
Cold-Start Problems in Data-Driven Prediction of Drug–Drug Interaction Effects



Abstract
:1. Introduction
2. Results
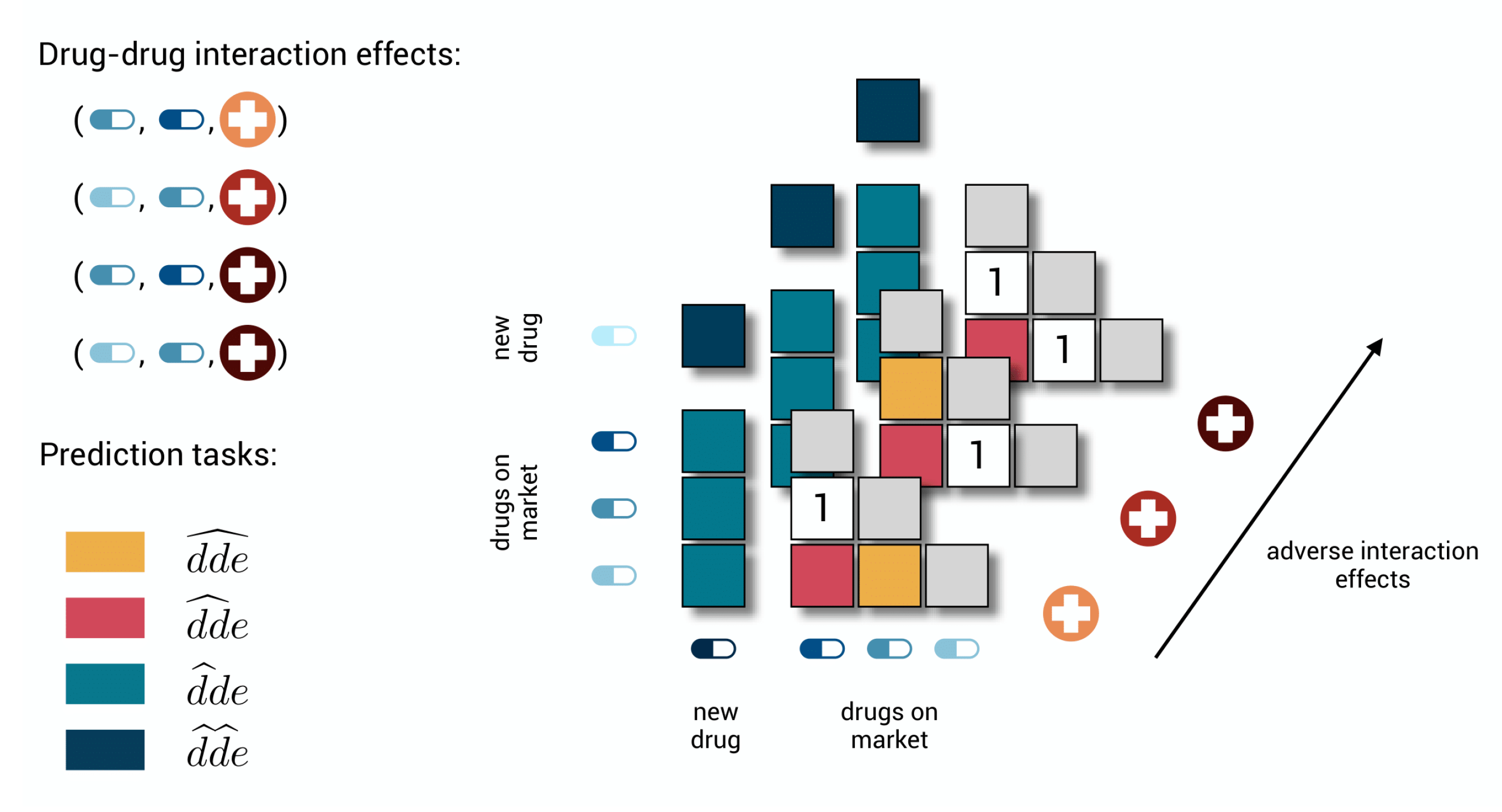
2.1. Formulation of the Prediction Subtasks
- 1.
- : unknown drug–drug-effect. Predict the occurrence of an effect for a drug–drug pair for which other effects are already known. This problem corresponds to regular tensor completion problems in machine learning.
- 2.
- : unknown drug–drug pair. Predict for a drug–drug pair for which no interaction effect is known. This is the first cold-start task.
- 3.
- : unknown drug. Predict for a new drug for which no effect is known in any combination with another drug. This is the second cold-start task.
- 4.
- : two unknown drugs. Predict for two new drugs for which no effect is known in any combination with another drug. This is the third cold-start task.
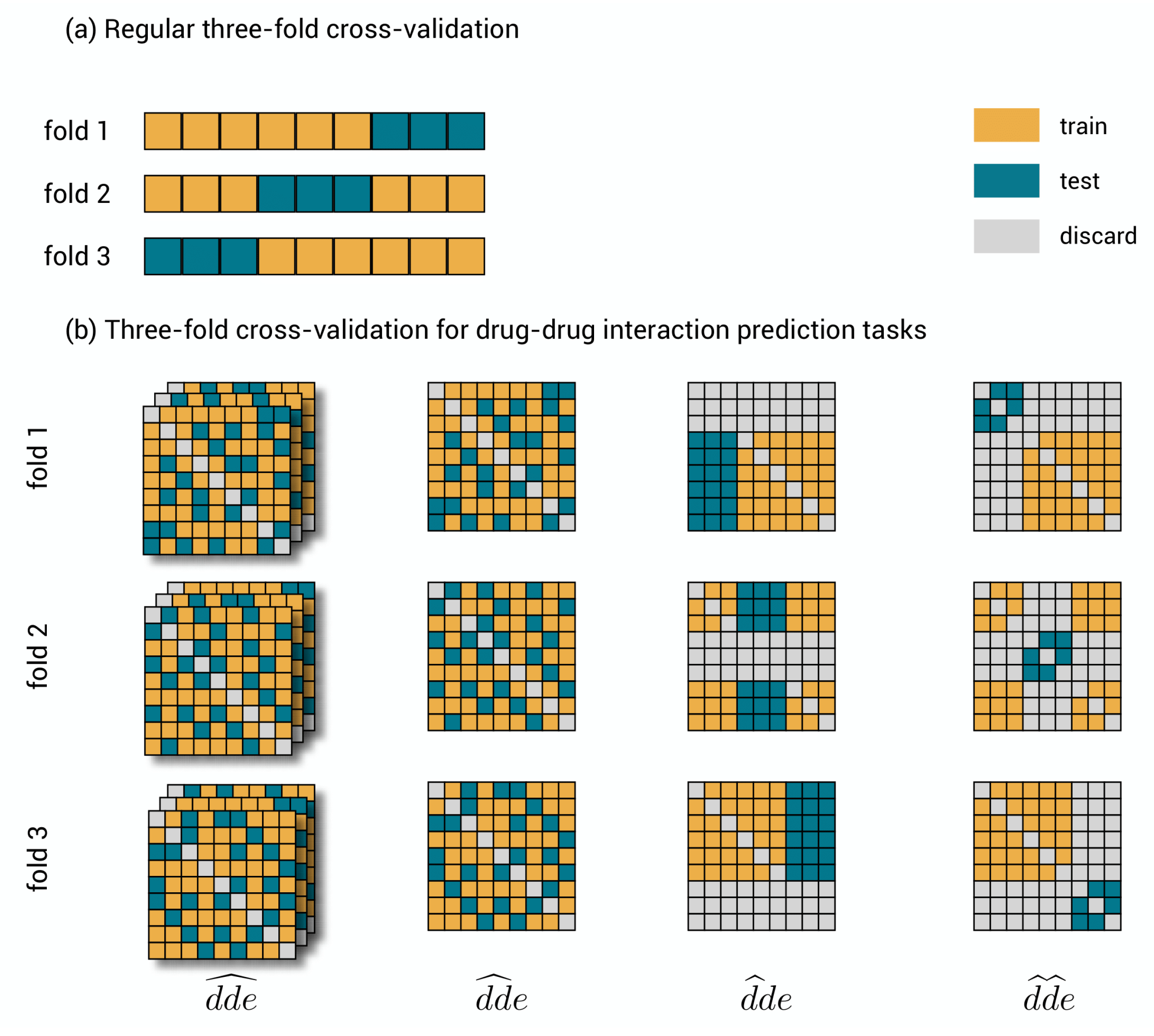
2.2. Validation Procedures for the Prediction Subtasks
- 1.
- : drug–drug-effect triplets are randomly assigned to the different test sets. Performance for a triplet is thus measured without any restriction on the availability of other triplets in the training data.
- 2.
- : drug–drug pairs are randomly assigned to the different test sets together with all the effects. Performance is thus measured with the restriction that for the drug–drug pair of a test triplet, not a single link with an effect is part of the training data.
- 3.
- : the first drugs are randomly assigned to the different test sets, together with all combinations with all other drugs and all effects. Performance is thus measured with the restriction that for the first drug of a test triplet, not a single effect from interaction with any other drug is part of the training data.
- 4.
- : drugs are assigned to the different test sets, at the same time for the first and second drug and for all effects. Prediction is thus measured with the restriction that for both drugs of a test triplet, not a single effect from interaction with any other drug is part of the training data.
2.3. Model Training and Validation
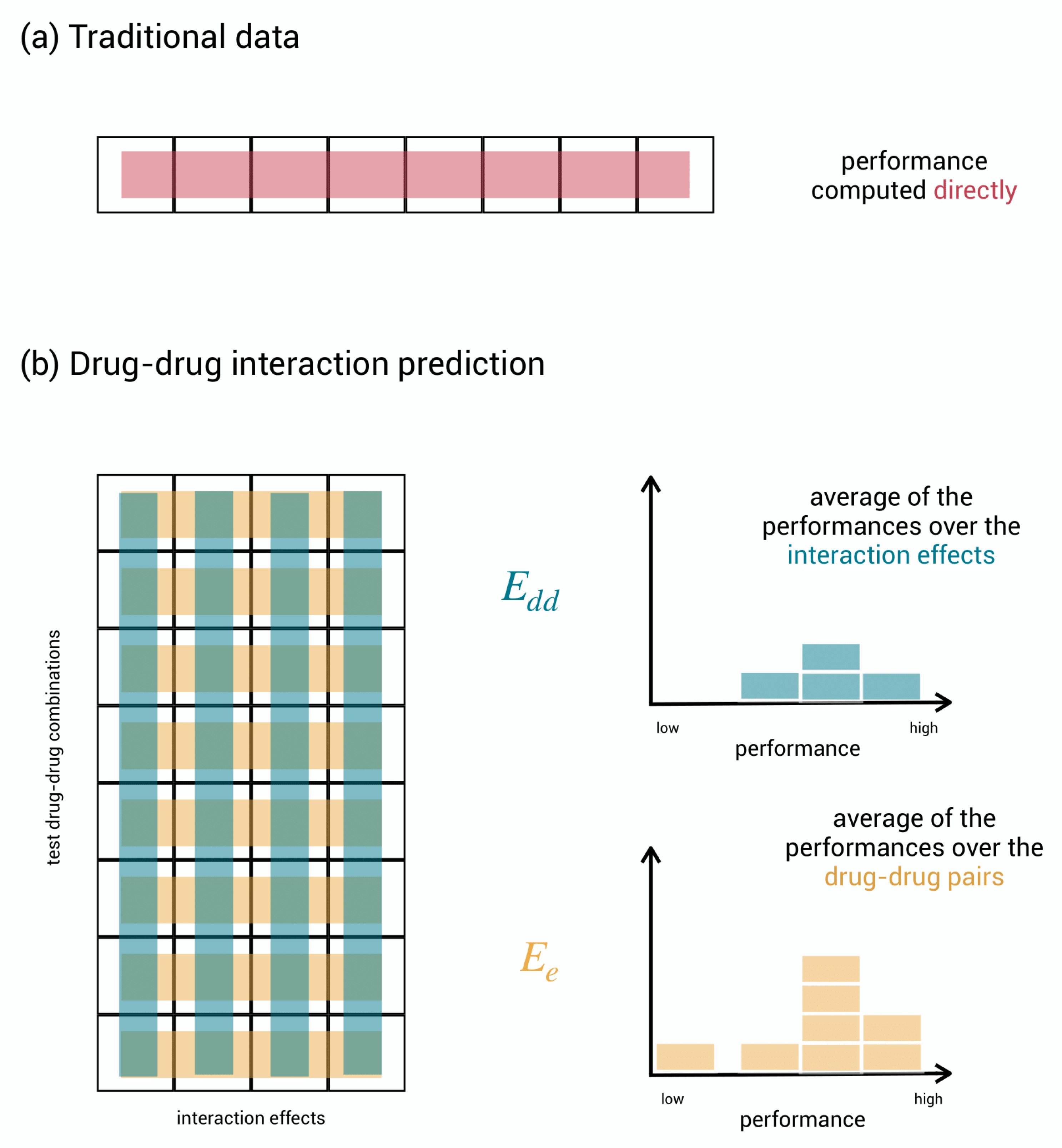
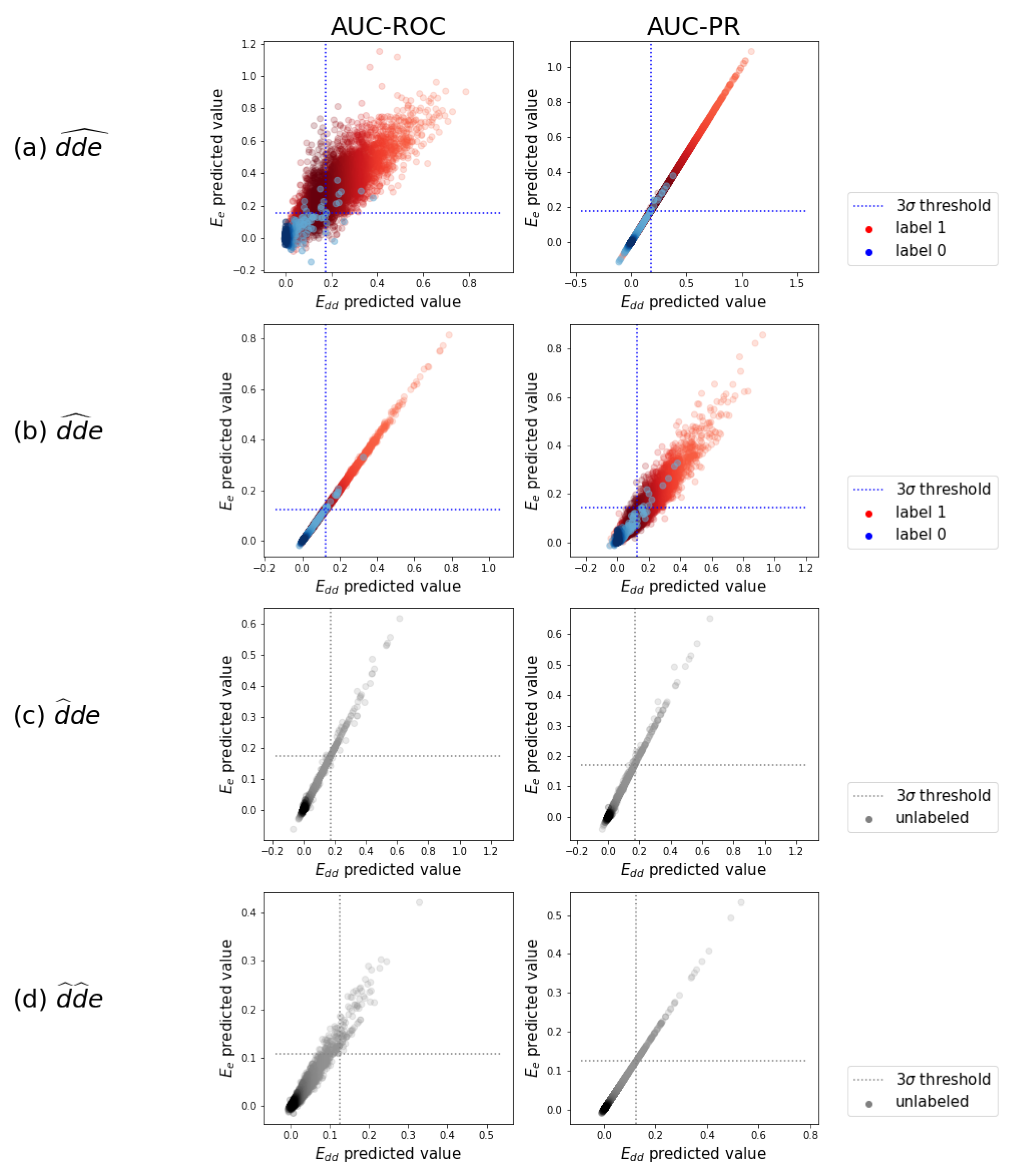
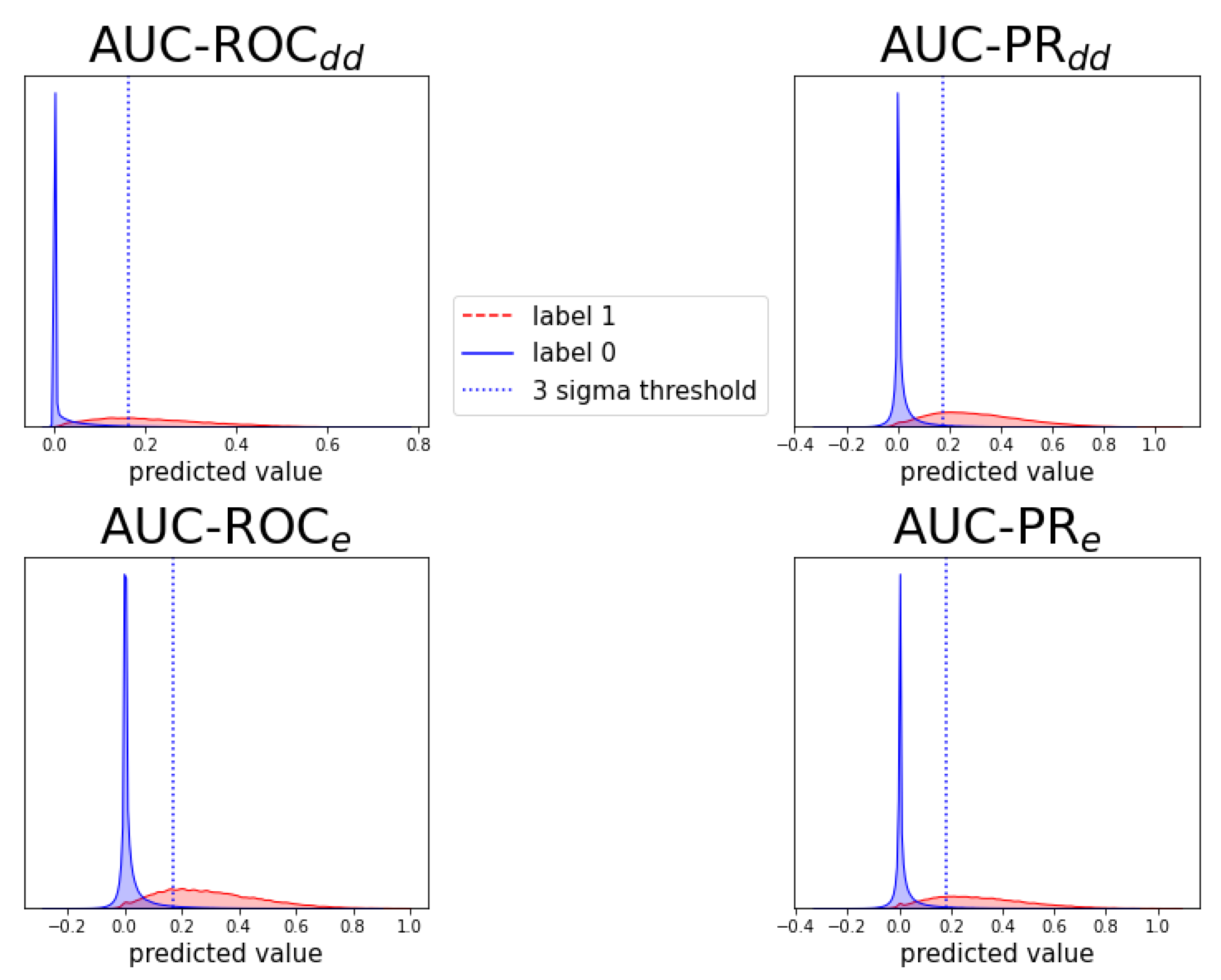
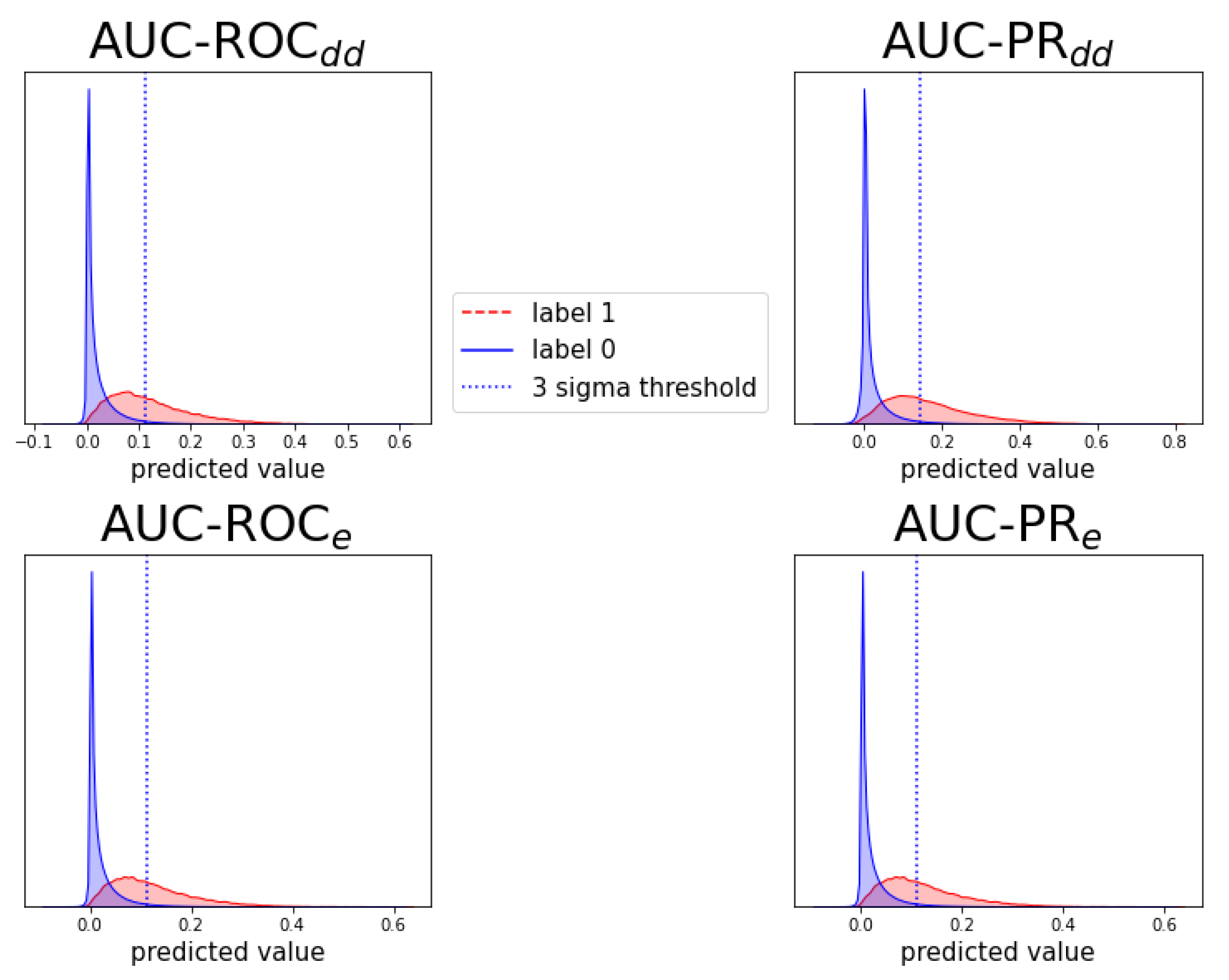
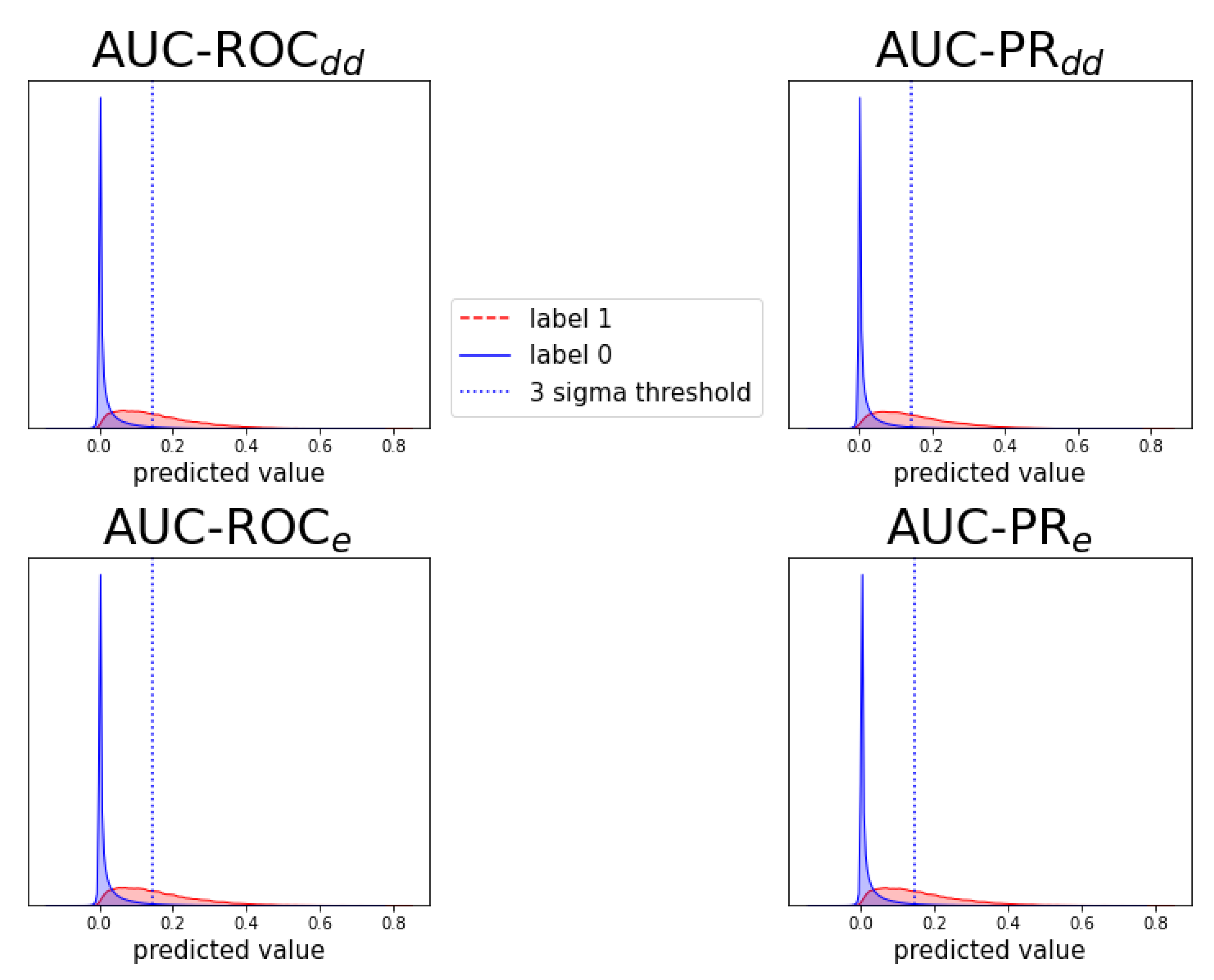
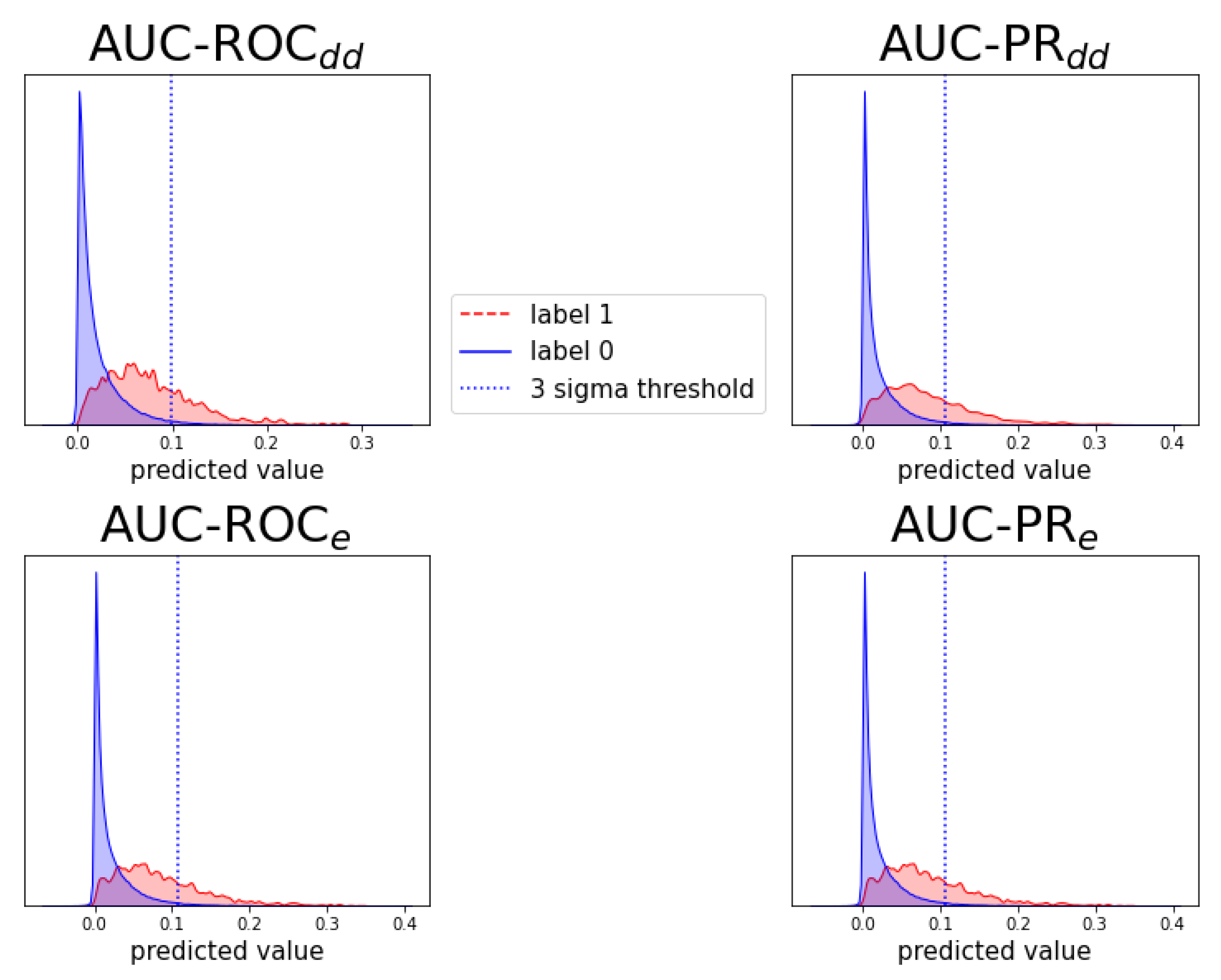
2.4. Detecting New Adverse Drug-Drug Interaction Effects
3. Discussion
4. Materials and Methods
4.1. Three-Step Kernel Ridge Regression
4.2. Data Set
4.3. Kernel Construction
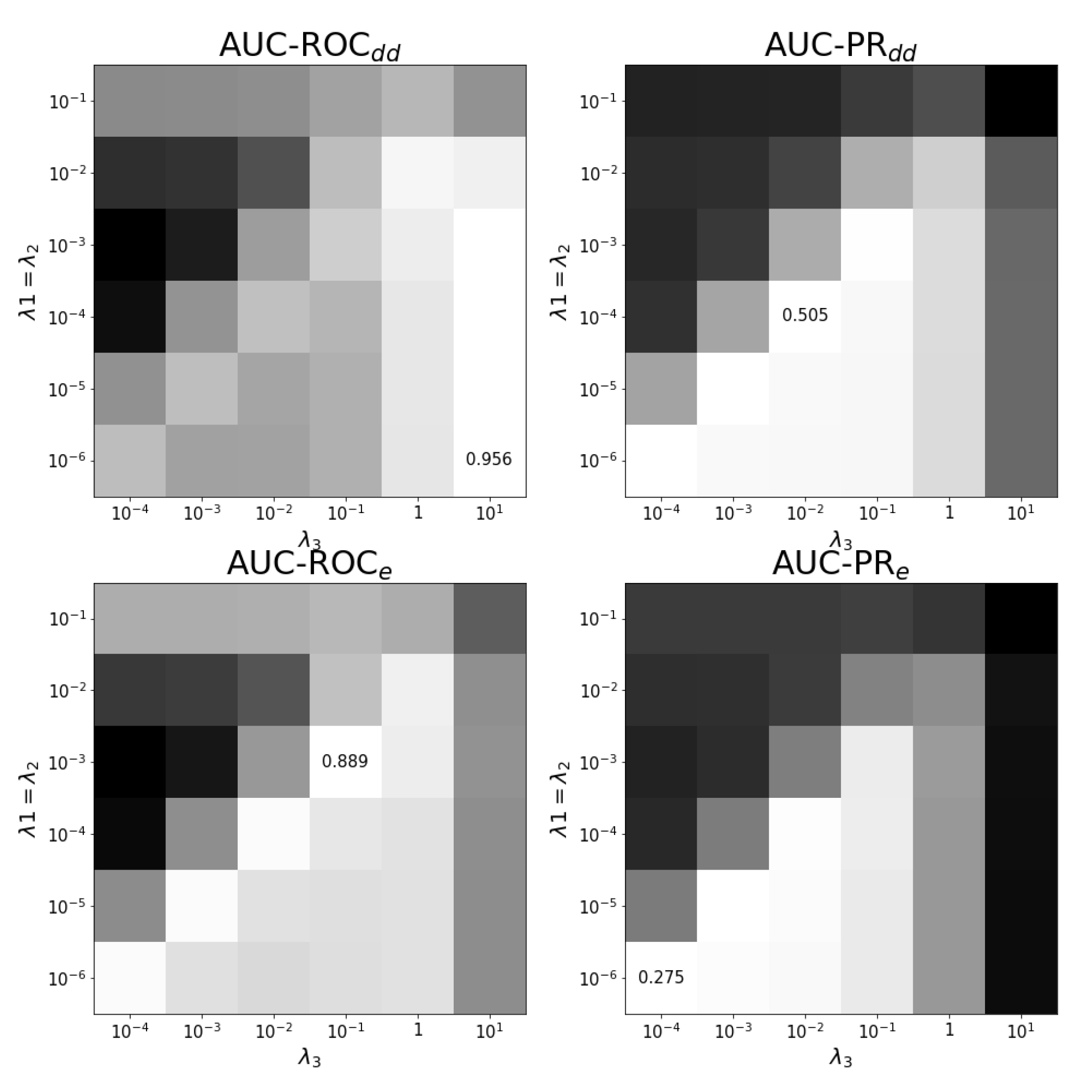
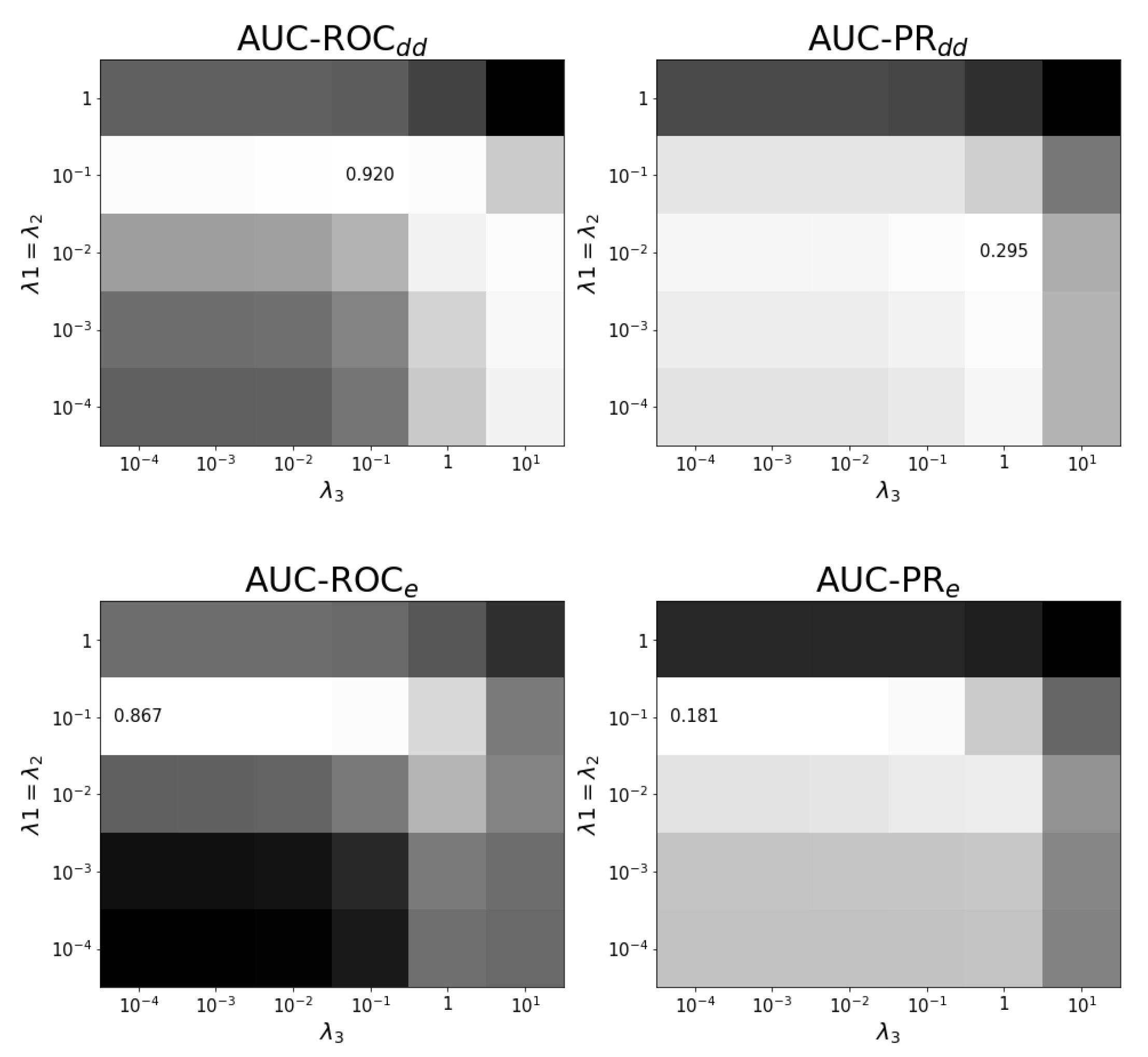
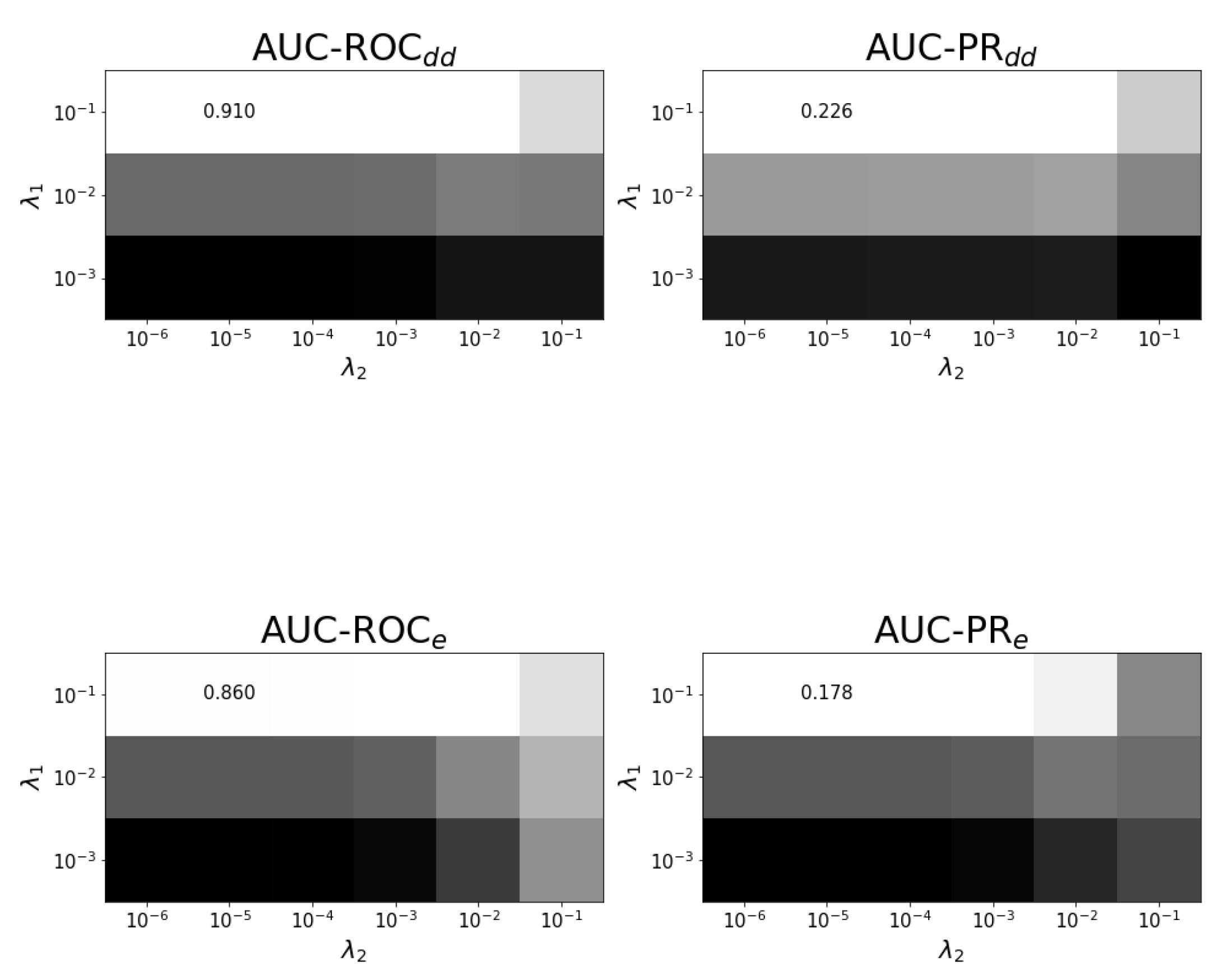
4.4. Experimental Setup and Tuning of Regularization Parameters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Three-Step Kernel Ridge Regression: Algebraic Expressions for Model Parameters and Cross-Validation Shortcuts
Appendix A.1. Kernel Ridge Regression and Shortcuts
Appendix A.2. Three-Step Kernel Ridge Regression and Shortcuts
- . As performs the regression for the first drug, we can simply replace by its leave-out variant . This makes sure that predictions for a certain first drug do not use the labels for that drug.
- . A similar reasoning applies, but now and are replaced by and , respectively.
- . The same strategy is followed, however, the situation is somewhat more difficult: the operation of subtracting the diagonal elements of the hat matrix and rescaling must now be applied on the combined tensor product of the matrices, instead of aplying it separately. Therefore, is replaced by a combined .
- . The same strategy is followed, however, the situation is somewhat more difficult: the operation of subtracting the diagonal elements of the hat matrix and rescaling must now be applied on the combined tensor product of the two drug matrices, instead of applying it separately. Therefore, is replaced by a combined .
Appendix B. Results on Hyper-Parametertuning





Appendix C. Individual Prediction Histograms




Appendix D. Notes on Computation of Auc-Pr in Evaluation Schemes and
References
- Pliakos, K.; Vens, C. Drug-target interaction prediction with tree-ensemble learning and output space reconstruction. BMC Bioinform. 2020, 21, 49. [Google Scholar] [CrossRef]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Krauss, R.M.; Roden, D.M.; Eichelbaum, M.; Hayden, M.R.; Nakamura, Y. When good drugs go bad. Nature 2007, 446, 975–977. [Google Scholar] [CrossRef]
- Sultana, J.; Cutroneo, P.; Trifirò, G. Clinical and economic burden of adverse drug reactions. J. Pharmacol. Pharmacother. 2013, 4, S73. [Google Scholar] [CrossRef] [Green Version]
- Edwards, I.R.; Aronson, J.K. Adverse drug reactions: Definitions, diagnosis, and management. Lancet 2000, 356, 1255–1259. [Google Scholar] [CrossRef]
- Bouvy, J.C.; De Bruin, M.L.; Koopmanschap, M.A. Epidemiology of adverse drug reactions in europe: A review of recent observational studies. Drug Saf. 2015, 38, 437–453. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Zhu, F.; Ma, X.; Cao, Z.W.; Li, Y.X.; Chen, Y.Z. Mechanisms of drug combinations: Interaction and network perspectives. Nat. Rev. Drug Discov. 2009, 8, 111–128. [Google Scholar] [CrossRef]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017, 35, 436. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.; Yang, J.; Karan, C.; Menden, M.P.; Costello, J.C.; Tang, H.; Xiao, G.; Li, Y.; Allen, J.; Zhong, R.; et al. A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol. 2014, 32, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sheng, Z.; Ma, C.; Tang, K.; Zhu, R.; Wu, Z.; Shen, R.; Feng, J.; Wu, D.; Huang, D.; et al. Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatonetti, N.P.; Patrick, P.Y.; Daneshjou, R.; Altman, R.B. Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012, 4, 125ra31. [Google Scholar] [CrossRef] [Green Version]
- Kantor, E.D.; Rehm, C.D.; Haas, J.S.; Chan, A.T.; Giovannucci, E.L. Trends in prescription drug use among adults in the United States from 1999–2012. JAMA 2015, 314, 1818–1830. [Google Scholar] [CrossRef]
- Percha, B.; Garten, Y.; Altman, R.B. Discovery and explanation of drug–drug interactions via text mining. In Biocomputing 2012; World Scientific: Singapore, 2012; pp. 410–421. [Google Scholar]
- Charlesworth, C.J.; Smit, E.; Lee, D.S.; Alramadhan, F.; Odden, M.C. Polypharmacy among adults aged 65 years and older in the united states: 1988–2010. J. Gerontol. Ser. A Biomed. Med. Sci. 2015, 70, 989–995. [Google Scholar] [CrossRef] [Green Version]
- National Center for Health Statistics. Health, United States, 2016, with Chartbook on Long-Term Trends in Health; Government Printing Office: Washington, DC, USA, 2017.
- US Food and Drug Administration. FDA Adverse Event Reporting System (FAERS) Public Dashboard; US Food and Drug Administration: Silver Spring, MD, USA, 2018.
- Vilar, S.; Friedman, C.; Hripcsak, G. Detection of drug–drug interactions through data mining studies using clinical sources, scientific literature and social media. Briefings Bioinform. 2018, 19, 863–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percha, B.; Altman, R.B. Informatics confronts drug–drug interactions. Trends Pharmacol. Sci. 2013, 34, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Liebler, D.C.; Guengerich, F.P. Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug Discov. 2005, 4, 410–420. [Google Scholar] [CrossRef]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Briefings Bioinform. 2016, 17, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Ryall, K.A.; Tan, A.C. Systems biology approaches for advancing the discovery of effective drug combinations. J. Cheminformatics 2015, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [Green Version]
- Malone, B.; García-Durán, A.; Niepert, M. Knowledge graph completion to predict polypharmacy side effects. In International Conference on Data Integration in the Life Sciences; Springer: Cham, Switzerland, 2018; pp. 144–149. [Google Scholar]
- Burkhardt, H.A.; Subramanian, D.; Mower, J.; Cohen, T. Predicting adverse drug–drug interactions with neural embedding of semantic predications. bioRxiv 2019, 752022. [Google Scholar] [CrossRef]
- Xu, H.; Sang, S.; Lu, H. Tri-graph information propagation for polypharmacy side effect prediction. arXiv 2020, arXiv:2001.10516. [Google Scholar]
- Nováček, V.; Mohamed, S.K. Predicting polypharmacy side-effects using knowledge graph embeddings. AMIA Summits Transl. Sci. 2020, 2020, 449. [Google Scholar]
- Julkunen, H.; Cichonska, A.; Gautam, P.; Szedmak, S.; Douat, J.; Pahikkala, T.; Aittokallio, T.; Rousu, J. Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Chen, H.; Li, J. Drugcom: Synergistic discovery of drug combinations using tensor decomposition. In Proceedings of the 2018 IEEE International Conference on Data Mining (ICDM), Singapore, 17–20 November 2018; pp. 899–904. [Google Scholar]
- Sun, Z.; Huang, S.; Jiang, P.; Hu, P. Dtf: Deep tensor factorization for predicting anticancer drug synergy. Bioinformatics 2020, 36, 4483–4489. [Google Scholar] [CrossRef]
- Stock, M.; Pahikkala, T.; Airola, A.; De Baets, B.; Waegeman, W. A comparative study of pairwise learning methods based on kernel ridge regression. Neural Comput. 2018, 30, 2245–2283. [Google Scholar] [CrossRef] [Green Version]
- Niculescu-Mizil, A.; Caruana, R. Predicting good probabilities with supervised learning. In Proceedings of the 22nd International Conference on Machine Learning, Bonn, Germany, 7–11 August 2005; Association for Computing Machinery: New York, NY, USA, 2005; pp. 625–632. [Google Scholar]
- Wang, W.; Zheng, V.W.; Yu, H.; Miao, C. A survey of zero-shot learning: Settings, methods, and applications. ACM Trans. Intell. Syst. Technol. (TIST) 2019, 10, 1–37. [Google Scholar] [CrossRef]
- Norgeot, B.; Quer, G.; Beaulieu-Jones, B.K.; Torkamani, A.; Dias, R.; Gianfrancesco, M.; Arnaout, R.; Kohane, I.S.; Saria, S.; Topol, E.; et al. Minimum information about clinical artificial intelligence modeling: The mi-claim checklist. Nat. Med. 2020, 26, 1320–1324. [Google Scholar] [CrossRef]
- Stock, M. Exact and Efficient Algorithms for Pairwise Learning. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2017. [Google Scholar]
- Romera-Paredes, B.; Torr, P. An embarrassingly simple approach to zero-shot learning. In Proceedings of the International Conference on Machine Learning (PMLR 2015), Lille, France, 6–11 July 2015; pp. 2152–2161. [Google Scholar]
- Stock, M.; Pahikkala, T.; Airola, A.; Waegeman, W.; De Baets, B. Algebraic shortcuts for leave-one-out cross-validation in supervised network inference. Briefings Bioinform. 2020, 21, 262–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, J.; Ueng, S. The effect of patient-specific drug–drug interaction alerting on the frequency of alerts: A pilot study. Ann. Pharmacother. 2019, 53, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Burridge, P.W.; Yu, K.H.; Ahrens, J.H.; Termglinchan, V.; Wu, H.; Liu, C.; Shukla, P.; Sayed, N.; Churko, J.M.; et al. Transcriptome profiling of patient-specific human ipsc-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 2016, 19, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Kuzmin, E.; VanderSluis, B.; Wang, W.; Tan, G.; Deshpande, R.; Chen, Y.; Usaj, M.; Balint, A.; Usaj, M.M.; Van Leeuwen, J.; et al. Systematic analysis of complex genetic interactions. Science 2018, 360, eaao1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitnik, M.; Leskovec, J. Predicting multicellular function through multi-layer tissue networks. Bioinformatics 2017, 33, i190–i198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitnik, M.; Sosič, R.; Maheshwari, S.; Leskovec, J. BioSNAP Datasets: Stanford Biomedical Network Dataset Collection. 2018. Available online: http://snap.stanford.edu/biodata (accessed on 12 June 2020).
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [Green Version]
- Wahba, G. Spline Models for Observational Data; SIAM: Philadelphia, PA, USA, 1990. [Google Scholar]
- Schrynemackers, M.; Küffner, R.; Geurts, P. On protocols and measures for the validation of supervised methods for the inference of biological networks. Front. Genet. 2013, 4, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AUC-ROC | AUC-PR | |||
|---|---|---|---|---|
| (No-Skill = 0.5) | (No-Skill = 0.02) | |||
| 0.957 | 0.888 | 0.557 | 0.257 | |
| 0.919 | 0.865 | 0.286 | 0.179 | |
| 0.910 | 0.859 | 0.221 | 0.176 | |
| 0.843 | 0.834 | 0.112 | 0.144 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dewulf, P.; Stock, M.; De Baets, B. Cold-Start Problems in Data-Driven Prediction of Drug–Drug Interaction Effects. Pharmaceuticals 2021, 14, 429. https://doi.org/10.3390/ph14050429
Dewulf P, Stock M, De Baets B. Cold-Start Problems in Data-Driven Prediction of Drug–Drug Interaction Effects. Pharmaceuticals. 2021; 14(5):429. https://doi.org/10.3390/ph14050429
Chicago/Turabian StyleDewulf, Pieter, Michiel Stock, and Bernard De Baets. 2021. "Cold-Start Problems in Data-Driven Prediction of Drug–Drug Interaction Effects" Pharmaceuticals 14, no. 5: 429. https://doi.org/10.3390/ph14050429
APA StyleDewulf, P., Stock, M., & De Baets, B. (2021). Cold-Start Problems in Data-Driven Prediction of Drug–Drug Interaction Effects. Pharmaceuticals, 14(5), 429. https://doi.org/10.3390/ph14050429