
Descriptors of Cytochrome Inhibitors and Useful Machine Learning Based Methods for the Design of Safer Drugs

 ,
,
Abstract
:1. Introduction
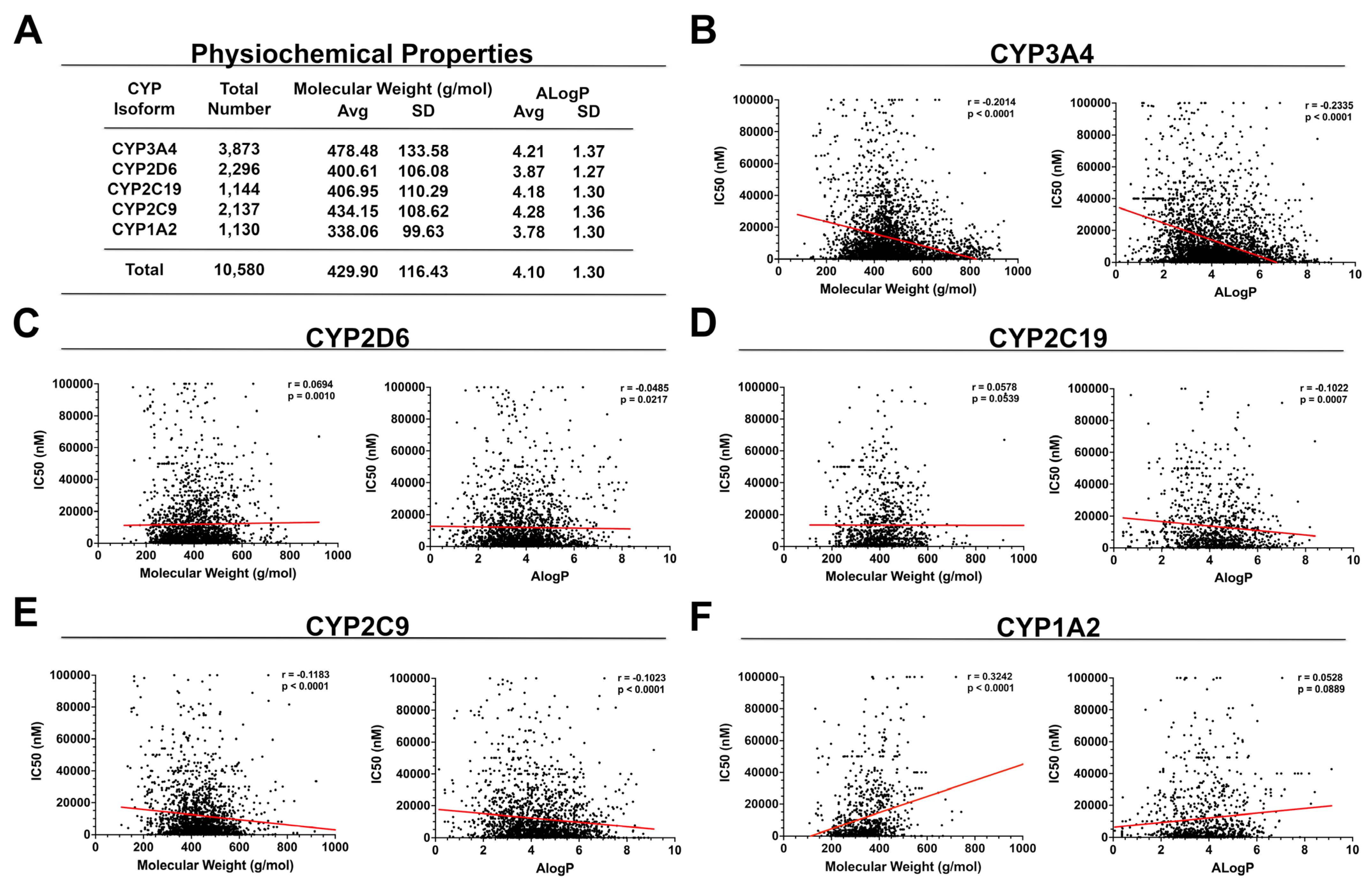
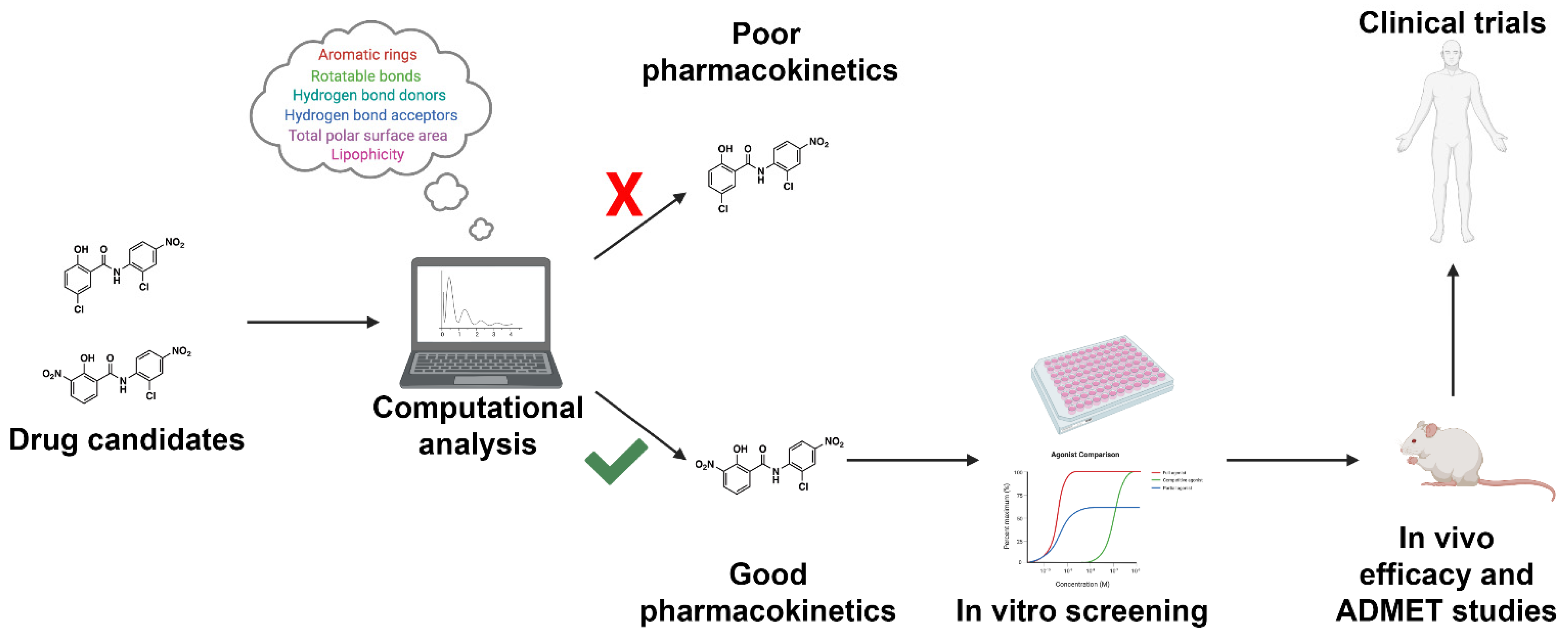
2. Physiochemical Properties
3. Structural Properties
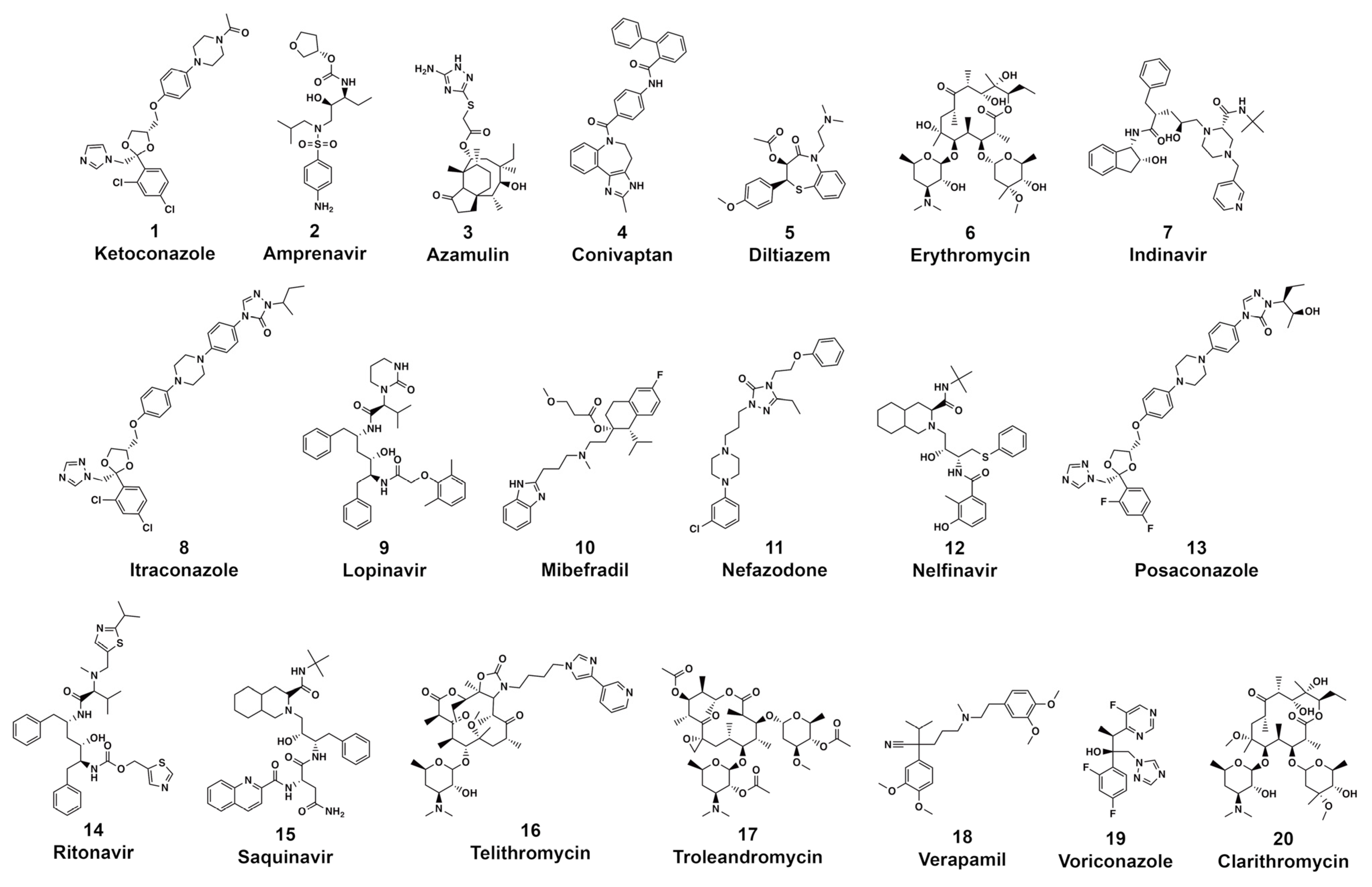
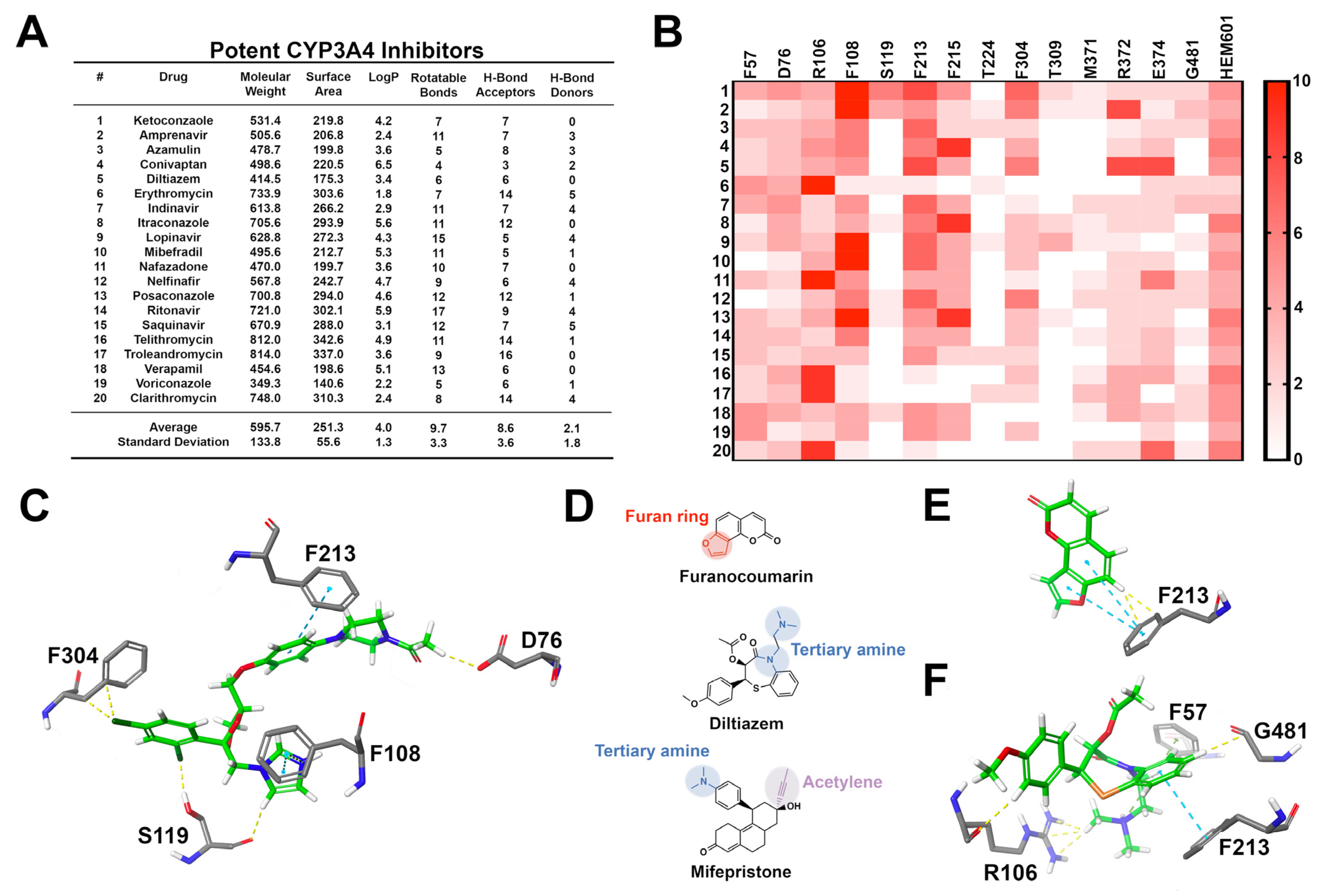
3.1. CYP3A4
3.2. CYP2D6
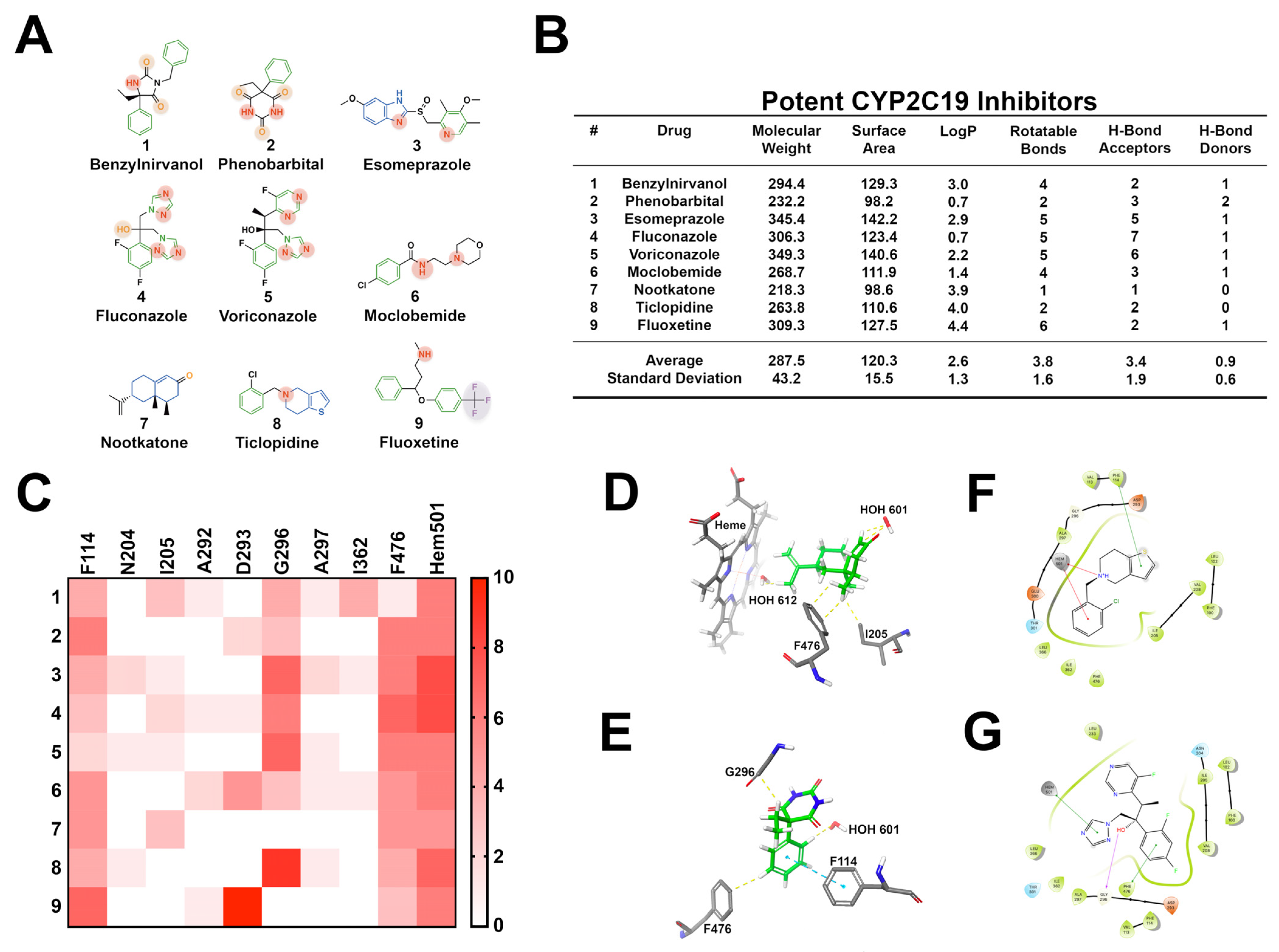
3.3. CYP2C19
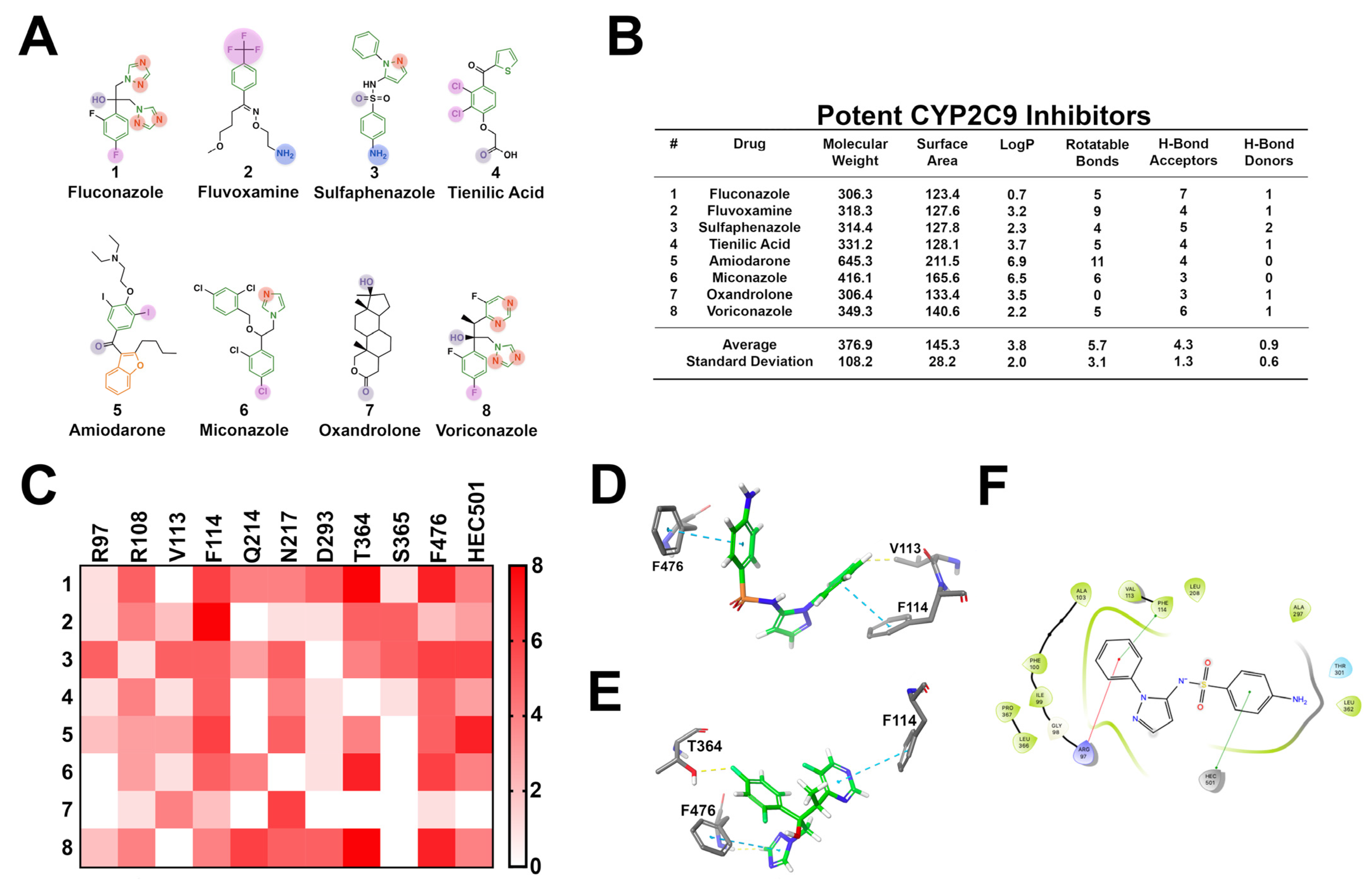
3.4. CYP2C9
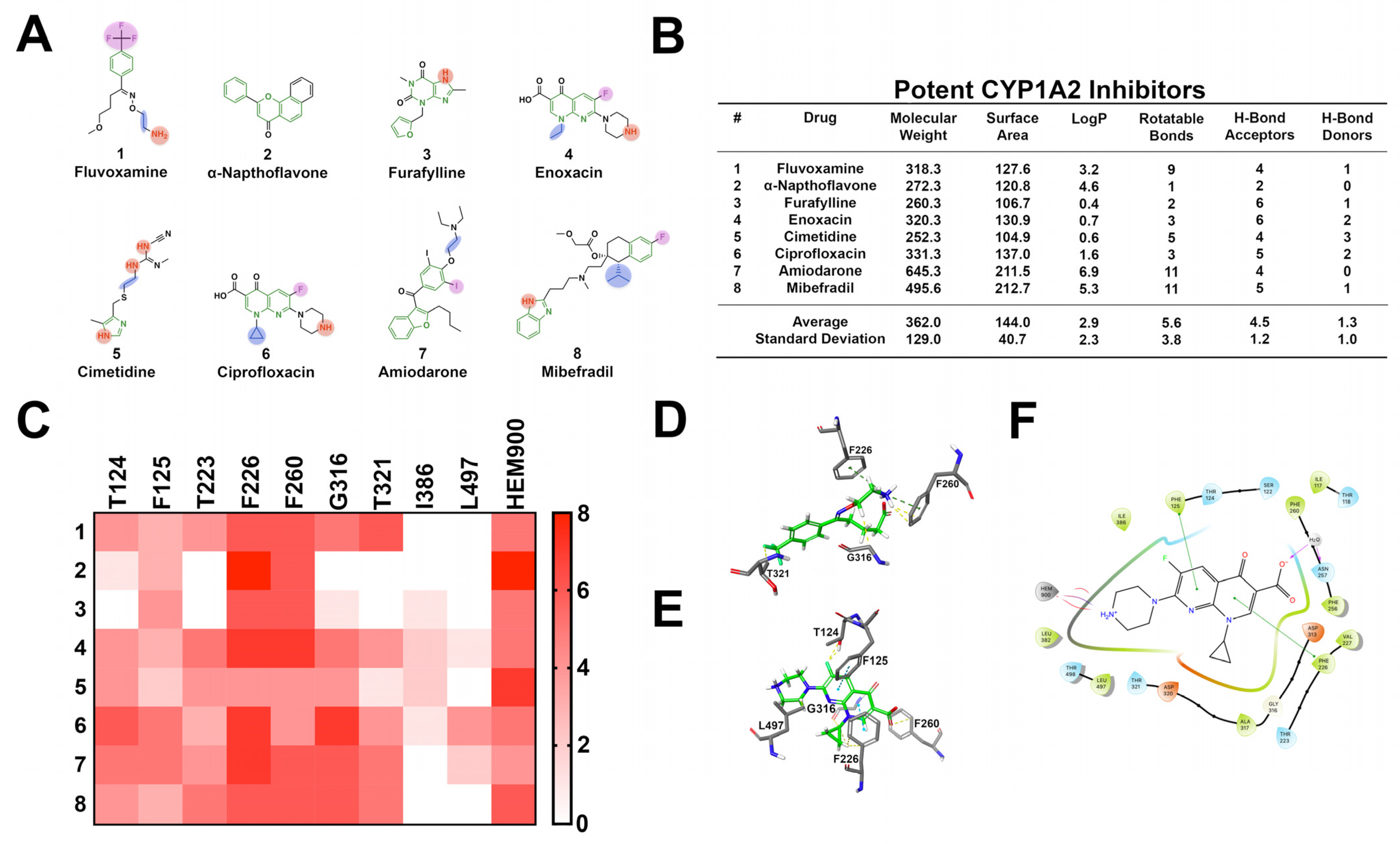
3.5. CYP1A2
3.6. Summary
4. Machine Learning-Based Methods
4.1. pkCSM
4.2. DeepCyp
4.3. SuperCYPsPred
4.4. vNN-ADMET
4.5. AdmetSAR 2.0
4.6. SwissADME
4.7. CypRules
4.8. CypReact
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Therapeutic Drug Use. Available online: https://www.cdc.gov/nchs/fastats/drug-use-therapeutic.htm (accessed on 28 February 2021).
- Mikulic, M. Prescription Drug Expenditure in the United States 1960–2020. Available online: https://www.statista.com/statistics/184914/prescription-drug-expenditures-in-the-us-since-1960/ (accessed on 28 January 2021).
- Donaldson, M.S.; Corrigan, J.M.; Kohn, L.T. (Eds.) In to Err Is Human: Building a Safer Health System; National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Lazarou, J.; Pomeranz, B.H.; Corey, P.N. Incidence of Adverse Drug Reactions in Hospitalized Patients. JAMA 1998, 279, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Preventable Adverse Drug Reactions: A Focus on Drug Interactions. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/preventable-adverse-drug-reactions-focus-drug-interactions (accessed on 2 February 2021).
- Gurwitz, J.H.; Field, T.S.; Avorn, J.; McCormick, D.; Jain, S.; Eckler, M.; Benser, M.; Edmondson, A.C.; Bates, D.W. Incidence and preventability of adverse drug events in nursing homes. Am. J. Med. 2000, 109, 87–94. [Google Scholar] [CrossRef]
- Banerjee, P.; Dunkel, M.; Kemmler, E.; Preissner, R. SuperCYPsPred—A web server for the prediction of cytochrome activity. Nucleic Acids Res. 2020, 48, W580–W585. [Google Scholar] [CrossRef] [PubMed]
- Redlich, G.; Zanger, U.M.; Riedmaier, S.; Bache, N.; Giessing, A.B.M.; Eisenacher, M.; Stephan, C.; Meyer, H.E.; Jensen, O.N.; Marcus, K. Distinction between Human Cytochrome P450 (CYP) Isoforms and Identification of New Phosphorylation Sites by Mass Spectrometry. J. Proteome Res. 2008, 7, 4678–4688. [Google Scholar] [CrossRef]
- Ogu, C.C.; Maxa, J.L. Drug Interactions Due to Cytochrome P450. In Baylor University Medical Center Proceedings; Informa UK Limited: London, UK, 2000; Volume 13, pp. 421–423. [Google Scholar]
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery. In A Comprehensive Guide to Toxicology in Preclinical Drug Development; Faqi, A.S., Ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef]
- Raunio, H.; Kuusisto, M.; Juvonen, R.O.; Pentikainen, O.T. Modeling of interactions between xenobiotics and cytochrome P450 (CYP) enzymes. Front. Pharmacol. 2015, 6, 123. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Kirchmair, J. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des. 2019, 93, 377–386. [Google Scholar] [CrossRef]
- Martiny, V.Y.; Carbonell, P.; Chevillard, F.; Moroy, G.; Nicot, A.B.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Integrated structure- and ligand-basedin silicoapproach to predict inhibition of cytochrome P450 2D6. Bioinformatics 2015, 31, 3930–3937. [Google Scholar] [CrossRef] [Green Version]
- Refsgaard, H.H.F.; Jensen, B.F.; Christensen, I.T.; Hagen, N.; Brockhoff, P.B. In silico prediction of cytochrome P450 inhibitors. Drug Dev. Res. 2006, 67, 417–429. [Google Scholar] [CrossRef]
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. MetaPASS: A Web Application for Analyzing the Biological Activity Spectrum of Organic Compounds Taking into Account their Biotransformation. Mol. Inform. 2021, 40, 2000231. [Google Scholar] [CrossRef]
- Kar, S. Recent Advances of Computational Modeling for Predicting Drug Metabolism: A Perspective. Curr. Drug Metab. 2018, 18, 1106–1122. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, S.; Chen, H.; Usharani, D.; Thiel, W. QM/MM Studies of Structure and Reactivity of Cytochrome P450 Enzymes: Methodology and Selected Applications. Early Drug Dev. 2014, 311, 133–178. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef]
- Kiani, Y.S.; Jabeen, I. Exploring the Chemical Space of Cytochrome P450 Inhibitors Using Integrated Physicochemical Parameters, Drug Efficiency Metrics and Decision Tree Models. Computation 2019, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Nassar, A.-E.F.; Kamel, A.M.; Clarimont, C. Improvingthe decision-making process in structural modification of drug candidates: Reducing toxicity. Drug Discov. Today 2004, 9, 1055–1064. [Google Scholar] [CrossRef]
- Lewis, D.F.V.; Dickins, M. Substrate SARs in human P450s. Drug Discov. Today 2002, 7, 918–925. [Google Scholar] [CrossRef]
- Lewis, D.F.V.; Dickins, M. Baseline Lipophilicity Relationships in Human Cytochromes P450 Associated with Drug Metabolism. Drug Metab. Rev. 2003, 35, 1–18. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a Set of Simple, Interpretable ADMET Rules of Thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Arnott, J.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Lewis, D.F.; Lake, B.G.; Ito, Y.; Dickins, M. Lipophilicity Relationships in Inhibitors of CYP2C9 and CYP2C19 Enzymes. J. Enzym. Inhib. Med. Chem. 2006, 21, 385–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.F.V.; Lake, B.G.; Dickins, M. Quantitative structure-activity relationships (QSARs) in inhibitors of various cytochromes P450: The importance of compound lipophilicity. J. Enzym. Inhib. Med. Chem. 2007, 22, 1–6. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, P.; Chamberlin, A.R.; Poulos, T.L.; Sevrioukova, I.F. Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J. Med. Chem. 2016, 59, 4210–4220. [Google Scholar] [CrossRef] [Green Version]
- Eagling, V.A.; Back, D.J.; Barry, M.G. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br. J. Clin. Pharmacol. 1997, 44, 190–194. [Google Scholar] [CrossRef] [Green Version]
- De Groot, M.J.; Vermeulen, N.P.E.; Kramer, J.D.; Van Acker, F.A.A.; Kelder, G.M.D.-O.D. A Three-Dimensional Protein Model for Human Cytochrome P450 2D6 Based on the Crystal Structures of P450 101, P450 102, and P450 108. Chem. Res. Toxicol. 1996, 9, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.A.; Flanagan, J.U.; Van Eldik, A.J.; Maréchal, J.-D.; Wolf, C.R.; Roberts, G.C.K.; Paine, M.J.I.; Sutcliffe, M.J. Validation of Model of Cytochrome P450 2D6: An in Silico Tool for Predicting Metabolism and Inhibition. J. Med. Chem. 2004, 47, 5340–5346. [Google Scholar] [CrossRef]
- Egnell, A.-C.; Eriksson, C.; Albertson, N.; Houston, B.; Boyer, S. Generation and Evaluation of a CYP2C9 Heteroactivation Pharmacophore. J. Pharmacol. Exp. Ther. 2003, 307, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S.; Bravi, G.; Binkley, S.; Gillespie, J.S.; Ring, B.J.; Wikel, J.H.; A Wrighton, S. Three- and four-dimensional-quantitative structure activity relationship (3D/4D-QSAR) analyses of CYP2C9 inhibitors. Drug Metab. Dispos. 2000, 28, 994–1002. [Google Scholar]
- Foti, R.S.; Rock, D.A.; Han, X.; Flowers, R.A.; Wienkers, L.C.; Wahlstrom, J.L. Ligand-Based Design of a Potent and Selective Inhibitor of Cytochrome P450 2C19. J. Med. Chem. 2012, 55, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wei, D.-Q.; Chou, K.-C. A pharmacophore model specific to active site of CYP1A2 with a novel molecular modeling explorer and CoMFA. Med. Chem. 2012, 8, 198–207. [Google Scholar] [PubMed]
- Korhonen, L.E.; Rahnasto, M.; Mähönen, N.J.; Wittekindt, C.; Poso, A.; Juvonen, R.O.; Raunio, H. Predictive Three-Dimensional Quantitative Structure−Activity Relationship of Cytochrome P450 1A2 Inhibitors. J. Med. Chem. 2005, 48, 3808–3815. [Google Scholar] [CrossRef]
- Gay, S.C.; Roberts, A.G.; Halpert, J.R. Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by X-ray crystallography and NMR. Future Med. Chem. 2010, 2, 1451–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef] [Green Version]
- Rock, B.M.; Hengel, S.M.; Rock, D.A.; Wienkers, L.C.; Kunze, K.L. Characterization of Ritonavir-Mediated Inactivation of Cytochrome P450 3A4. Mol. Pharmacol. 2014, 86, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Woolf, T.F.; Hollenberg, P.F. Mechanism-based inactivation of cytochrome P-450-3A4 by mifepristone (RU486). J. Pharmacol. Exp. Ther. 1999, 288, 791–797. [Google Scholar]
- Zhou, S.; Chan, E.; Lim, L.; Boelsterli, U.; Li, S.; Wang, J.; Zhang, Q.; Huang, M.; Xu, A. Therapeutic Drugs that Behave as Mechanism-Based Inhibitors of Cytochrome P450 3A4. Curr. Drug Metab. 2004, 5, 415–442. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Šarić Mustapić, D.; Debeljak, Ž.; Maleš, Ž.; Bojić, M. The Inhibitory Effect of Flavonoid Aglycones on the Metabolic Activity of CYP3A4 Enzyme. Molecules 2018, 23, 2553. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.; Tang, J.; Rose, R.; Hodgson, E.; Bienstock, R.J.; Mohrenweiser, H.W.; A Goldstein, J. Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J. Pharmacol. Exp. Ther. 2001, 299, 825–831. [Google Scholar]
- Ekroos, M.; Sjögren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Van Hoppe, S.; Rood, J.J.M.; Buil, L.; Wagenaar, E.; Sparidans, R.W.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein (MDR1/ABCB1) Restricts Brain Penetration of the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib, While Cytochrome P450-3A (CYP3A) Limits Its Oral Bioavailability. Mol. Pharm. 2018, 15, 5124–5134. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; Patki, K.C.; Von Moltke, L.L.; Shader, R.I. Drug Interactions with Grapefruit Juice: An Update. J. Clin. Psychopharmacol. 2001, 21, 357–359. [Google Scholar] [CrossRef]
- Vuppalanchi, R. Metabolism of Drugs and Xenobiotics; Saxena, R., Ed.; Elsevier: Cambridge, MA, USA, 2011. [Google Scholar]
- Rowland, P.; Blaney, F.E.; Smyth, M.G.; Jones, J.J.; Leydon, V.R.; Oxbrow, A.K.; Lewis, C.J.; Tennant, M.G.; Modi, S.; Eggleston, D.S.; et al. Crystal Structure of Human Cytochrome P450 2D6. J. Biol. Chem. 2006, 281, 7614–7622. [Google Scholar] [CrossRef] [Green Version]
- Cytochrome 2D6 Inhibition. Available online: https://www.cambridgemedchemconsulting.com/resources/ADME/cyp2d6inhibition.html (accessed on 9 February 2021).
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- VandenBrink, B.M.; Foti, R.S.; Rock, D.A.; Wienkers, L.C.; Wahlstrom, J.L. Prediction of CYP2D6 Drug Interactions from In Vitro Data: Evidence for Substrate-Dependent Inhibition. Drug Metab. Dispos. 2011, 40, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demkow, U. Next Generation Sequencing in Pharmacogenomics; Płoski, U.D.R., Ed.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Derayea, S.M.; Tsujino, H.; Oyama, Y.; Ishikawa, Y.; Yamashita, T.; Uno, T. Investigation on drug-binding in heme pocket of CYP2C19 with UV–visible and resonance Raman spectroscopies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 209, 209–216. [Google Scholar] [CrossRef]
- Cytochrome 2C19 Inhibition. Available online: https://www.cambridgemedchemconsulting.com/resources/ADME/cyp2c19inhibition.html (accessed on 2 February 2021).
- Ibeanu, G.C.; Goldstein, J.; Meyer, U.; Benhamou, S.; Bouchardy, C.; Dayer, P.; Ghanayem, B.; Blaisdell, J. Identification of new human CYP2C19 alleles (CYP2C19*6 and CYP2C19*2B) in a Caucasian poor metabolizer of mephenytoin. J. Pharmacol. Exp. Ther. 1998, 286, 1490–1495. [Google Scholar] [PubMed]
- Fukushima-Uesaka, H.; Saito, Y.; Maekawa, K.; Ozawa, S.; Hasegawa, R.; Kajio, H.; Kuzuya, N.; Yasuda, K.; Kawamoto, M.; Kamatani, N.; et al. Genetic Variations and Haplotypes of CYP2C19 in a Japanese Population. Drug Metab. Pharmacokinet. 2005, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.C.; Risinger, C.; Dahl, M.-L.; Aklillu, E.; Christensen, M.; Bertilsson, L.; Ingelman-Sundberg, M. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin. Pharmacol. Ther. 2006, 79, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Cytochrome 2C9 Inhibition. Available online: https://www.cambridgemedchemconsulting.com/resources/ADME/cyp2c9inhibition.html (accessed on 4 February 2021).
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Vinković, D.M.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef]
- Wester, M.R.; Yano, J.K.; Schoch, G.A.; Yang, C.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Cytochrome P450 2C9 Complexed with Flurbiprofen at 2.0-Å Resolution. J. Biol. Chem. 2004, 279, 35630–35637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, V.M.; Cavallari, L.H.; Del Tredici, A.L.; Hachad, H.; Ji, Y.; Moyer, A.M.; Scott, S.A.; Whirl-Carrillo, M.; Weck, K.E. Recommendations for Clinical CYP2C9 Genotyping Allele Selection: A Joint Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2019, 21, 746–755. [Google Scholar] [CrossRef] [PubMed]
- García-Martín, E.; Martínez, C.; Ladero, J.M.; Agúndez, J.A.G. Interethnic and Intraethnic Variability of CYP2C8 and CYP2C9 Polymorphisms in Healthy Individuals. Mol. Diagn. Ther. 2006, 10, 29–40. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Lamba, J.K. Pharmacogenomics; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Fontana, R.J.; Lown, K.S.; Paine, M.F.; Fortlage, L.; Santella, R.M.; Felton, J.S.; Knize, M.G.; Greenberg, A.; Watkins, P.B. Effects of a chargrilled meat diet on expression of CYP3A, CYP1A, and P-glycoprotein levels in healthy volunteers. Gastroenterology 1999, 117, 89–98. [Google Scholar] [CrossRef]
- Sanday, K. South Asains and Europeans React Differently to Common Drugs. Available online: https://medicalxpress.com/news/2011-10-south-asians-europeans-react-differently.html (accessed on 8 February 2021).
- Vedani, A.; Dobler, M.; Smieško, M. VirtualToxLab—A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 2012, 261, 142–153. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM Pharmacokinetics. Available online: http://biosig.unimelb.edu.au/pkcsm/ (accessed on 8 February 2021).
- Li, X.; Xu, Y.; Lai, L.; Pei, J. Prediction of Human Cytochrome P450 Inhibition Using a Multitask Deep Autoencoder Neural Network. Mol. Pharm. 2018, 15, 4336–4345. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2010, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Siramshetty, V.B.; Drwal, M.N.; Preissner, R. Computational methods for prediction of in vitro effects of new chemical structures. J Cheminform. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN Web Server for ADMET Predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2018, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Rostkowski, M.; Spjuth, O.; Rydberg, P. WhichCyp: Prediction of cytochromes P450 inhibition. Bioinformatics 2013, 29, 2051–2052. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.-Y.; Su, B.-H.; Tu, Y.-S.; Lin, C.; Lin, O.A.; Tseng, Y.J. CypRules: A rule-based P450 inhibition prediction server. Bioinformatics 2015, 31, 1869–1871. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Xie, Q.; Ge, W.; Qian, F.; Fang, H.; Shi, L.; Su, Z.; Perkins, R.; Tong, W. Mold2, Molecular Descriptors from 2D Structures for Chemoinformatics and Toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. [Google Scholar] [CrossRef]
- Tian, S.; Djoumbou-Feunang, Y.; Greiner, R.; Wishart, D.S.; Djoumbou, Y.; Greiner, R. CypReact: A Software Tool for in Silico Reactant Prediction for Human Cytochrome P450 Enzymes. J. Chem. Inf. Model. 2018, 58, 1282–1291. [Google Scholar] [CrossRef]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. XenoSite: Accurately Predicting CYP-Mediated Sites of Metabolism with Neural Networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef]
- Olsen, L.; Montefiori, M.; Tran, K.P.; Jørgensen, F.S. SMARTCyp 3.0: Enhanced cytochrome P450 site-of-metabolism prediction server. Bioinformatics 2019, 35, 3174–3175. [Google Scholar] [CrossRef]
- Ghosh, J.; Lawless, M.S.; Waldman, M.; Gombar, V.; Fraczkiewicz, R.; Benfenati, E. Modeling ADMET. Adv. Struct. Saf. Stud. 2016, 1425, 63–83. [Google Scholar] [CrossRef]
- Cleaned ChEMBL Data on Major Cytochrome Inhibitors. 2021. Available online: https://figshare.com/articles/dataset/Cleaned_ChEMBL_Data_on_Major_Cytochrome_Inhibitors/14272661 (accessed on 8 February 2021).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Inhibitors | |||
|---|---|---|---|---|
| CYP | Cavity Size (Å3) | Description | Description | Common Descriptors |
| 3A4 | 1173–2862 | Large, open, flexible, aromatic, diverse range of substrates, oxidation site is commonly a nitrogen or allylic position | Large, structurally diverse, lipophilic, aromatic, highly flexible, high hydrogen bond accepting capacity | One of more aromatic moieties, furan rings, tertiary amines, acetylene groups |
| 2D6 | 510 | Flat, restricted volume, acidic, aromatic, site of oxidation proximal to a primary or secondary amine | Flat, planar, aromatic structures capable of procuring a positive charge, with 2–3 hydrogen bond acceptors | One of more aromatic moieties, heterocycles, primary or secondary amines capable of carrying a positive charge |
| 2C19 | Not reported | Aromatic, moderately flexible, similar to CYP2C9 | Medium sized molecules, variable lipophilicity, aromatic, oxidation site close to two hydrogen bond acceptors | Several aromatic moieties, heterocycles, carbonyl groups, and aromatic nitrogen atoms |
| 2C9 | 978–1271 | Larger cavity volume, moderately flexible | Aromatic, lipophilic, moderately flexible, several hydrogen bond acceptors | Aromatic, heterocycles, aromatic nitrogens, primary amines, and halogens |
| 1A2 | 375–390 | Small cavity volume, planar, rigid | Small, planar, aromatic, lipophilic, slightly acidic | Several aromatic moieties, heterocycles, secondary amines, and halogens |
| Name | CYPs | Prediction Type | ML Method | No. Structures | Avg. Accuracy 1 | Additional Features |
|---|---|---|---|---|---|---|
| pkCSM | 3A4, 2D6, 2C19, 2C9, 1A2 | Inhibition | Graph-based signatures | 18,000 | 0.810 (0.780–0.853) | Comprehensive ADMET predictions (23 total) |
| DeepCYP | 3A4, 2D6, 2C19, 2C9, 1A2 | Inhibition | Multitask autoencoder deep neural network | 13,000 | 0.864 (0.809–0.968) | Assigns probabilities for CYP inhibition |
| SuperCYPs-Pred | 3A4, 2D6, 2C19, 2C9, 1A2 | Inhibition | Random forests | 41,963 2 | 0.930 (0.840–0.970) | Assigns probabilities for CYP inhibition |
| vNN-ADMET | 3A4, 2D6, 2C19, 2C9, 1A2 | Inhibition | Variable nearest neighbors | 6261 | 0.890 (0.870–0.910) | |
| AdmetSAR 2.0 | 3A4, 2D6, 2C19, 2C9, 1A2 | Inhibition | Random forests, support vector machines, k-nearest neighbors | 96,000 3 | 0.784 (0.645–0.855) | Comprehensive ADMET predictions (47 total); ADMETopt for lead optimization |
| SwissADME | 3A4, 2D6l 2C19, 2C9, 1A2 | Inhibition | Support vector machines | 16,561 4 | 0.794 (0.720–0.800) | Predictions of physicochemical properties, pharmacokinetics, and drug likeness; high throughput |
| CypRules | 3A4, 2D6, 2C19, 2C9, 1A2 | Inhibition | Decision trees | 16,561 | 0.812 (0.730–0.900) | High throughput |
| CypReact | 3A4, 2E1, 2D6,2C19, 2C9, 2C8, 2B6, 2A6, 1A2 | Sites of metabolism | LBM learning algorithm | 2685 | Unavailable | Metabolite predictions; Additional CYPs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beck, T.C.; Beck, K.R.; Morningstar, J.; Benjamin, M.M.; Norris, R.A. Descriptors of Cytochrome Inhibitors and Useful Machine Learning Based Methods for the Design of Safer Drugs. Pharmaceuticals 2021, 14, 472. https://doi.org/10.3390/ph14050472
Beck TC, Beck KR, Morningstar J, Benjamin MM, Norris RA. Descriptors of Cytochrome Inhibitors and Useful Machine Learning Based Methods for the Design of Safer Drugs. Pharmaceuticals. 2021; 14(5):472. https://doi.org/10.3390/ph14050472
Chicago/Turabian StyleBeck, Tyler C., Kyle R. Beck, Jordan Morningstar, Menny M. Benjamin, and Russell A. Norris. 2021. "Descriptors of Cytochrome Inhibitors and Useful Machine Learning Based Methods for the Design of Safer Drugs" Pharmaceuticals 14, no. 5: 472. https://doi.org/10.3390/ph14050472
APA StyleBeck, T. C., Beck, K. R., Morningstar, J., Benjamin, M. M., & Norris, R. A. (2021). Descriptors of Cytochrome Inhibitors and Useful Machine Learning Based Methods for the Design of Safer Drugs. Pharmaceuticals, 14(5), 472. https://doi.org/10.3390/ph14050472