
Investigation of the Impact of CYP3A5 Polymorphism on Drug–Drug Interaction between Tacrolimus and Schisantherin A/Schisandrin A Based on Physiologically-Based Pharmacokinetic Modeling

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
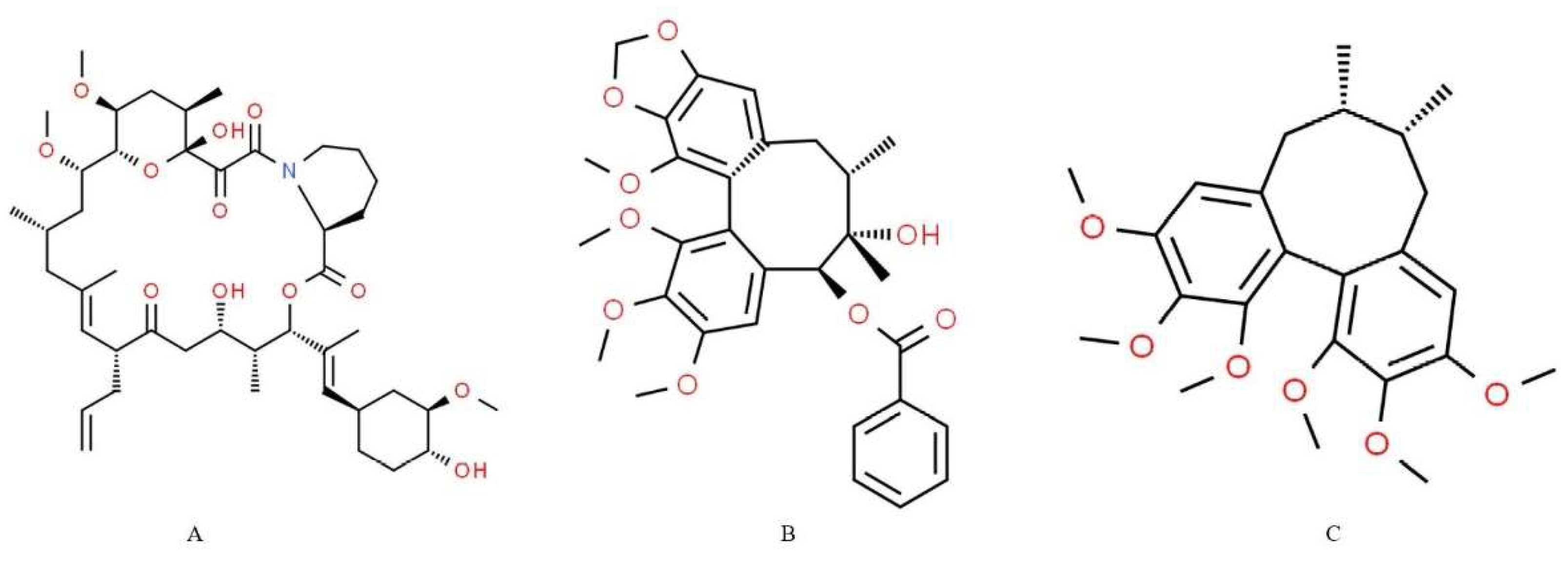
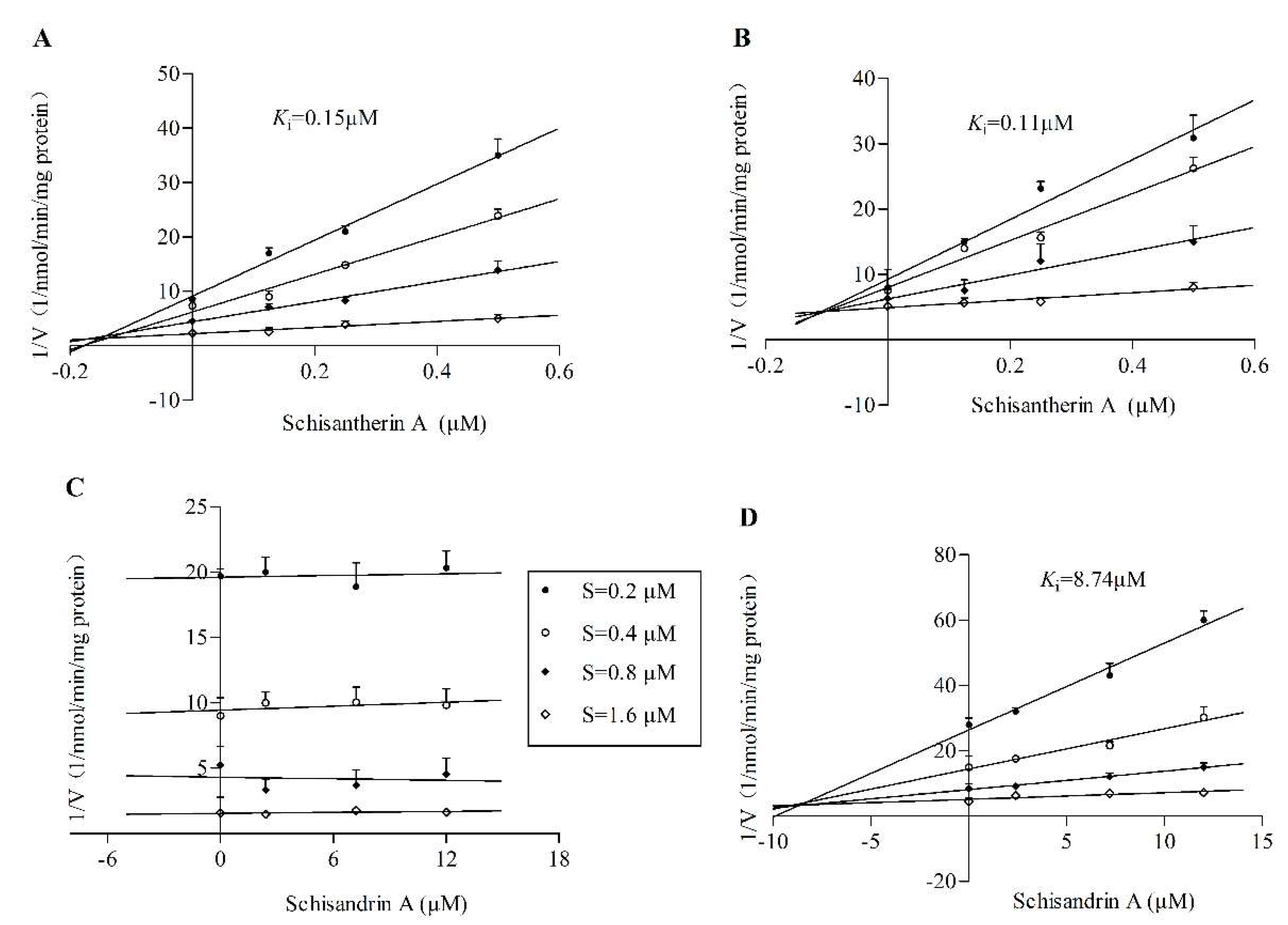
2.1. RI Assay
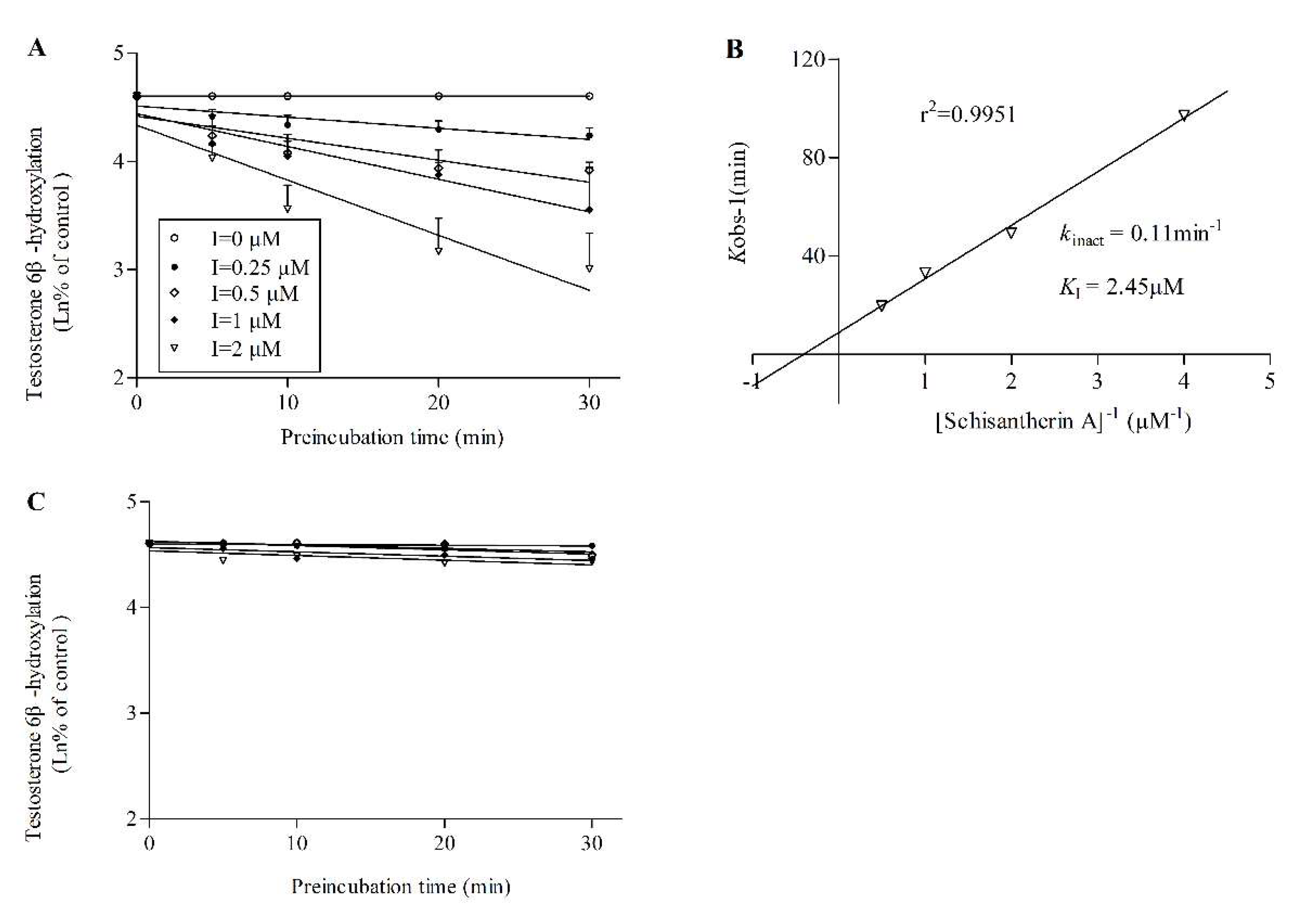
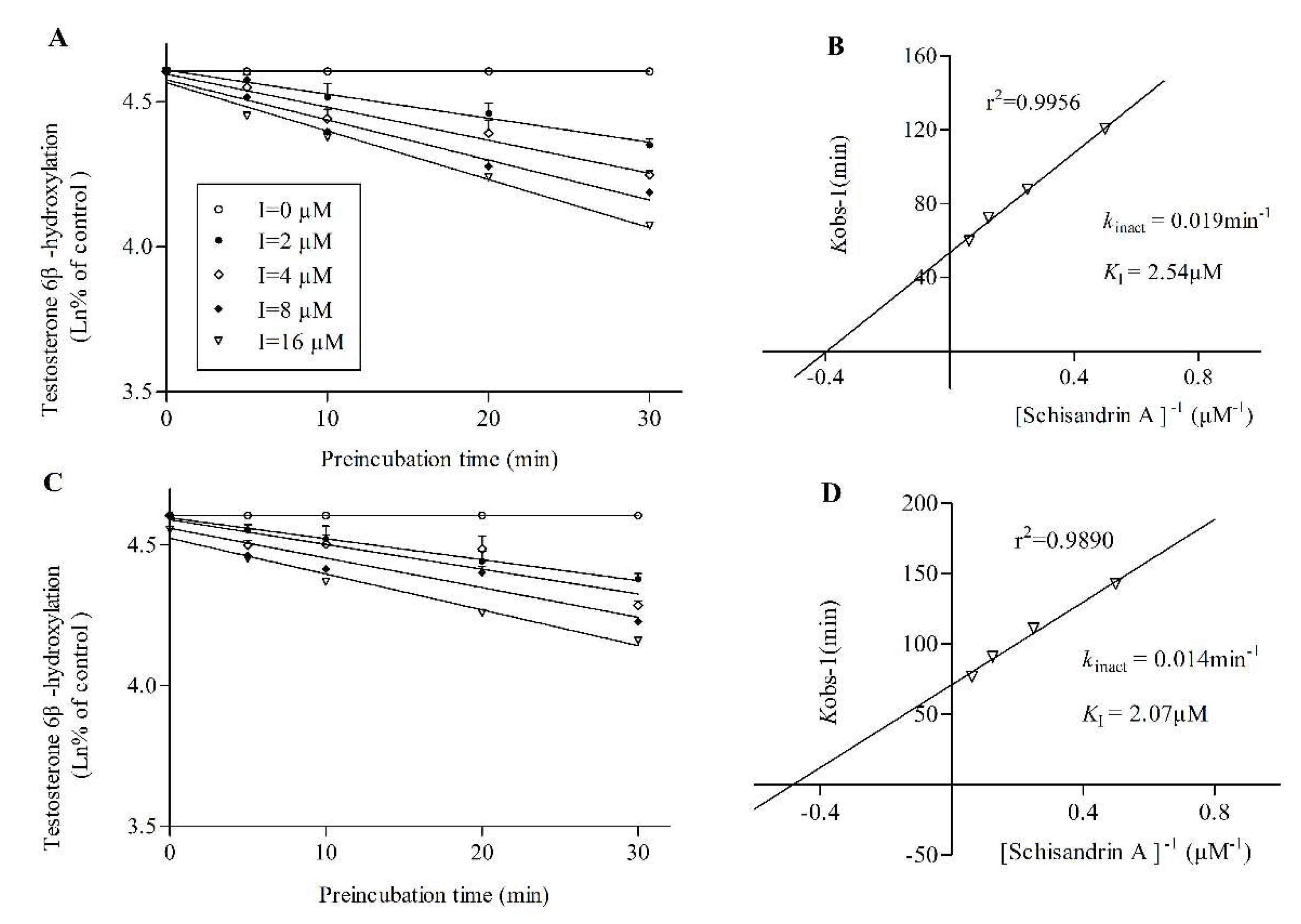
2.2. TDI assay
2.3. Model Development and Verification
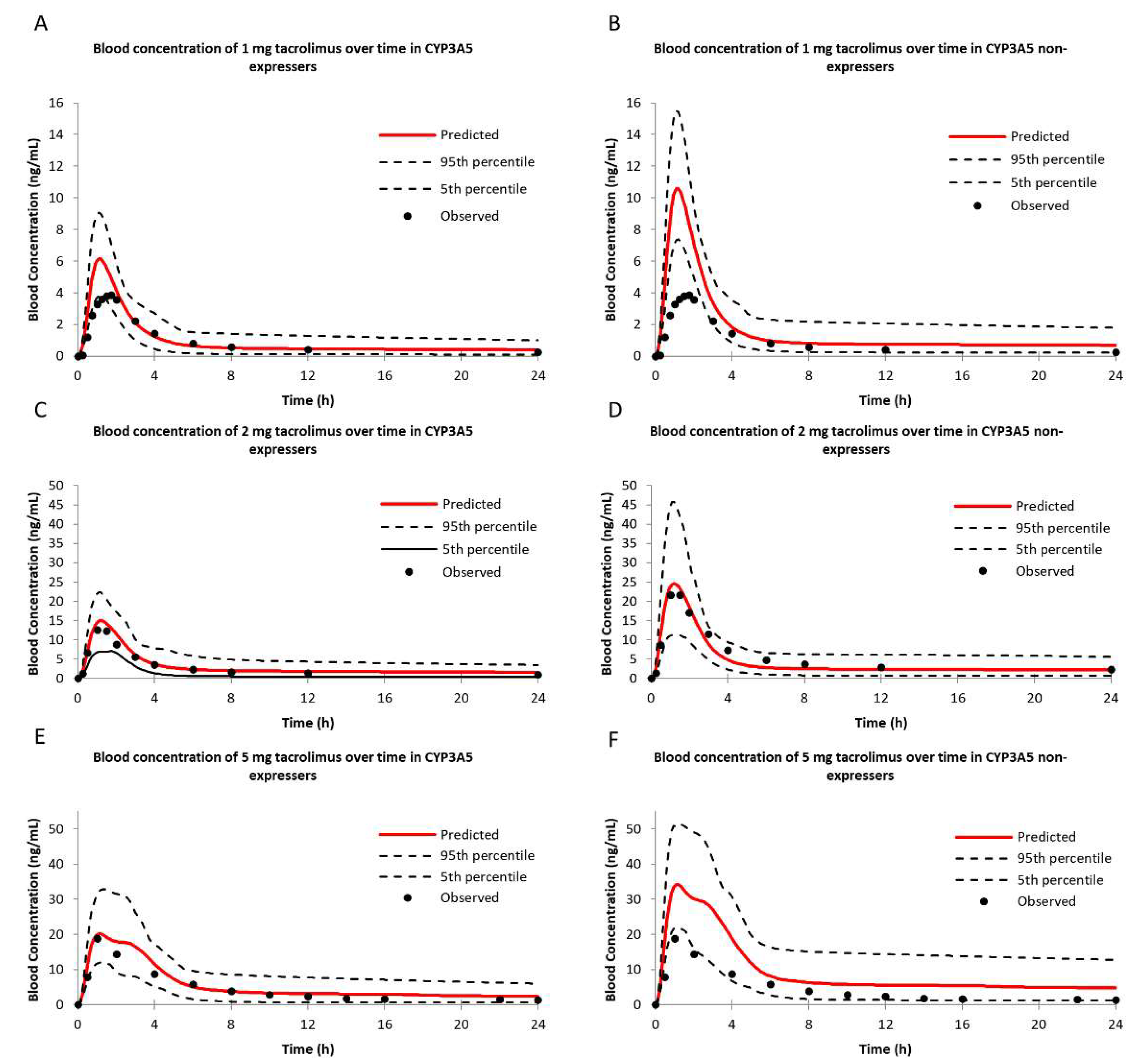
2.3.1. Tacrolimus Pharmacokinetics in CYP3A5 Expressers and Non-Expressers
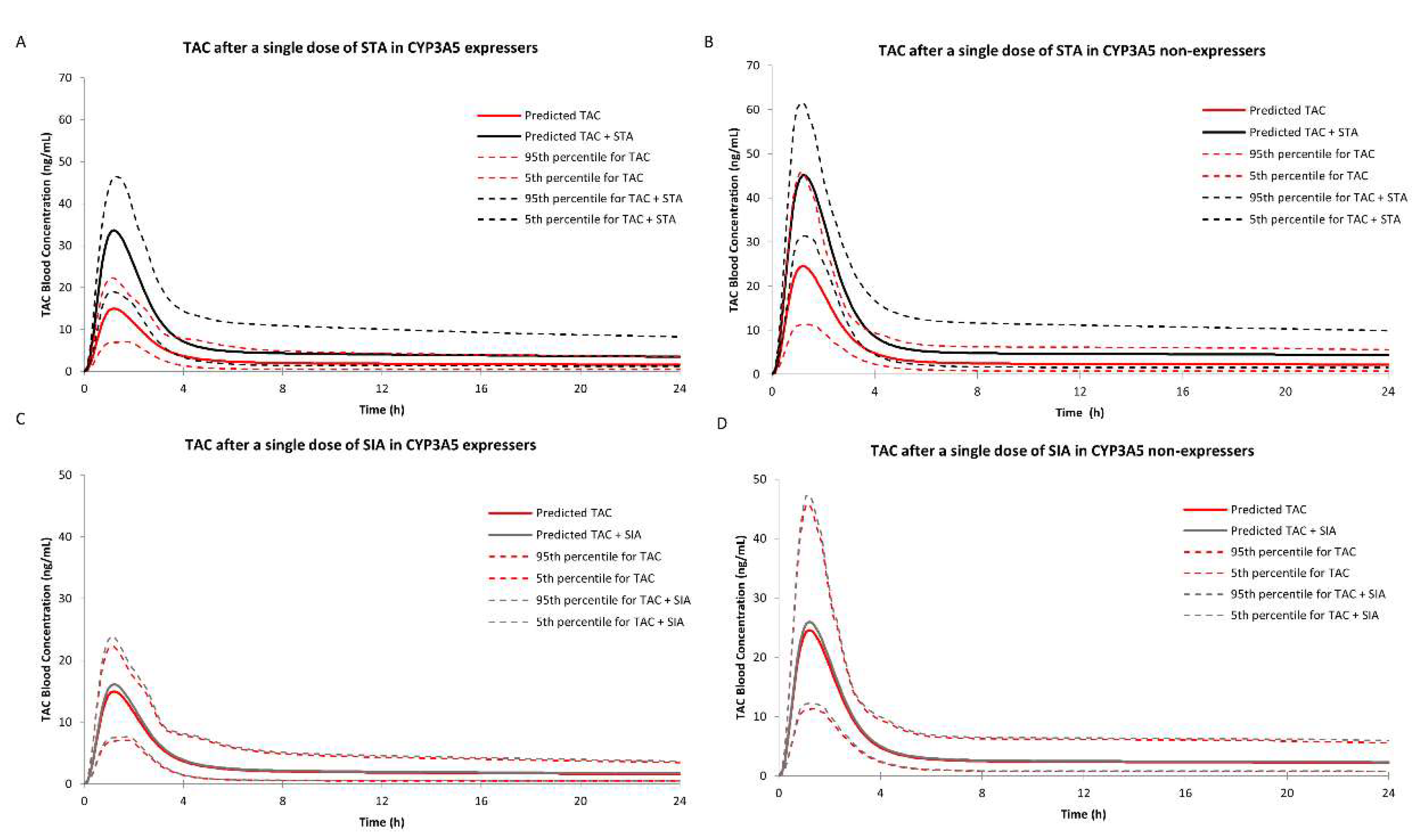
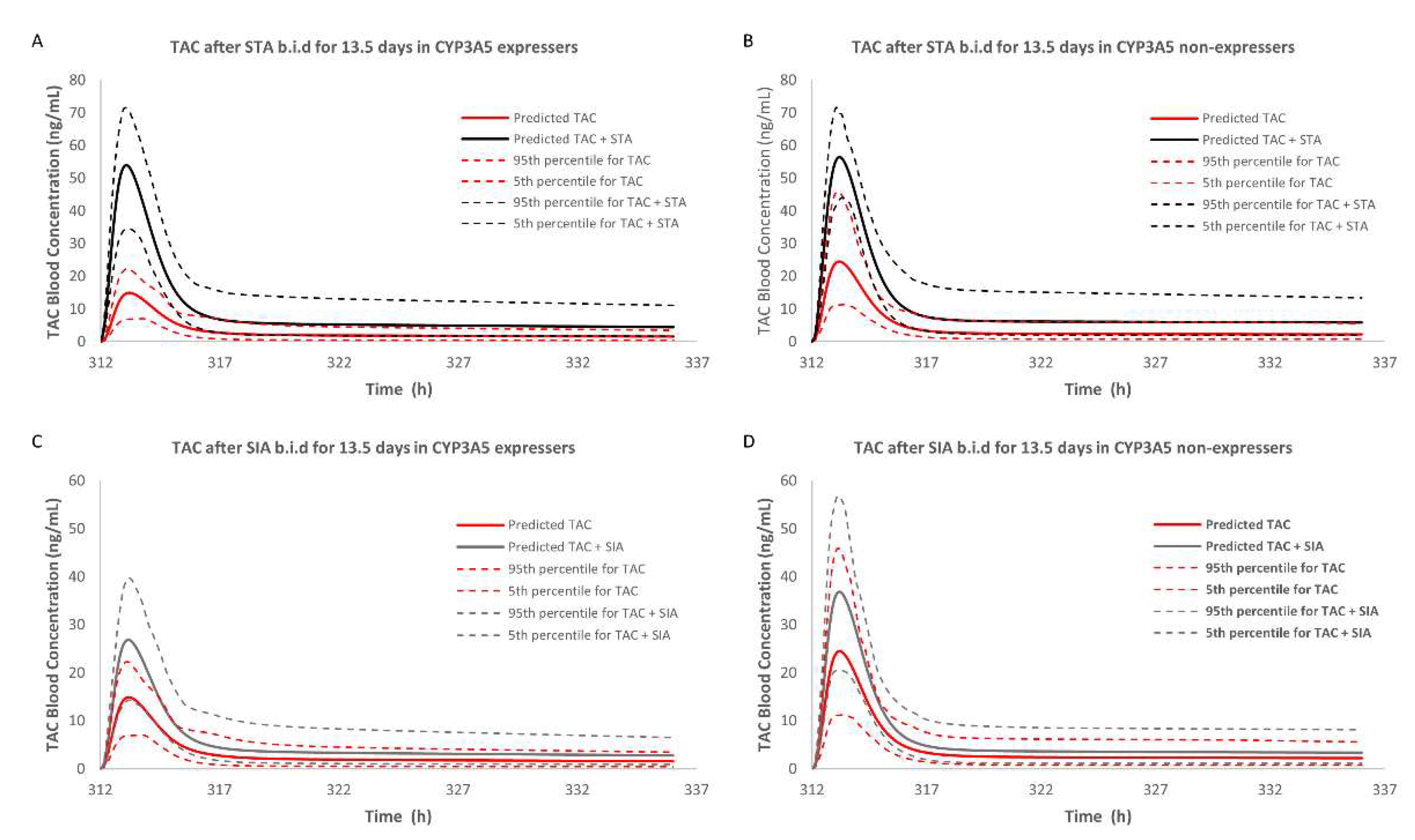
2.3.2. DDI Prediction in CYP3A5 Expressers
2.3.3. DDI Prediction in CYP3A5 Non-Expressers
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. HPLC-MS/MS Method
4.3. Reversible Inhibition (RI) Assay of STA/SIA on CYP3A4 and CYP3A5
4.4. Time-Dependent Inhibition (TDI) Assay of STA/SIA on CYP3A4 and CYP3A5
4.5. PBPK Model Development of Tacrolimus, STA and SIA
4.6. Simcyp® Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, Y.; Jiang, F.; An, H.; Park, H.J.; Choi, J.H.; Lee, H. A pharmacogenomic study on the pharmacokinetics of tacrolimus in healthy subjects using the DMETTM Plus platform. Pharm. J. 2017, 17, 174–179. [Google Scholar] [CrossRef]
- Coto, E.; Tavira, B.; Suárez-Álvarez, B.; López-Larrea, C.; Díaz-Corte, C.; Ortega, F.; Álvarez, V. Pharmacogenetics of tacrolimus: Ready for clinical translation? Kidney Int. 2011, 1, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Hebert, M.F.; Isoherranen, N.; Davis, C.L.; Marsh, C.; Shen, D.D.; Thummel, K.E. Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in vitro. Drug Metab. Dispos. 2006, 34, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.T.; Andrews, L.M.; van Gelder, T.; Shi, Y.Y.; van Schaik, R.H.N.; Wang, L.L.; Hesselink, D.A. Pharmacogenetic aspects of the use of tacrolimus in renal transplantation: Recent developments and ethnic considerations. Expert Opin. Drug Metab. Toxicol. 2016, 12, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Tasnif, Y.; Hebert, M.F.; Davis, C.L.; Shitara, Y.; Calamia, J.C.; Lin, Y.S.; Shen, D.D.; Thummel, K.E. Measurement and compartmental modeling of the effect of CYP3A5 gene variation on systemic and intrarenal tacrolimus disposition. Clin. Pharmacol. Ther. 2012, 92, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Obach, R.S.; Walsky, R.L.; Venkatakrishnan, K. Mechanism-based inactivation of human cytochrome P450 enzymes and the prediction of drug-drug interactions. Drug Metab. Dispos. 2007, 35, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.-W.; Wu, X.-C.; Li, Q.; Yu, A.-R.; Zhu, M.; Shen, Y.; Su, D.; Xiong, L. Effects of Schisandra sphenanthera extract on the pharmacokinetics of tacrolimus in healthy volunteers. Br. J. Clin. Pharmacol. 2007, 64, 469–475. [Google Scholar] [CrossRef] [Green Version]
- ChemSpider. Available online: http://www.chemspider.com/ (accessed on 18 February 2021).
- Iwata, H.; Tezuka, Y.; Kadota, S.; Hiratsuka, A.; Watabe, T. Identification and characterization of potent CYP3A4 inhibitors in schisandra fruit extract. Drug Metab. Dispos. 2004, 32, 1351–1358. [Google Scholar] [CrossRef]
- Lai, L.; Hao, H.; Wang, Q.; Zheng, C.; Zhou, F.; Liu, Y.; Wang, Y.; Yu, G.; Kang, A.; Peng, Y.; et al. Effects of short-term and long-term pretreatment of schisandra lignans on regulating hepatic and intestinal CYP3A in Rats. Drug Metab. Dispos. 2009, 37, 2399–2407. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-L.; Xin, H.-W.; Su, M.-W. Inhibitory Effects of Continuous Ingestion of Schisandrin A on CYP3A in the Rat. Basic Clin. Pharmacol. Toxicol. 2012, 110, 187–192. [Google Scholar] [CrossRef]
- Qin, X.L.; Chen, X.; Wang, Y.; Xue, X.P.; Wang, Y.; Li, J.L.; Wang, X.D.; Zhong, G.P.; Wang, C.X.; Yang, H.; et al. In vivo to in vitro effects of six bioactive lignans of Wuzhi tablet (schisandra sphenanthera extract) on the CYP3A/P-glycoprotein–mediated absorption and metabolism of tacrolimus. Drug Metab. Dispos. 2014, 42, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Lu, J. Hyperkalemia induced by tacrolimus combined with Wuzhi-capsule following renal transplantation: One case report (in Chinese). J. Clin. Rehabil. Tissue Eng. Res. 2012, 15, 8341–8343. [Google Scholar]
- Zhang, H.; Bu, F.; Li, L.; Jiao, Z.; Ma, G.; Cai, W.; Zhuang, X.; Lin, H.-S.; Shin, J.-G.; Xiang, X. Prediction of drug-drug interaction between tacrolimus and principal ingredients of Wuzhi capsule in chinese healthy volunteers using physiologically-based pharmacokinetic modelling. Basic Clin. Pharmacol. Toxicol. 2018, 122, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Fukuen, S.; Maune, H.; Ikenaga, Y.; Yamamoto, I.; Inaba, T.; Azuma, J. Novel detection assay by PCR–RFLP and frequency of the CYP3A5 SNPs, CYP3A5*3 and *6, in a Japanese population. Pharmacogenetics 2002, 12, 331–334. [Google Scholar] [CrossRef]
- Birdwell, K.; Decker, B.; Barbarino, J.; Peterson, J.; Stein, C.; Sadee, W.; Wang, D.; Vinks, A.; He, Y.; Swen, J.; et al. Clinical pharmacogenetics implementation consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin. Pharmacol. Toxicol. 2015, 98, 19–24. [Google Scholar]
- Tseng, E.; Walsky, R.L.; Luzietti, R.A.; Harris, J.J.; Kosa, R.E.; Goosen, T.C.; Zientek, M.A.; Obach, R.S. Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A-cleared drugs assessed in vitro using a CYP3A4-selective inactivator (CYP3cide). Drug Metab. Dispos. 2014, 42, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Walsky, R.L.; Obach, R.S.; Hyland, R.; Kang, P.; Zhou, S.; West, M.; Geoghegan, K.F.; Helal, C.J.; Walker, G.S.; Goosen, T.C.; et al. Selective mechanism-based inactivation of CYP3A4 by CYP3cide (PF-04981517) and its utility as an in vitro tool for delineating the relative roles of CYP3A4 versus CYP3A5 in the metabolism of drugs. Drug Metab. Dispos. 2012, 40, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Zientek, M.A.; Goosen, T.C.; Tseng, E.; Lin, J.; Bauman, J.N.; Walker, G.S.; Kang, P.; Jiang, Y.; Freiwald, S.; Neul, D.; et al. In vitro kinetic characterization of axitinib metabolism. Drug Metab. Dispos. 2015, 44, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Johnson, T.R.; Smith, B.J. Prediction of drug-drug interactions with crizotinib as the CYP3A substrate using a physiologically based pharmacokinetic model. Drug Metab. Dispos. 2015, 43, 1417–1429. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Loi, C.M.; Hoffman, J.; Wang, D. Physiologically based pharmacokinetic modeling of palbociclib. J. Clin. Pharmacol. 2017, 57, 173–184. [Google Scholar] [CrossRef]
- Djebli, N.; Fabre, D.; Boulenc, X.; Fabre, G.; Sultan, E.; Hurbin, F. Physiologically based pharmacokinetic modeling for sequential metabolism: Effect of cyp2c19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab. Dispos. 2015, 43, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Emoto, C.; Fukuda, T.; Venkatasubramanian, R.; Vinks, A.A. The impact of CYP3A5*3 polymorphism on sirolimus pharmacokinetics: Insights from predictions with a physiologically-based pharmacokinetic model. Br. J. Clin. Pharmacol. 2015, 80, 1438–1446. [Google Scholar] [CrossRef] [Green Version]
- Yeo, K.R.; Kenny, J.R.; Rostami-Hodjegan, A. Application of in vitro-in vivo extrapolation (IVIVE) and physiologically based pharmacokinetic (PBPK) modelling to investigate the impact of the CYP2C8 polymorphism on rosiglitazone exposure. Eur. J. Clin. Pharmacol. 2013, 69, 1311–1320. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Zhang, H.; Ding, X.-L.; Ma, S.; Miao, L.-Y. Effect of the P450 oxidoreductase *28 polymorphism on the pharmacokinetics of tacrolimus in Chinese healthy male volunteers. Eur. J. Clin. Pharmacol. 2013, 69, 807–812. [Google Scholar] [CrossRef]
- Shen, L.; Fitzloff, J.F.; Cook, C.S. Differential enantioselectivity and product-dependent activation and inhibition in metabolism of verapamil by human CYP3AS. Drug Metab. Dispos. 2004, 32, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, F.; Xiong, L.; Li, W.; Yu, A. Study on the association of synergistic effects of Wuzhi capsules on tacrolimus with CYP3A5*3 gene polymorphism (in Chinese). China Pharm. 2017, 28, 581–585. [Google Scholar]
- Zuo, X.-C.; Zhou, Y.-N.; Zhang, B.-K.; Yang, G.-P.; Cheng, Z.-N.; Yuan, H.; Ouyang, D.-S.; Liu, S.-K.; Barrett, J.S.; Li, P.-J.; et al. Effect of CYP3A5*3 Polymorphism on Pharmacokinetic Drug Interaction between Tacrolimus and Amlodipine. Drug Metab. Dispos. 2013, 28, 398–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Peng, H.; Xia, Z. Influence of Wuzhi-capsule on blood drug concentration of tacrolimus in nephrotic syndrome children with different CYP3A5 genotypes (in Chinese). Jiangsu Med. 2016, 42, 98–103. [Google Scholar]
- Chandel, N.; Aggarwal, P.K.; Minz, M.; Sakhuja, V.; Kohli, K.K.; Jha, V. CYP3A5*1/*3 genotype influences the blood concentration of tacrolimus in response to metabolic inhibition by ketoconazole. Pharm. Genom. 2009, 19, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Barter, Z.E.; Perrett, H.F.; Yeo, K.R.; Allorge, D.; Lennard, M.S.; Rostami-Hodjegan, A. Determination of a quantitative relationship between hepatic CYP3A5*1/*3 and CYP3A4 expression for use in the prediction of metabolic clearance in virtual populations. Biopharm. Drug Dispos. 2010, 31, 516–532. [Google Scholar] [CrossRef]
- Tamura, S.; Tokunaga, Y.; Ibuki, R.; Amidon, G.L.; Sezaki, H.; Yamashita, S. The site-specific transport and metabolism of tacrolimus in rat small intestine. J. Pharmacol. Exp. Ther. 2003, 306, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Wu, X.; He, Y.; Yu, A.; Xiong, L.; Xiong, Y. Evaluation the effects and cost on the application of tacrolimus combination with Wuzhi-capsule in renal transplanted recipients. Chin. J. Clin. Pharmacol. 2011, 27, 295–298. (In Chinese) [Google Scholar]
- Hollenberg, P.F.; Kent, U.M.; Bumpus, N.N. Mechanism-based inactivation of human cytochromes P450s: Experimental characterization, reactive intermediates, and clinical implications. Chem. Res. Toxicol. 2008, 21, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Emoto, C.; Fukuda, T.; Cox, S.; Christians, U.; Vinks, A. Development of a physiologically-based pharmacokinetic model for sirolimus: Predicting bioavailability based on intestinal CYP3A content. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Lancia, P.; Jacqz-Aigrain, E.; Zhao, W. Choosing the right dose of tacrolimus. Arch. Dis. Child. 2015, 100, 406–413. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Cmax (ng/mL) | Tmax (h) | AUC (ng/mL·h) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dose | Population | Pre 3 | Obs 4 | FE 5 | Pre | Obs | FE | Pre | Obs | FE |
| 1 mg | Exp 1 (n = 16) | 5.74 | 3.88 | 1.48 | 1.08 | 1.75 | 1.61 | 22.50 | 19.34 | 1.16 |
| Non-exp 2 (n = 26) | 9.93 | 5.52 | 1.80 | 1.08 | 1.5 | 1.38 | 37.47 | 30.34 | 1.24 | |
| 2 mg | Exp (n = 31) | 14.50 | 14.09 | 1.03 | 1.08 | 1.26 | 1.16 | 71.53 | 60.83 | 1.18 |
| Non-exp (n = 40) | 23.87 | 24.28 | 1.02 | 1.08 | 1.35 | 1.25 | 102.54 | 119.02 | 1.16 | |
| 5 mg | Exp (n = 12) | 21.41 | 20.8 | 1.03 | 0.84 | 1.40 | 1.67 | 131.07 | 90.40 | 1.45 |
| Non-exp (n = 12) | 36.73 | 27.90 | 1.32 | 0.84 | 1.30 | 1.55 | 227.07 | 134.77 | 1.68 | |
| AUCR | Inhibitors | Dose Regimen | RI Case#1 | TDI Case#2 | RI and TDI Case#3 | |
|---|---|---|---|---|---|---|
| Population | ||||||
| CYP3A5 Non-expresser | STA | Single dose | 102.80 1/152.07 2 1.48 3 | 102.80/154.70 1.50 | 102.80/195.23 1.90 | |
| Multidose | 102.81/156.70 1.52 | 102.81/233.02 2.27 | 102.81/247.91 2.41 | |||
| SIA | Single dose | — | 102.80/113.25 1.10 | — | ||
| Multidose | — | 102.81/142.93 1.39 | — | |||
| CYP3A5 Expresser | STA | Single dose | 72.04/127.91 1.78 | 72.04/65.67 1.33 | 72.04/156.05 2.17 | |
| Multidose | 72.05/134.10 1.86 | 72.05/126.94 1.76 | 72.05/193.91 2.70 | |||
| SIA | Single dose | 72.04/73.20 1.02 | 72.04/80.99 1.12 | 72.04/82.24 1.14 | ||
| Multidose | 72.05/73.20 1.02 | 72.05/111.45 1.55 | 72.05/112.85 1.57 | |||
| Elimination | Parameters | Value | Source |
|---|---|---|---|
| CYP3A5 Expresser | CYP3A4/5 13-DMT 1 Vmax 2 | 8/17 pmol/min/pmol | [3] |
| CYP3A4/5 13-DMT Km,u 3 | 0.21/0.21 μM | ||
| CYP3A4/5 12-HT 4 Vmax | 0.6/1.4 pmol/min/pmol | [3] | |
| CYP3A4/5 12-HT Km,u | 0.29/0.35 μM | [3] | |
| CYP3A5 Non-expresser | CYP3A4 13-DMT/12 HT Vmax | 8/0.6 pmol/min/pmol | [3] |
| CYP3A4 13-DMT/12-HT Km,u | 0.21/0.29 μM | [3] | |
| CYP3A4/5 ISEF 5 | 0.24 (BD Supersomes) | Simcyp® |
| Population | CYP3A4/5 | Caucasian | Chinese |
|---|---|---|---|
| CYP3A5 expressers | CYP3A4 in liver | 137 | 120 |
| CYP3A5 in liver | 103 | 82 | |
| CYP3A4 in intestine | 66.2 | 58 | |
| CYP3A5 in intestine | 24.6 | 21.5 | |
| CYP3A5 non-expressers | CYP3A4 in liver | 137 | 120 |
| CYP3A4 in intestine | 66.2 | 58 | |
| CYP3A5 in liver and intestine | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Bu, F.; Zhang, H.; Wang, Q.; Tang, Z.; Yuan, J.; Lin, H.-S.; Xiang, X. Investigation of the Impact of CYP3A5 Polymorphism on Drug–Drug Interaction between Tacrolimus and Schisantherin A/Schisandrin A Based on Physiologically-Based Pharmacokinetic Modeling. Pharmaceuticals 2021, 14, 198. https://doi.org/10.3390/ph14030198
He Q, Bu F, Zhang H, Wang Q, Tang Z, Yuan J, Lin H-S, Xiang X. Investigation of the Impact of CYP3A5 Polymorphism on Drug–Drug Interaction between Tacrolimus and Schisantherin A/Schisandrin A Based on Physiologically-Based Pharmacokinetic Modeling. Pharmaceuticals. 2021; 14(3):198. https://doi.org/10.3390/ph14030198
Chicago/Turabian StyleHe, Qingfeng, Fengjiao Bu, Hongyan Zhang, Qizhen Wang, Zhijia Tang, Jing Yuan, Hai-Shu Lin, and Xiaoqiang Xiang. 2021. "Investigation of the Impact of CYP3A5 Polymorphism on Drug–Drug Interaction between Tacrolimus and Schisantherin A/Schisandrin A Based on Physiologically-Based Pharmacokinetic Modeling" Pharmaceuticals 14, no. 3: 198. https://doi.org/10.3390/ph14030198
APA StyleHe, Q., Bu, F., Zhang, H., Wang, Q., Tang, Z., Yuan, J., Lin, H. -S., & Xiang, X. (2021). Investigation of the Impact of CYP3A5 Polymorphism on Drug–Drug Interaction between Tacrolimus and Schisantherin A/Schisandrin A Based on Physiologically-Based Pharmacokinetic Modeling. Pharmaceuticals, 14(3), 198. https://doi.org/10.3390/ph14030198