G.; Troxler, R.F. Localization of the genes for histatins to human chromosome 4q13 and tissue distribution of the mRNAs. Am. J. Hum. Genet. 1989, 45, 381–387. [Google Scholar] [PubMed]
- Xu, T.; Levitz, S.M.; Diamond, R.D.; Oppenheim, F.G. Anticandidal activity of major human salivary histatins. Infect. Immun. 1991, 59, 2549–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, S.; Edgerton, M. How Does It Kill?: Understanding the Candidacidal Mechanism of Salivary Histatin 5. Eukaryot. Cell 2014, 13, 958. [Google Scholar] [CrossRef] [Green Version]
- Conklin, S.E.; Bridgman, E.C.; Su, Q.; Riggs-Gelasco, P.; Haas, K.L.; Franz, K.J. Specific Histidine Residues Confer Histatin Peptides with Copper-Dependent Activity against Candida albicans. Biochemistry 2017, 56, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Juliano, S.A.; Pierce, S.; de Mayo, J.A.; Balunas, M.J.; Angeles-Boza, A.M. Exploration of the Innate Immune System of Styela clava: Zn2+ Binding Enhances the Antimicrobial Activity of the Tunicate Peptide Clavanin A. Biochemistry 2017, 56, 1403–1414. [Google Scholar] [CrossRef]
- Rydengård, V.; Andersson Nordahl, E.; Schmidtchen, A. Zinc potentiates the antibacterial effects of histidine-rich peptides against Enterococcus faecalis. FEBS J. 2006, 273, 2399–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.; Aisenbrey, C.; Harmouche, N.; Raya, J.; Bertani, P.; Voievoda, N.; Süss, R.; Bechinger, B. pH-Dependent Membrane Interactions of the Histidine-Rich Cell-Penetrating Peptide LAH4-L1. Biophys. J. 2017, 113, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, B.; Miles, A.J.; Salnikov, E.S.; Wallace, B.A.; Bechinger, B. Lipid interactions of LAH4, a peptide with antimicrobial and nucleic acid transfection activities. Eur. Biophys. J. 2014, 43, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T.C.; Bechinger, B. The interactions of histidine-containing amphipathic helical peptide antibiotics with lipid bilayers. The effects of charges and pH. J. Biol. Chem. 1999, 274, 29115–29121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, A.J.; Moussaoui, W.; Abdelrahman, T.; Boukhari, A.; Bertani, P.; Marquette, A.; Shooshtarizaheh, P.; Moulay, G.; Boehm, N.; Guerold, B.; et al. Structural determinants of antimicrobial and antiplasmodial activity and selectivity in histidine-rich amphipathic cationic peptides. J. Biol. Chem. 2009, 284, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Lointier, M.; Aisenbrey, C.; Marquette, A.; Tan, J.H.; Kichler, A.; Bechinger, B. Membrane pore-formation correlates with the hydrophilic angle of histidine-rich amphipathic peptides with multiple biological activities. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183212. [Google Scholar] [CrossRef]
- Hitchner, M.A.; Santiago-Ortiz, L.E.; Necelis, M.R.; Shirley, D.J.; Palmer, T.J.; Tarnawsky, K.E.; Vaden, T.D.; Caputo, G.A. Activity and characterization of a pH-sensitive antimicrobial peptide. Biochim. Biophys. Acta Biomembr. 2019, 1861, 182984. [Google Scholar] [CrossRef]
- Tu, Z.; Volk, M.; Shah, K.; Clerkin, K.; Liang, J.F. Constructing bioactive peptides with pH-dependent activities. Peptides 2009, 30, 1523–1528. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, D.A. Cation-π Interactions in Chemistry and Biology: A New View of Benzene, Phe, Tyr, and Trp. Science 1996, 271, 163. [Google Scholar] [CrossRef]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The preference of tryptophan for membrane interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef] [Green Version]
- Senes, A.; Chadi, D.C.; Law, P.B.; Walters, R.F.S.; Nanda, V.; DeGrado, W.F. Ez, a Depth-dependent Potential for Assessing the Energies of Insertion of Amino Acid Side-chains into Membranes: Derivation and Applications to Determining the Orientation of Transmembrane and Interfacial Helices. J. Mol. Biol. 2007, 366, 436–448. [Google Scholar] [CrossRef]
- Eisenberg, D. Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 1984, 53, 595–623. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Matheron, L.; Cribier, S.; Chaignepain, S.; Jobin, M.L.; Sagan, S.; Alves, I.D. Direct translocation of cell-penetrating peptides in liposomes: A combined mass spectrometry quantification and fluorescence detection study. Anal. Biochem. 2013, 438, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Bauzá, A.; Girardet, C.; Alves, I.D.; Lecomte, S.; Illien, F.; Cardon, S.; Chaianantakul, N.; Pallerla, M.; Burlina, F.; et al. Ionpair-π interactions favor cell penetration of arginine/tryptophan-rich cell-penetrating peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183098. [Google Scholar] [CrossRef]
- Aliste, M.P.; MacCallum, J.L.; Tieleman, D.P. Molecular Dynamics Simulations of Pentapeptides at Interfaces: Salt Bridge and Cation−π Interactions. Biochemistry 2003, 42, 8976–8987. [Google Scholar] [CrossRef]
- Jing, W.; Demcoe, A.R.; Vogel, H.J. Conformation of a Bactericidal Domain of Puroindoline a: Structure and Mechanism of Action of a 13-Residue Antimicrobial Peptide. J. Bacteriol. 2003, 185, 4938. [Google Scholar] [CrossRef] [Green Version]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-H.; Chen, C.; Jou, M.-L.; Lee, A.Y.-L.; Lin, Y.-C.; Yu, Y.-P.; Huang, W.-T.; Wu, S.-H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Zarzosa-Moreno, D.; Avalos-Gómez, C.; Ramírez-Texcalco, L.S.; Torres-López, E.; Ramírez-Mondragón, R.; Hernández-Ramírez, J.O.; Serrano-Luna, J.; de la Garza, M. Lactoferrin and Its Derived Peptides: An Alternative for Combating Virulence Mechanisms Developed by Pathogens. Molecules 2020, 25, 5763. [Google Scholar] [CrossRef]
- Blochet, J.-E.; Chevalier, C.; Forest, E.; Pebay-Peyroula, E.; Gautier, M.-F.; Joudrier, P.; Pézolet, M.; Marion, D. Complete amino acid sequence of puroindoline, a new basic and cystine-rich protein with a unique tryptophan-rich domain, isolated from wheat endosperm by Triton X-114 phase partitioning. FEBS Lett. 1993, 329, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Ong, Z.Y.; Wiradharma, N.; Yang, Y.Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv. Drug Deliv. Rev. 2014, 78, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, L.S.; Lan, Y.; Abbate, V.; Ruh, E.; Bui, T.T.; Wilkinson, L.J.; Kanno, T.; Jumagulova, E.; Kozlowska, J.; Patel, J.; et al. Conformational flexibility determines selectivity and antibacterial, antiplasmodial, and anticancer potency of cationic α-helical peptides. J. Biol. Chem. 2012, 287, 34120–34133. [Google Scholar] [CrossRef] [Green Version]
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-rich antimicrobial peptides: Converging to a non-lytic mechanism of action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Welch, N.G.; Li, W.; Hossain, M.A.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. (Re)Defining the Proline-Rich Antimicrobial Peptide Family and the Identification of Putative New Members. Front. Chem. 2020, 8, 1157. [Google Scholar] [CrossRef]
- Otvos, L.; Rogers, M.E.; Consolvo, P.J.; Condie, B.A.; Lovas, S.; Bulet, P.; Blaszczyk-Thurin, M. Interaction between Heat Shock Proteins and Antimicrobial Peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar] [CrossRef] [PubMed]
- Kragol, G.; Hoffmann, R.; Chattergoon, M.A.; Lovas, S.; Cudic, M.; Bulet, P.; Condie, B.A.; Rosengren, K.J.; Montaner, L.J.; Otvos, L., Jr. Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin. Eur. J. Biochem. 2002, 269, 4226–4237. [Google Scholar] [CrossRef]
- Runti, G.; Lopez Ruiz, M.d.C.; Stoilova, T.; Hussain, R.; Jennions, M.; Choudhury, H.G.; Benincasa, M.; Gennaro, R.; Beis, K.; Scocchi, M. Functional Characterization of SbmA, a Bacterial Inner Membrane Transporter Required for Importing the Antimicrobial Peptide Bac7(1-35). J. Bacteriol. 2013, 195, 5343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; O’Brien-Simpson, N.M.; Tailhades, J.; Pantarat, N.; Dawson, R.M.; Otvos, L., Jr.; Reynolds, E.C.; Separovic, F.; Hossain, M.A.; Wade, J.D. Multimerization of a Proline-Rich Antimicrobial Peptide, Chex-Arg20, Alters Its Mechanism of Interaction with the Escherichia coli Membrane. Chem. Biol. 2015, 22, 1250–1258. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E.; Litt, J.; Kane, R.S.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of Action of the Antimicrobial Peptide Buforin II: Buforin II Kills Microorganisms by Penetrating the Cell Membrane and Inhibiting Cellular Functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Pawar, A.P.; DuBay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “Aggregation-prone” and “Aggregation-susceptible” Regions in Proteins Associated with Neurodegenerative Diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef]
- Creasey, R.G.; Voelcker, N.H.; Schultz, C.J. Investigation of self-assembling proline- and glycine-rich recombinant proteins and peptides inspired by proteins from a symbiotic fungus using atomic force microscopy and circular dichroism spectroscopy. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Sharma, M.; Zhou, H.-X.; Cross, T.A. Glycines: Role in α-Helical Membrane Protein Structures and a Potential Indicator of Native Conformation. Biochemistry 2012, 51, 4779–4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J. The roles of GxxxG motif and gamma-secretase components in APP processing. Interdiscip. Bio Cent. 2009, 1, 1. [Google Scholar] [CrossRef]
- Fonte, V.; Dostal, V.; Roberts, C.M.; Gonzales, P.; Lacor, P.; Magrane, J.; Dingwell, N.; Fan, E.Y.; Silverman, M.A.; Stein, G.H.; et al. A glycine zipper motif mediates the formation of toxic β-amyloid oligomers in vitro and in vivo. Mol. Neurodegener. 2011, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruston, F.; Lacombe, C.; Zimmermann, K.; Piesse, C.; Nicolas, P.; El Amri, C. Structural malleability of plasticins: Preorganized conformations in solution and relevance for antimicrobial activity. Biopolymers 2007, 86, 42–56. [Google Scholar] [CrossRef]
- Carlier, L.; Joanne, P.; Khemtémourian, L.; Lacombe, C.; Nicolas, P.; El Amri, C.; Lequin, O. Investigating the role of GXXXG motifs in helical folding and self-association of plasticins, Gly/Leu-rich antimicrobial peptides. Biophys. Chem. 2015, 196, 40–52. [Google Scholar] [CrossRef]
- Petkov, P.; Lilkova, E.; Ilieva, N.; Litov, L. Self-Association of Antimicrobial Peptides: A Molecular Dynamics Simulation Study on Bombinin. Int. J. Mol. Sci. 2019, 20, 5450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangger, K.; Gößler, R.; Khatai, L.; Lohner, K.; Jilek, A. Structures of the glycine-rich diastereomeric peptides bombinin H2 and H4. Toxicon 2008, 52, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.C.; Berto, R.F.; Gois, E.A.; Fontenele-Cardi, N.C.; Honório-Júnior, J.E.R.; Konno, K.; Richardson, M.; Rocha, M.F.G.; Camargo, A.A.C.M.; Pimenta, D.C.; et al. Leptoglycin: A new Glycine/Leucine-rich antimicrobial peptide isolated from the skin secretion of the South American frog Leptodactylus pentadactylus (Leptodactylidae). Toxicon 2009, 54, 23–32. [Google Scholar] [CrossRef]
- Verdon, J.; Coutos-Thevenot, P.; Rodier, M.-H.; Landon, C.; Depayras, S.; Noel, C.; La Camera, S.; Moumen, B.; Greve, P.; Bouchon, D.; et al. Armadillidin H, a Glycine-Rich Peptide from the Terrestrial Crustacean Armadillidium vulgare, Displays an Unexpected Wide Antimicrobial Spectrum with Membranolytic Activity. Front. Microbiol. 2016, 7, 1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, J.P.; Nguyen, G.K.T.; Loo, S.; Wang, S.; Yang, D.; Kam, A. Ginsentides: Cysteine and Glycine-rich Peptides from the Ginseng Family with Unusual Disulfide Connectivity. Sci. Rep. 2018, 8, 16201. [Google Scholar] [CrossRef]
- Finkina, E.I.; Ovchinnikova, T.V. Plant Defensins: Structure, Functions, Biosynthesis, and the Role in the Immune Response. Russ. J. Bioorganic Chem. 2018, 44, 261–278. [Google Scholar] [CrossRef]
- Sahl, H.-G.; Pag, U.; Bonness, S.; Wagner, S.; Antcheva, N.; Tossi, A. Mammalian defensins: Structures and mechanism of antibiotic activity. J. Leukoc. Biol. 2005, 77, 466–475. [Google Scholar] [CrossRef]
- Slavokhotova, A.A.; Shelenkov, A.A.; Andreev, Y.A.; Odintsova, T.I. Hevein-like antimicrobial peptides of plants. Biochemistry 2017, 82, 1659–1674. [Google Scholar] [CrossRef]
- Kini, S.G.; Wong, K.H.; Tan, W.L.; Xiao, T.; Tam, J.P. Morintides: Cargo-free chitin-binding peptides from Moringa oleifera. BMC Plant Biol. 2017, 17, 68. [Google Scholar] [CrossRef] [Green Version]
- Driessen, A.J.; van den Hooven, H.W.; Kuiper, W.; van de Kamp, M.; Sahl, H.G.; Konings, R.N.; Konings, W.N. Mechanistic studies of lantibiotic-induced permeabilization of phospholipid vesicles. Biochemistry 1995, 34, 1606–1614. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, P.; Wilmes, M.; Pugnière, M.; Aumelas, A.; Bachère, E.; Sahl, H.G.; Schneider, T.; Destoumieux-Garzón, D. Insight into invertebrate defensin mechanism of action: Oyster defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J. Biol. Chem. 2010, 285, 29208–29216. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kim, B.S. Antimicrobial Cyclic Peptides for Plant Disease Control. Plant Pathol. J. 2015, 31, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.-J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta Biomembr. 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmschneider, J.P. Charged Antimicrobial Peptides Can Translocate across Membranes without Forming Channel-like Pores. Biophys. J. 2017, 113, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, C.H.; Hu, D.; Ulmschneider, M.B.; Ulmschneider, J.P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016, 7, 13535. [Google Scholar] [CrossRef] [Green Version]
- Dias, S.A.; Freire, J.M.; Pérez-Peinado, C.; Domingues, M.M.; Gaspar, D.; Vale, N.; Gomes, P.; Andreu, D.; Henriques, S.T.; Castanho, M.A.R.B.; et al. New Potent Membrane-Targeting Antibacterial Peptides from Viral Capsid Proteins. Front. Microbiol. 2017, 8, 775. [Google Scholar] [CrossRef]
- Gregory, S.M.; Cavenaugh, A.; Journigan, V.; Pokorny, A.; Almeida, P.F.F. A Quantitative Model for the All-or-None Permeabilization of Phospholipid Vesicles by the Antimicrobial Peptide Cecropin A. Biophys. J. 2008, 94, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Oreopoulos, J.; Epand, R.F.; Epand, R.M.; Yip, C.M. Peptide-Induced Domain Formation in Supported Lipid Bilayers: Direct Evidence by Combined Atomic Force and Polarized Total Internal Reflection Fluorescence Microscopy. Biophys. J. 2010, 98, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Tamba, Y.; Ariyama, H.; Levadny, V.; Yamazaki, M. Kinetic Pathway of Antimicrobial Peptide Magainin 2-Induced Pore Formation in Lipid Membranes. J. Phys. Chem. B 2010, 114, 12018–12026. [Google Scholar] [CrossRef]
- Sani, M.A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-Stave Model or Toroidal Model? A Case Study on Melittin Pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Hale, J.D.F.; Hancock, R.E.W. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti Infect. Ther. 2007, 5, 951–959. [Google Scholar] [CrossRef]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W.; Chen, F.-Y.; Lee, M.-T. Molecular Mechanism of Peptide-Induced Pores in Membranes. Phys. Rev. Lett. 2004, 92, 198304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, S.; Wang, W.; Yang, L.; Huang, H.W. Structure of the Alamethicin Pore Reconstructed by X-Ray Diffraction Analysis. Biophys. J. 2008, 94, 3512–3522. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K.; Yoneyama, S.; Miyajima, K. Pore formation and translocation of melittin. Biophys. J. 1997, 73, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Wimley, W.C.; Hristova, K. Antimicrobial Peptides: Successes, Challenges and Unanswered Questions. J. Membr. Biol. 2011, 239, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Rotem, S.; Mor, A.; Berno, B.; Epand, R.F. Bacterial Membranes as Predictors of Antimicrobial Potency. J. Am. Chem. Soc. 2008, 130, 14346–14352. [Google Scholar] [CrossRef]
- Joanne, P.; Galanth, C.; Goasdoué, N.; Nicolas, P.; Sagan, S.; Lavielle, S.; Chassaing, G.; El Amri, C.; Alves, I.D. Lipid reorganization induced by membrane-active peptides probed using differential scanning calorimetry. Biochim. Biophys. Acta 2009, 1788, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K. New strategies for novel antibiotics: Peptides targeting bacterial cell membranes. Gen. Physiol. Biophys. 2009, 28, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Marrink, S.J.; Melo, M.N. Localization Preference of Antimicrobial Peptides on Liquid-Disordered Membrane Domains. Front. Cell Dev. Biol. 2020, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Krämer, U.; et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, E1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakowska, P.D.; Jiang, H.; Ray, S.; Pyne, A.; Lamarre, B.; Carr, M.; Judge, P.J.; Ravi, J.M.; Gerling, U.I.; Koksch, B.; et al. Nanoscale imaging reveals laterally expanding antimicrobial pores in lipid bilayers. Proc. Natl. Acad. Sci. USA 2013, 110, 8918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambroggio, E.E.; Separovic, F.; Bowie, J.H.; Fidelio, G.D.; Bagatolli, L.A. Direct Visualization of Membrane Leakage Induced by the Antibiotic Peptides: Maculatin, Citropin, and Aurein. Biophys. J. 2005, 89, 1874–1881. [Google Scholar] [CrossRef] [Green Version]
- Strandberg, E.; Tremouilhac, P.; Wadhwani, P.; Ulrich, A.S. Synergistic transmembrane insertion of the heterodimeric PGLa/magainin 2 complex studied by solid-state NMR. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1667–1679. [Google Scholar] [CrossRef] [Green Version]
- Hong, R.W.; Shchepetov, M.; Weiser, J.N.; Axelsen, P.H. Transcriptional Profile of the Escherichia coli Response to the Antimicrobial Insect Peptide Cecropin A. Antimicrob. Agents Chemother. 2003, 47, 1. [Google Scholar] [CrossRef] [Green Version]
- Ablan, F.D.; Spaller, B.L.; Abdo, K.I.; Almeida, P.F. Charge Distribution Fine-Tunes the Translocation of α -Helical Amphipathic Peptides across Membranes. Biophys. J. 2016, 111, 1738–1749. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Hristova, K.; Wimley, W.C. A Highly Charged Voltage-Sensor Helix Spontaneously Translocates across Membranes. Angew. Chem. Int. Ed. 2012, 51, 7150–7153. [Google Scholar] [CrossRef] [Green Version]
- Marxer, M.; Vollenweider, V.; Schmid-Hempel, P. Insect antimicrobial peptides act synergistically to inhibit a trypanosome parasite. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef]
- Cokol, M.; Chua, H.N.; Tasan, M.; Mutlu, B.; Weinstein, Z.B.; Suzuki, Y.; Nergiz, M.E.; Costanzo, M.; Baryshnikova, A.; Giaever, G.; et al. Systematic exploration of synergistic drug pairs. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Tamma, P.D.; Cosgrove, S.E.; Maragakis, L.L. Combination Therapy for Treatment of Infections with Gram-Negative Bacteria. Clin. Microbiol. Rev. 2012, 25, 450–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthington, R.J.; Melander, C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol. 2013, 31, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chait, R.; Craney, A.; Kishony, R. Antibiotic interactions that select against resistance. Nature 2007, 446, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.J.; Hegreness, M.J.; Aiden, A.P.; Kishony, R. Drug interactions and the evolution of antibiotic resistance. Nat. Rev. Microbiol. 2009, 7, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Huang, Y.; Chen, M.; Li, G.; Chen, Y. Functional synergy of α-helical antimicrobial peptides and traditional antibiotics against Gram-negative and Gram-positive bacteria in vitro and in vivo. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 34, 197–204. [Google Scholar] [CrossRef]
- Ruden, S.; Rieder, A.; Schwartz, T.; Mikut, R.; Hilpert, K. Synergy pattern of short cationic antimicrobial peptides against multidrug-resistant Pseudomonas aeruginosa. bioRxiv 2019, 639286. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Casanova, Y.; Rodríguez-Mayor, A.V.; Cardenas, K.J.; Leal-Castro, A.L.; Muñoz-Molina, L.C.; Fierro-Medina, R.; Rivera-Monroy, Z.J.; García-Castañeda, J.E. Synergistic bactericide and antibiotic effects of dimeric, tetrameric, or palindromic peptides containing the RWQWR motif against Gram-positive and Gram-negative strains. RSC Adv. 2019, 9, 7239–7245. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, Z.; Li, X.; Tian, Y.; Fan, Y.; Yu, C.; Zhou, B.; Liu, Y.; Xiang, R.; Yang, L. Synergistic effects of antimicrobial peptide DP7 combined with antibiotics against multidrug-resistant bacteria. Drug Des. Devel. Ther. 2017, 11, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Huang, Y.; Chen, M.; Hu, C.; Chen, Y. Functional Synergy of Antimicrobial Peptides and Chlorhexidine Acetate against Gram-Negative/Gram-Positive Bacteria and A Fungus In Vitro And In Vivo. Infect. Drug Resist. 2019, 12, 3227–3239. [Google Scholar] [CrossRef] [Green Version]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial Peptides: Interaction with Model and Biological Membranes and Synergism with Chemical Antibiotics. Front. Chem. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Combination Effects of Antimicrobial Peptides. Antimicrob. Agents Chemother. 2016, 60, 1717–1724. [Google Scholar] [CrossRef] [Green Version]
- Bevins, C.L.; Zasloff, M. Peptides from Frog Skin. Annu. Rev. Biochem. 1990, 59, 395–414. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.R.; Makarova, O.; Rolff, J. Inducible Defenses Stay Up Late: Temporal Patterns of Immune Gene Expression inTenebrio molitor. Genes Genomes Genet. 2014, 4, 947–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, Y.; Barra, D.; Simmaco, M.; Shai, Y.; Mangoni, M.L. A Synergism between Temporins toward Gram-negative Bacteria Overcomes Resistance Imposed by the Lipopolysaccharide Protective Layer. J. Biol. Chem. 2006, 281, 28565–28574. [Google Scholar] [CrossRef] [Green Version]
- Pino-Angeles, A.; Leveritt, J.M., III; Lazaridis, T. Pore Structure and Synergy in Antimicrobial Peptides of the Magainin Family. PLoS Comput. Biol. 2016, 12, e1004570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K.; Mitani, Y.; Akada, K.-Y.; Murase, O.; Yoneyama, S.; Zasloff, M.; Miyajima, K. Mechanism of Synergism between Antimicrobial Peptides Magainin 2 and PGLa. Biochemistry 1998, 37, 15144–15153. [Google Scholar] [CrossRef]
- Strandberg, E.; Zerweck, J.; Wadhwani, P.; Ulrich, A.S. Synergistic Insertion of Antimicrobial Magainin-Family Peptides in Membranes Depends on the Lipid Spontaneous Curvature. Biophys. J. 2013, 104, L9–L11. [Google Scholar] [CrossRef] [Green Version]
- Zerweck, J.; Strandberg, E.; Bürck, J.; Reichert, J.; Wadhwani, P.; Kukharenko, O.; Ulrich, A.S. Homo- and heteromeric interaction strengths of the synergistic antimicrobial peptides PGLa and magainin 2 in membranes. Eur. Biophys. J. 2016, 45, 535–547. [Google Scholar] [CrossRef]
- Zerweck, J.; Strandberg, E.; Kukharenko, O.; Reichert, J.; Bürck, J.; Wadhwani, P.; Ulrich, A.S. Molecular mechanism of synergy between the antimicrobial peptides PGLa and magainin 2. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Han, E.; Lee, H. Synergistic effects of magainin 2 and PGLa on their heterodimer formation, aggregation, and insertion into the bilayer. RSC Adv. 2015, 5, 2047–2055. [Google Scholar] [CrossRef]
- Rzepiela, A.J.; Sengupta, D.; Goga, N.; Marrink, S.J. Membrane poration by antimicrobial peptides combining atomistic and coarse-grained descriptions. Faraday Discuss. 2010, 144, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, H.-J.; Wallqvist, A. Spontaneous Buckling of Lipid Bilayer and Vesicle Budding Induced by Antimicrobial Peptide Magainin 2: A Coarse-Grained Simulation Study. J. Phys. Chem. B 2011, 115, 8122–8129. [Google Scholar] [CrossRef]
- Bechinger, B.; Juhl, D.W.; Glattard, E.; Aisenbrey, C. Revealing the Mechanisms of Synergistic Action of Two Magainin Antimicrobial Peptides. Front. Med Technol. 2020, 2. [Google Scholar] [CrossRef]
- Fields, F.R.; Freed, S.D.; Carothers, K.E.; Hamid, M.N.; Hammers, D.E.; Ross, J.N.; Kalwajtys, V.R.; Gonzalez, A.J.; Hildreth, A.D.; Friedberg, I.; et al. Novel antimicrobial peptide discovery using machine learning and biophysical selection of minimal bacteriocin domains. Drug Dev. Res. 2020, 81, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Ghimire, J.; Wu, E.; Wimley, W.C. Mechanistic Landscape of Membrane-Permeabilizing Peptides. Chem. Rev. 2019, 119, 6040–6085. [Google Scholar] [CrossRef] [PubMed]
- Hai Nan, Y.; Jacob, B.; Kim, Y.; Yub Shin, S. Linear bactenecin analogs with cell selectivity and anti-endotoxic activity. J. Pept. Sci. 2012, 18, 740–747. [Google Scholar] [CrossRef]
- Haney, E.F.; Brito-Sánchez, Y.; Trimble, M.J.; Mansour, S.C.; Cherkasov, A.; Hancock, R.E.W. Computer-aided Discovery of Peptides that Specifically Attack Bacterial Biofilms. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Hilpert, K.; Volkmer-Engert, R.; Walter, T.; Hancock, R.E.W. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 2005, 23, 1008–1012. [Google Scholar] [CrossRef]
- Lee, E.Y.; Lee, M.W.; Fulan, B.M.; Ferguson, A.L.; Wong, G.C.L. What can machine learning do for antimicrobial peptides, and what can antimicrobial peptides do for machine learning? Interface Focus 2017, 7. [Google Scholar] [CrossRef]
- Li, H.; Nantasenamat, C. Toward insights on determining factors for high activity in antimicrobial peptides via machine learning. PeerJ 2019, 7. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veltri, D.; Kamath, U.; Shehu, A.; Hancock, J. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnepolsky, B.; Pirtskhalava, M. Prediction of Linear Cationic Antimicrobial Peptides Based on Characteristics Responsible for Their Interaction with the Membranes. J. Chem. Inf. Model. 2014, 54, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Hasper, H.E.; Kramer, N.E.; Smith, J.L.; Hillman, J.D.; Zachariah, C.; Kuipers, O.P.; de Kruijff, B.; Breukink, E. An Alternative Bactericidal Mechanism of Action for Lantibiotic Peptides That Target Lipid II. Science 2006, 313, 1636–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, H.; Nagaraj, R. Human β-Defensin 4 with Non-Native Disulfide Bridges Exhibit Antimicrobial Activity. PLoS ONE 2015, 10, e0119525. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Wang, P.; Lin, W.-Z.; Jia, J.-H.; Chou, K.-C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-R.; Jhong, J.-H.; Wang, Z.; Chen, S.; Wan, Y.; Horng, J.-T.; Lee, T.-Y. Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms. Int. J. Mol. Sci. 2020, 21, 986. [Google Scholar] [CrossRef] [Green Version]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]
- Yoshida, M.; Hinkley, T.; Tsuda, S.; Abul-Haija, Y.M.; McBurney, R.T.; Kulikov, V.; Mathieson, J.S.; Galiñanes Reyes, S.; Castro, M.D.; Cronin, L. Using Evolutionary Algorithms and Machine Learning to Explore Sequence Space for the Discovery of Antimicrobial Peptides. Chem 2018, 4, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Landreh, M.; Johansson, J.; Jörnvall, H. Separate Molecular Determinants in Amyloidogenic and Antimicrobial Peptides. J. Mol. Biol. 2014, 426, 2159–2166. [Google Scholar] [CrossRef]
- El Amri, C.; Nicolas, P. Plasticins: Membrane-damaging peptides with ‘chameleon-like’ properties. Cell. Mol. Life Sci. 2007, 65, 895–909. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Managadze, G.; Grigolava, M.; Makhatadze, G.I.; Pirtskhalava, M. Predictive Model of Linear Antimicrobial Peptides Active against Gram-Negative Bacteria. J. Chem. Inf. Model. 2018, 58, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Zaalishvili, G.; Karapetian, M.; Nasrashvili, T.; Kuljanishvili, N.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Grigolava, M.; et al. De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals 2019, 12, 82. [Google Scholar] [CrossRef] [Green Version]
- Cutrona, K.J.; Kaufman, B.A.; Figureueroa, D.M.; Elmore, D.E. Role of arginine and lysine in the antimicrobial mechanism of histone-derived antimicrobial peptides. FEBS Lett. 2015, 589, 3915–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirtskhalava, M.; Vishnepolsky, B.; Grigolava, M.; Managadze, G. Physicochemical Features and Peculiarities of Interaction of AMP with the Membrane. Pharmaceuticals 2021, 14, 471. https://doi.org/10.3390/ph14050471
Pirtskhalava M, Vishnepolsky B, Grigolava M, Managadze G. Physicochemical Features and Peculiarities of Interaction of AMP with the Membrane. Pharmaceuticals. 2021; 14(5):471. https://doi.org/10.3390/ph14050471
Chicago/Turabian StylePirtskhalava, Malak, Boris Vishnepolsky, Maya Grigolava, and Grigol Managadze. 2021. "Physicochemical Features and Peculiarities of Interaction of AMP with the Membrane" Pharmaceuticals 14, no. 5: 471. https://doi.org/10.3390/ph14050471
APA StylePirtskhalava, M., Vishnepolsky, B., Grigolava, M., & Managadze, G. (2021). Physicochemical Features and Peculiarities of Interaction of AMP with the Membrane. Pharmaceuticals, 14(5), 471. https://doi.org/10.3390/ph14050471