
Comparison of Solution Chemical Properties and Biological Activity of Ruthenium Complexes of Selected β-Diketone, 8-Hydroxyquinoline and Pyrithione Ligands

 , ,
, ,  , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
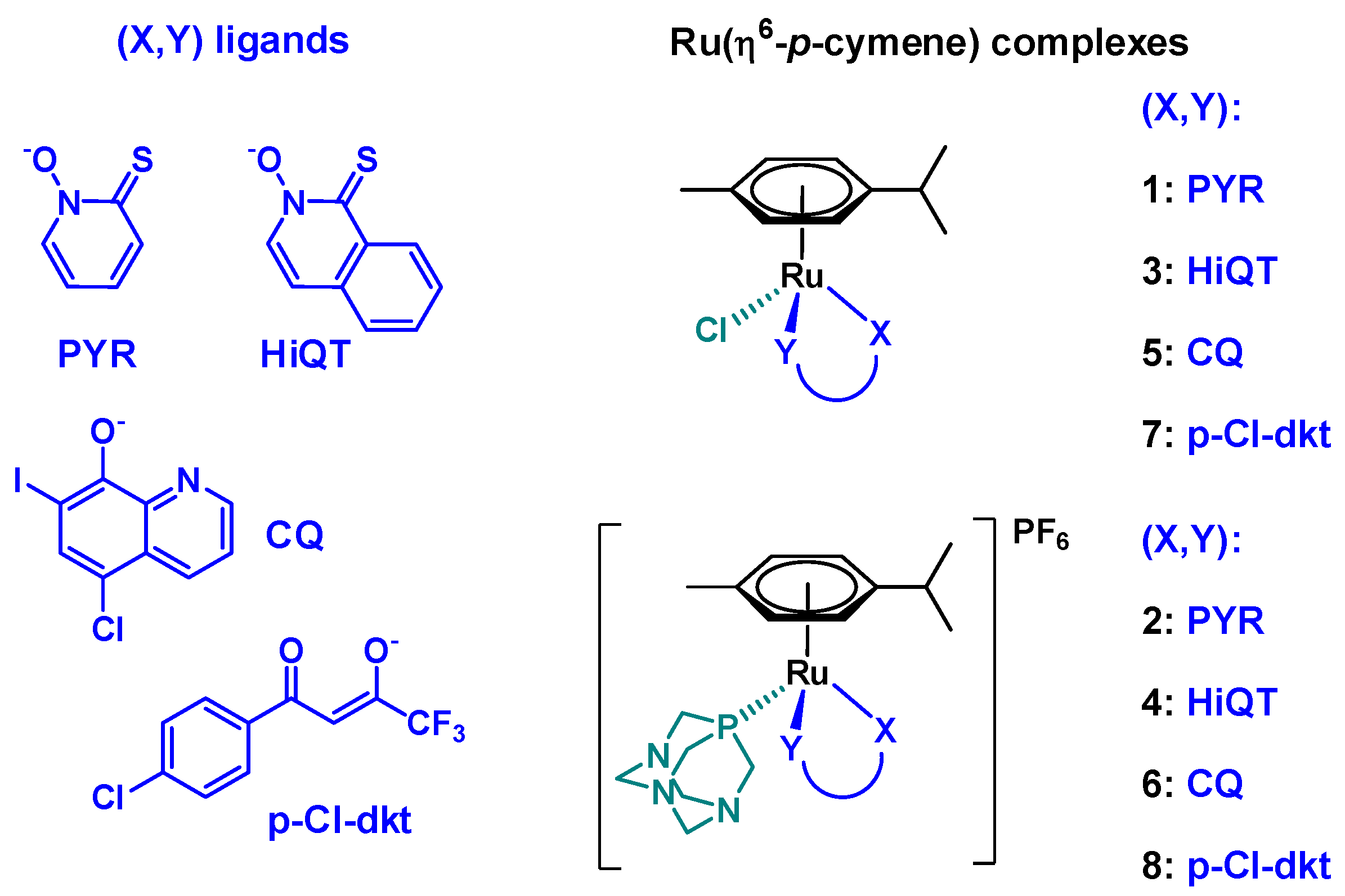
2.1. Synthesis and Characterization of the Complexes
2.2. Pharmacological Activity of the Complexes 1–8
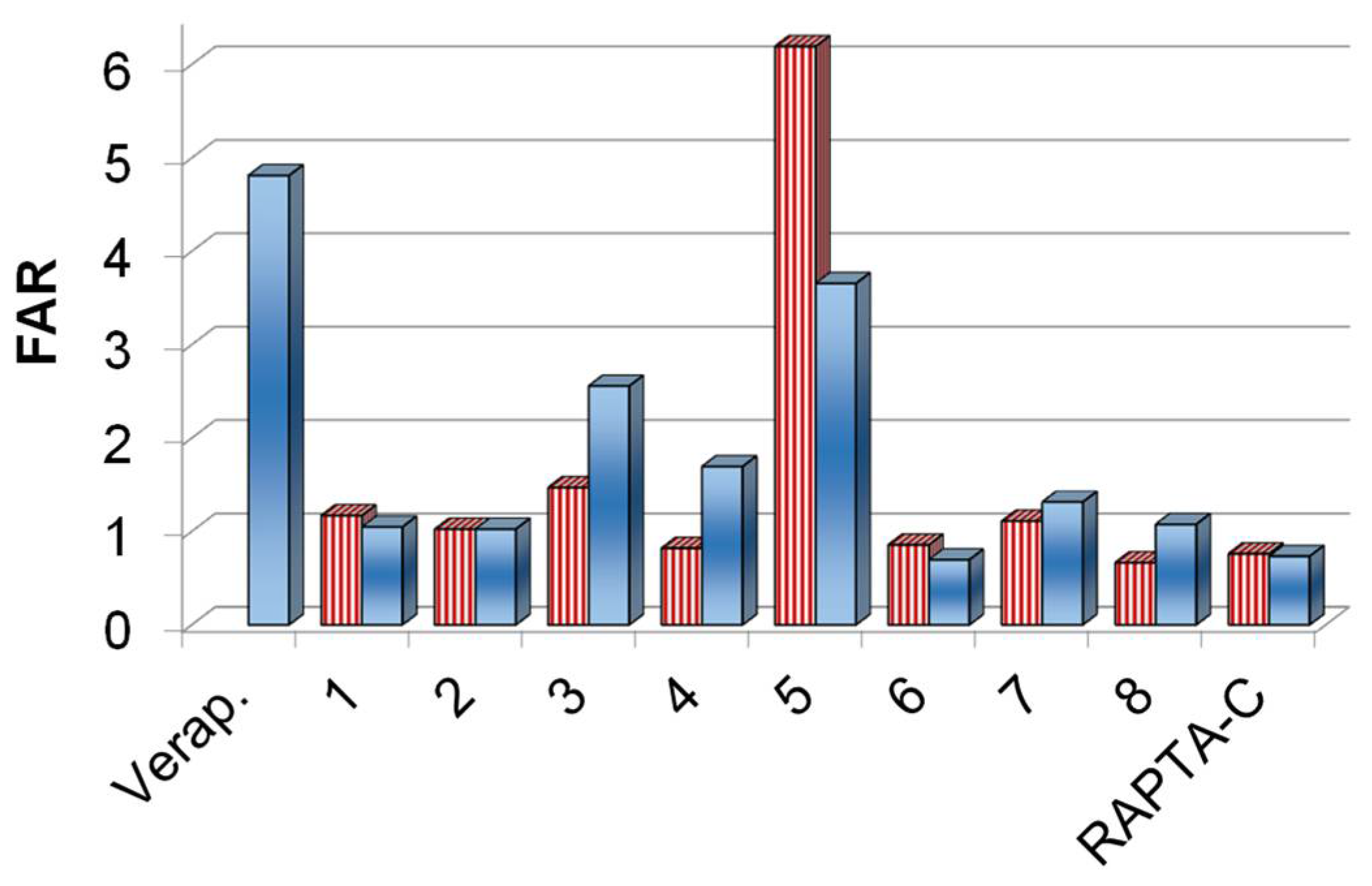
2.2.1. In Vitro Cytotoxicity of the Complexes 1–8 on Cancer cells and Inhibition of the ABCB1 Efflux Pump
2.2.2. Antibacterial Effect of the Complexes 1–8
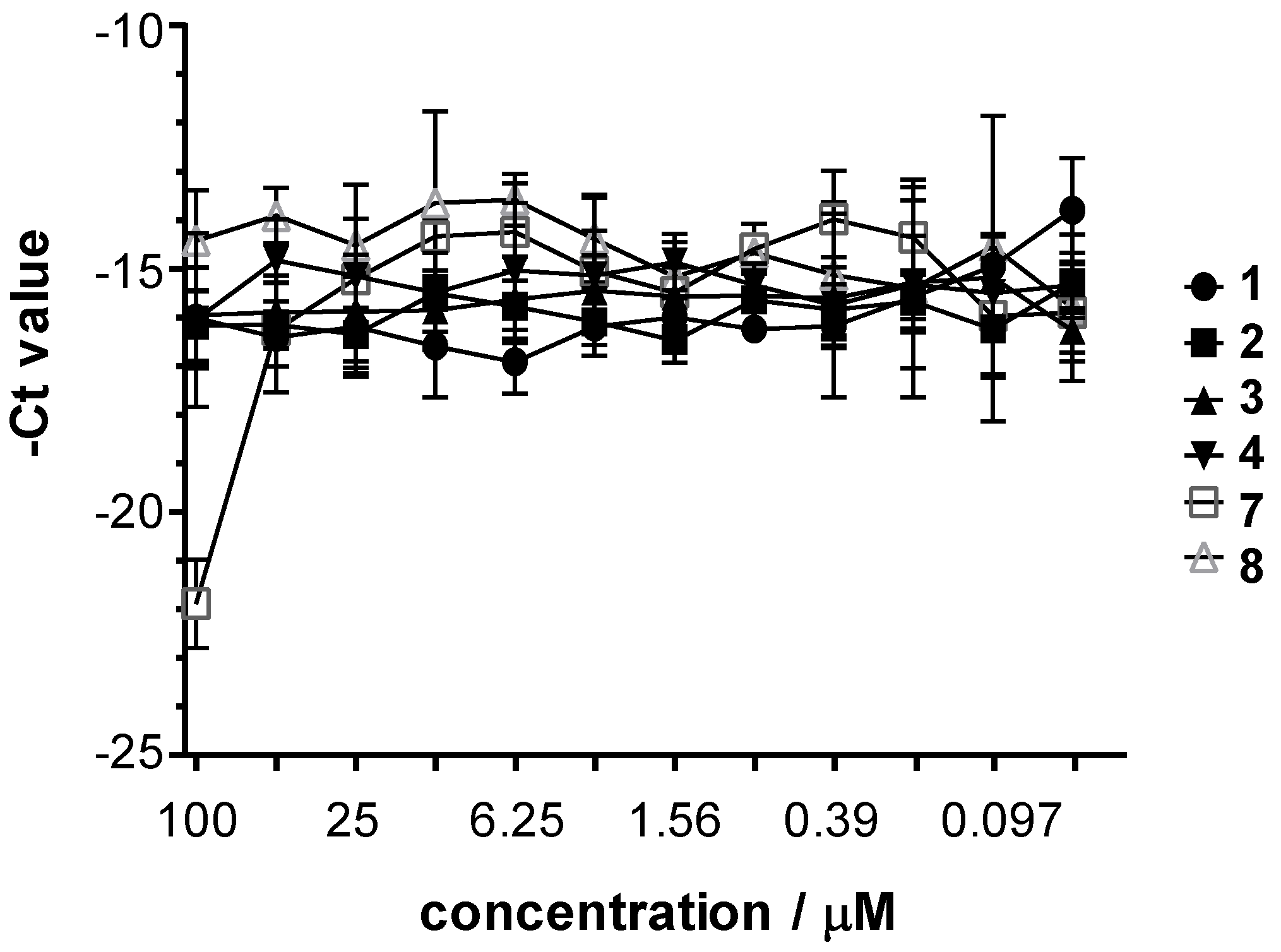
2.2.3. Antichlamydia Activity of the Complexes 1–8
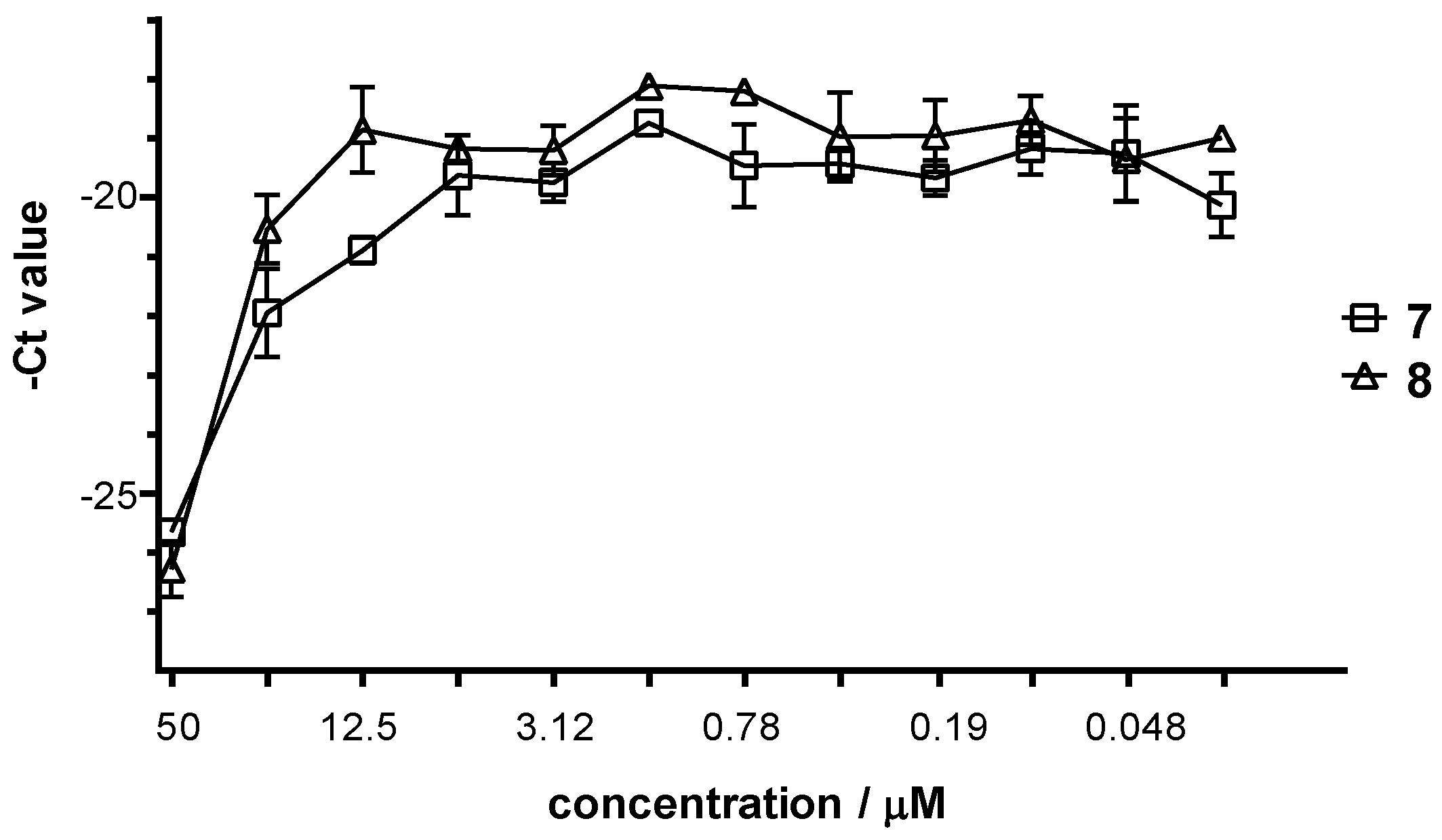
2.2.4. Antiviral Activity of the Complexes 1–8
2.3. Solution Speciation of Complexes 1–8
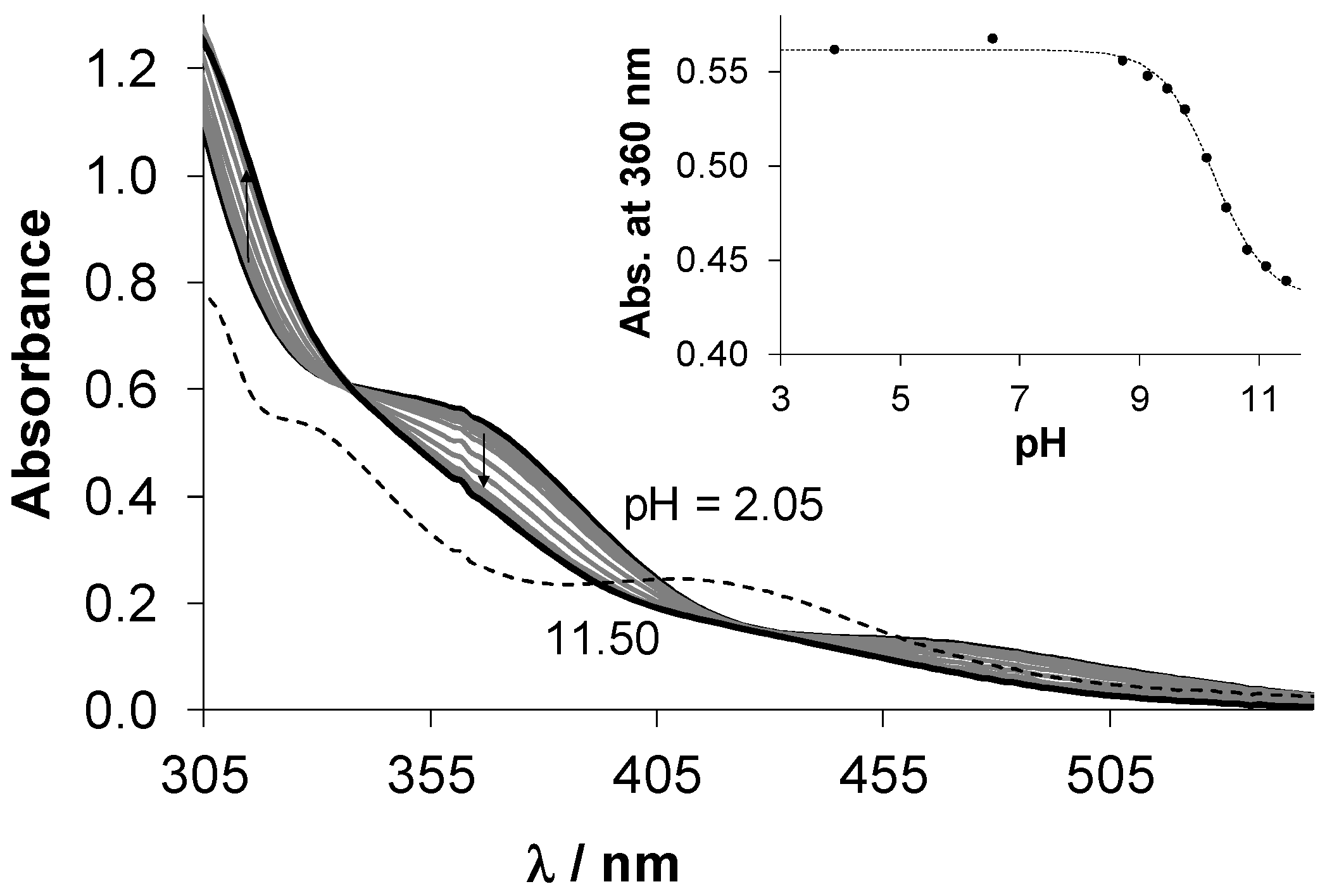
2.3.1. Solution Chemical Properties of β-Diketone and 8-Hydroxyquinoline Complexes 5–8
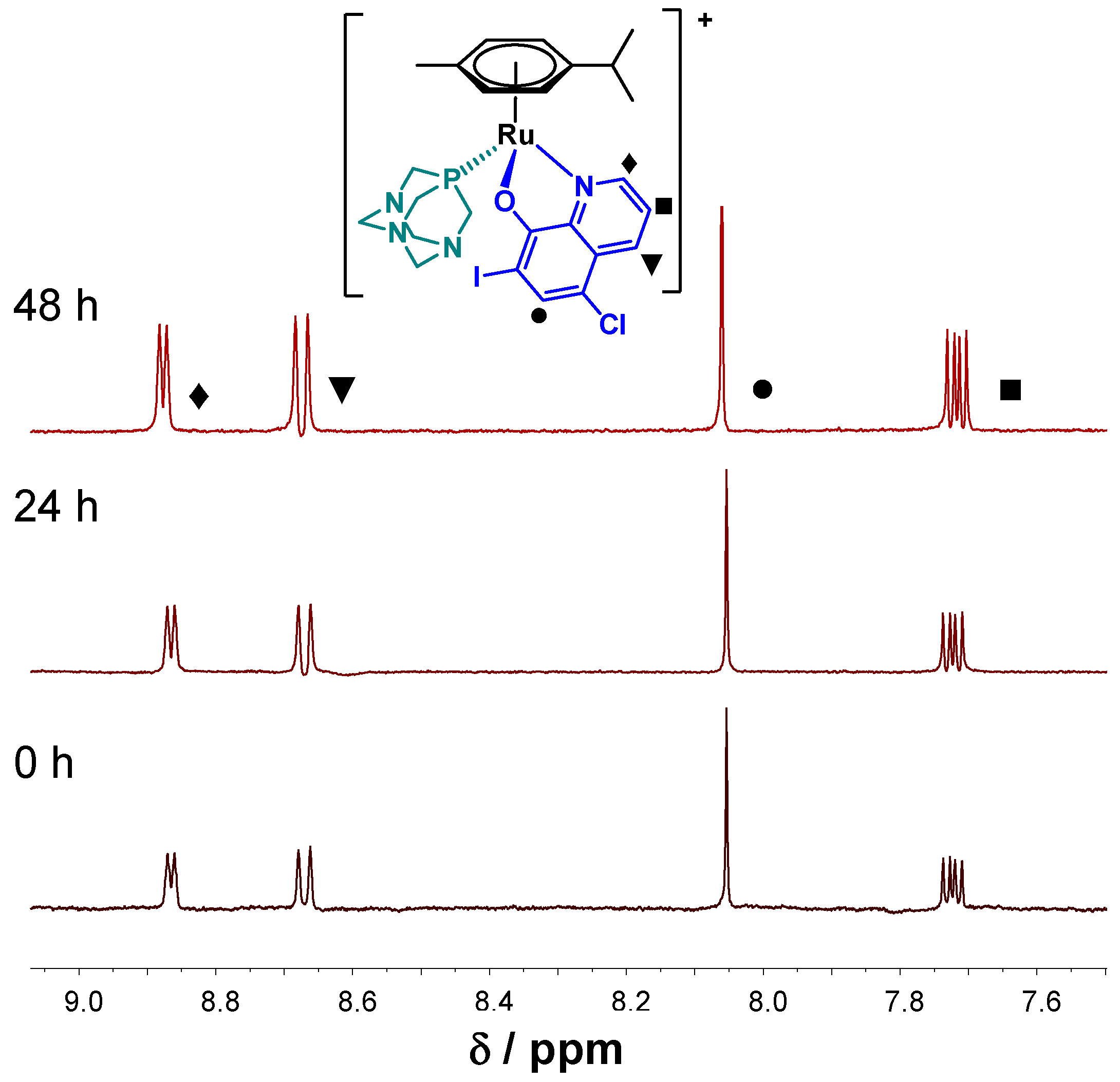
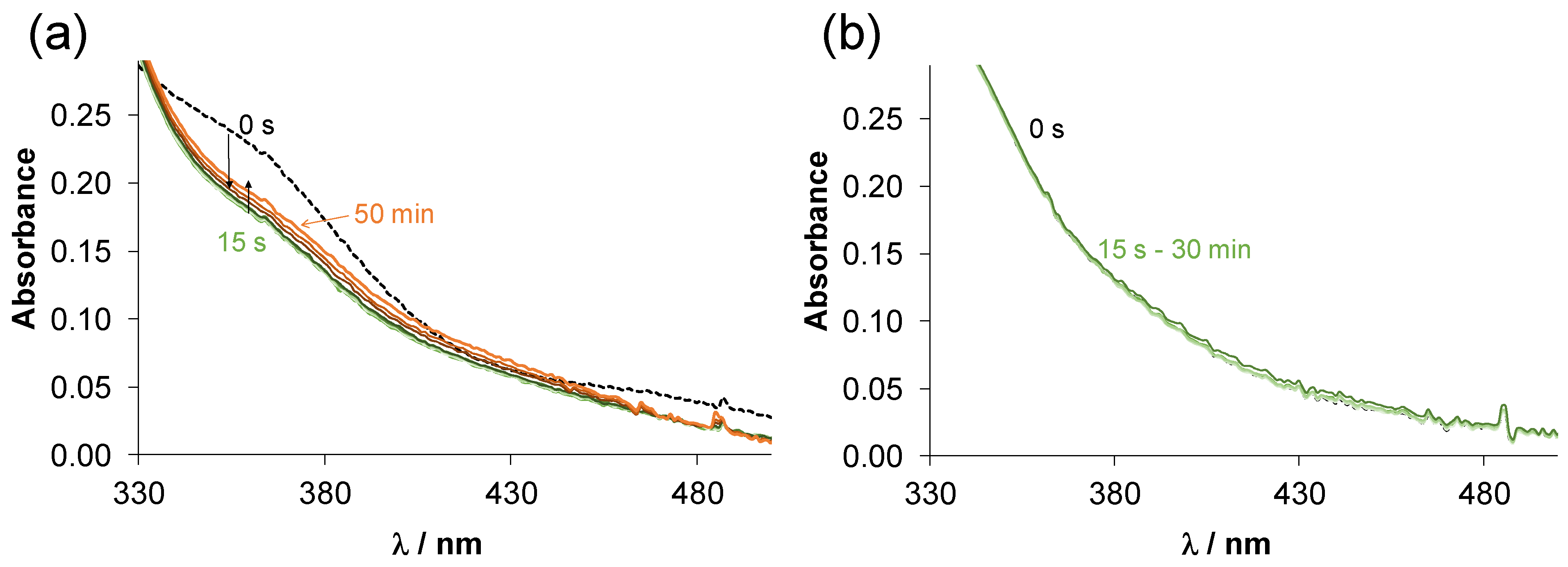
2.3.2. Solution Chemical Properties of Pyrithione-Type Complexes 1–4
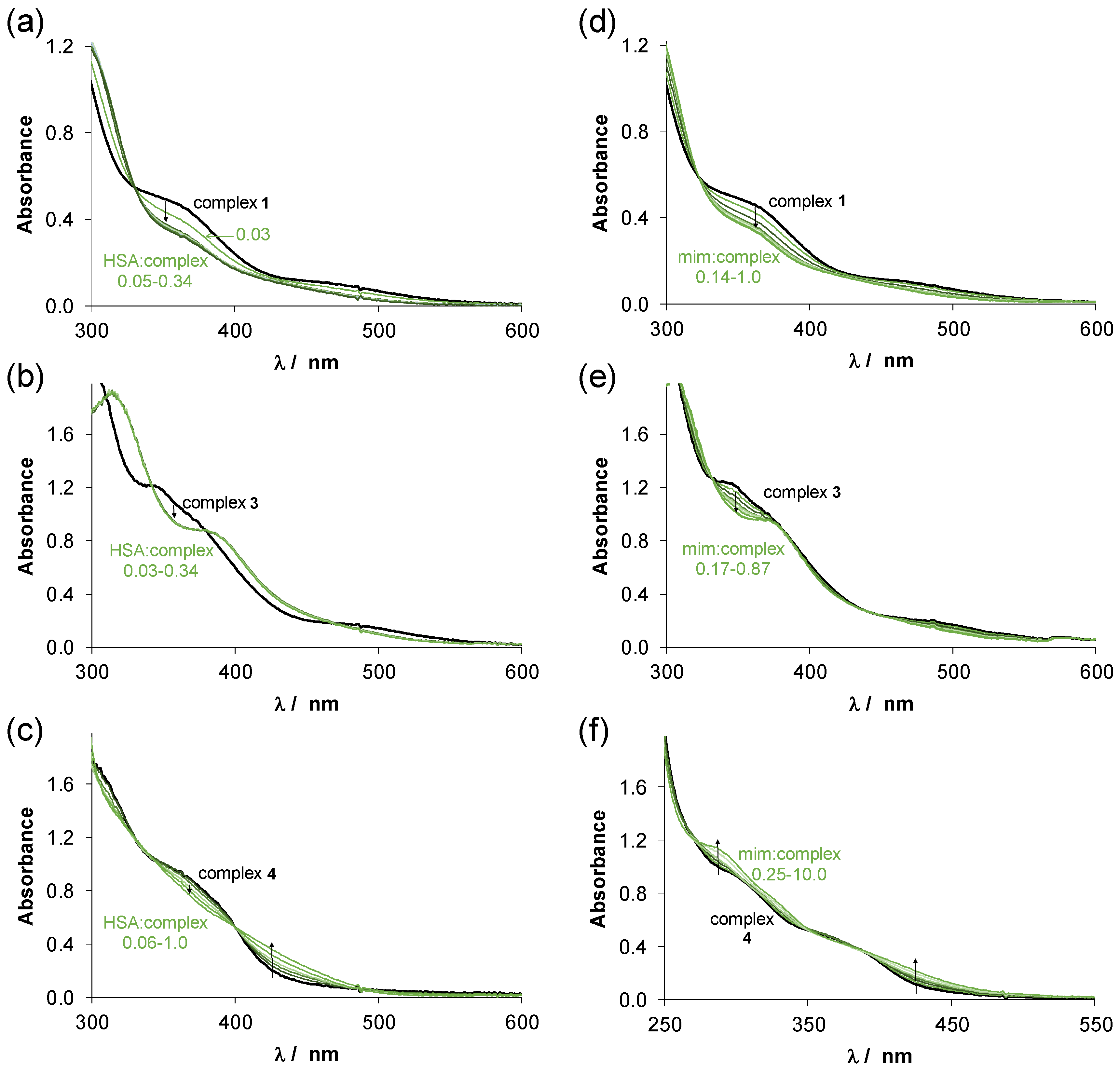
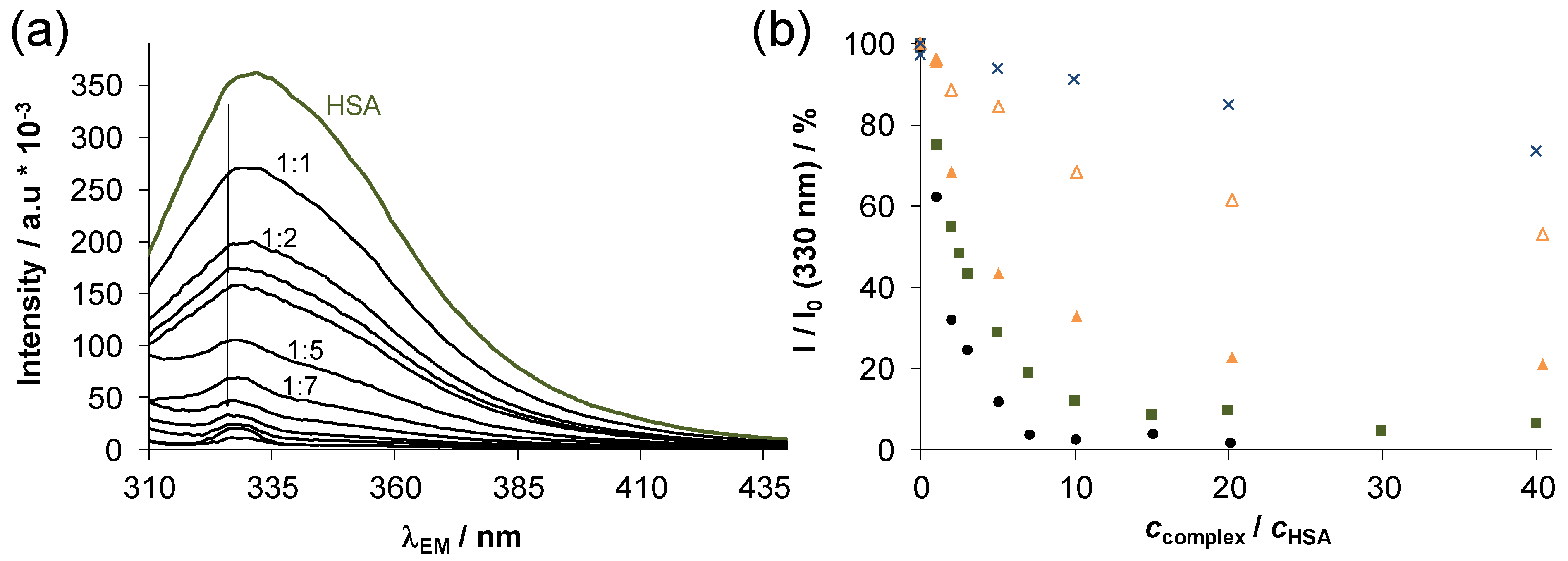
2.4. Interaction of Complexes 1–4 with Human Serum Albumin
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis and Characterization of Complexes
3.3. Solution Studies: pH-potentiometry, UV-vis Spectrophotometry and 1H NMR Spectroscopy
3.4. Lipophilicity and PAMPA Measurements
3.5. HSA Binding Studies: UV-vis and Fluorometry
3.6. In Vitro Cytotoxicity Studies
3.6.1. Cell Lines and Culture Conditions
3.6.2. MTT Assay for Cytotoxic Effect
3.7. Rhodamine 123 Uptake/Retention Fluorescence Assay
3.8. Antibacterial Effect: Bacterial Cell Culture and MIC Determination
3.9. Antichlamydia Activity: Growth in Hela Cells, Cultivation and Quantification
3.10. Antiviral Activity: Growth in Vero Cells, Cultivation and Quantification
3.11. Chlamydia Trachomatis and HSV-2 Growth Monitoring by Direct Quantitative PCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alessio, E. Thirty years of the drug candidate NAMI-A and the myths in the field of ruthenium anticancer compounds: A personal perspective. Eur. J. Inorg. Chem. 2017, 12, 1549–1560. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents—Towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Intravesical Photodynamic Therapy (PDT) in BCG Refractory High-Risk Non-Muscle Invasive Bladder Cancer (NMIBC) Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03053635?term=tld-1433 (accessed on 18 April 2021).
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Hayward, R.L.; Schornagel, Q.C.; Tente, R.; Macpherson, J.S.; Aird, R.E.; Guichard, S.; Habtemariam, A.; Sadler, P.; Jodrell, D.I. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organometallic agent, RM175. Cancer Chemother. Pharmacol. 2005, 55, 577–583. [Google Scholar] [CrossRef]
- Bergamo, A.; Masi, A.; Peacock, A.F.A.; Habtemariam, A.; Sadler, P.J.; Sava, G. In vivo tumour and metastasis reduction and in vitro effects on invasion assays of the ruthenium RM175 and osmium AFAP51 organometallics in the mammary cancer model. J. Inorg. Biochem. 2010, 104, 79–86. [Google Scholar] [CrossRef]
- Kaluderovic, N.G.; Paschke, R. Anticancer metallotherapeutics in preclinical development. Curr. Med. Chem. 2011, 18, 4738–4752. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.-S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Habtemariam, A.; Melchart, M.; Fernández, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure−activity relationships for cytotoxic ruthenium(II) arene complexes containing N,N-, N,O-, and O,O-chelating Ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef]
- Banerjee, S.; Sadler, P.J. Transfer hydrogenation catalysis in cells. RSC Chem. Biol. 2021, 2, 12–29. [Google Scholar] [CrossRef]
- Süss-Fink, G. Areneruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef]
- Chow, M.J.; Babak, M.V.; Tan, K.W.; Cheong, M.C.; Pastorin, G.; Gaiddon, C.; Ang, W.H. Induction of the endoplasmic reticulum stress pathway by highly cytotoxic organoruthenium Schiff-base complexes. Mol. Pharm. 2018, 15, 3020–3031. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Alonso, M.; Busto, N.; Jalon, F.A.; Manzano, B.R.; Leal, J.M.; Rodriguez, A.M.; García, B.; Espino, G. Derivation of structure–activity relationships from the anticancer properties of ruthenium (II) arene complexes with 2-aryldiazole ligands. Inorg. Chem. 2014, 53, 11274–11288. [Google Scholar] [CrossRef]
- Liu, S.; Wu, K.; Zheng, W.; Zhao, Y.; Luo, Q.; Xiong, S.; Wang, F. Identification and discrimination of binding sites of an organoruthenium anticancer complex to single-stranded oligonucleotides by mass spectrometry. Analyst 2014, 139, 4491–4496. [Google Scholar] [CrossRef] [PubMed]
- Kljun, J.; Anko, M.; Traven, K.; Sinreih, M.; Pavlič, R.; Peršič, Š.; Ude, Z.; Codina, E.E.; Stojan, J.; Rizner, T.L.; et al. Pyrithione-based ruthenium complexes as inhibitors of aldo–keto reductase 1C enzymes and anticancer agents. Dalton Trans. 2016, 45, 11791–11800. [Google Scholar] [CrossRef] [Green Version]
- Kladnik, J.; Kljun, J.; Burmeister, H.; Ott, I.; Romero-Canelón, I.; Turel, I. Towards identification of essential structural elements of organoruthenium(II)-pyrithionato complexes for anticancer activity. Chem. Eur. J. 2019, 25, 14169–14182. [Google Scholar] [CrossRef]
- Gobec, M.; Kljun, J.; Sosič, I.; Mlinarič-Raščan, I.; Uršič, M.; Gobec, S.; Turel, I. Structural characterization and biological evaluation of a clioquinol–ruthenium complex with copper-independent antileukaemic activity. Dalton Trans. 2014, 43, 9045–9051. [Google Scholar] [CrossRef] [Green Version]
- Seršen, S.; Kljun, J.; Požgan, F.; Štefane, B.; Turel, I. Novel organoruthenium(II) β-diketonates as catalysts for ortho arylation via C−H activation. Organometallics 2013, 32, 609–616. [Google Scholar] [CrossRef]
- Seršen, S.; Kljun, J.; Kryeziu, K.; Panchuk, R.; Alte, B.; Körner, W.; Heffeter, P.; Berger, W.; Turel, I. Structure-related mode-of-action differences of anticancer organoruthenium complexes with β-diketonates. J. Med. Chem. 2015, 58, 3986–3996. [Google Scholar] [CrossRef]
- Kladnik, J.; Ristovski, S.; Kljun, J.; Defant, A.; Mancini, I.; Sepčić, K.; Turel, I. Structural isomerism and enhanced lipophilicity of pyrithione ligands of organoruthenium(II) complexes increase inhibition on AChE and BuChE. Int. J. Mol. Sci. 2020, 21, 5628. [Google Scholar] [CrossRef]
- Kladnik, J.; Coverdale, J.P.C.; Kljun, J.; Burmeister, H.; Lippman, P.; Ellis, F.G.; Jones, A.M.; Ott, I.; Romero-Canelón, I.; Turel, I. Organoruthenium complexes with benzo-fused pyrithiones overcome platinum resistance in ovarian cancer cells. Cancers 2021, 13, 2493. [Google Scholar] [CrossRef]
- Uršič, M.; Lipec, T.; Meden, A.; Turel, I. Synthesis and structural evaluation of organo-ruthenium complexes with β-diketonates. Molecules 2017, 22, 326. [Google Scholar] [CrossRef] [Green Version]
- Kljun, J.; Leon, I.E.; Peršič, Š.; Cadavid-Vargas, J.F.; Etcheverry, S.B.; He, W.; Bai, Y.; Turel, I. Synthesis and biological characterization of organoruthenium complexes with 8-hydroxyquinolines. J. Inorg. Biochem. 2018, 186, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer cell lines are useful model systems for medical research. Cancers 2019, 11, 1098. [Google Scholar] [CrossRef] [Green Version]
- Gulve, N.; Rudel, T. Chlamydia trachomatis and human herpesvirus 6 infections in ovarian cancer—Casual or causal? PLoS Pathog. 2019, 15, e1008055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Rizner, T.L.; Penning, T.M. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Bal, A.M.; David, M.Z.; Garau, J.; Gottlieb, T.; Mazzei, T.; Scaglione, F.; Tattevin, P.; Gould, I.M. Future trends in the treatment of meticillin-resistant Staphylococcus aureus (MRSA) infection: An in-depth review of newer antibiotics active against an enduring pathogen. J. Glob. Antimicrob. Resist. 2015, 10, 295–303. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, C.M.; Ferone, M.E. Chlamydia trachomatis genital infections. Microb. Cell 2016, 3, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.J. Chlamydia trachomatis in the United Kingdom: A systematic review and analysis of prevalence studies. Sex. Transm. Infect. 2004, 80, 354–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanov, A.; Janovák, L.; Lantos, I.; Endrész, V.; Sebők, D.; Szabó, T.; Dékány, I.; Deák, J.; Rázga, Z.; Burián, K.; et al. Nonactivated titanium-dioxide nanoparticles promote the growth of Chlamydia trachomatis and decrease the antimicrobial activity of silver nanoparticles. J. Appl. Microbiol. 2017, 123, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.J.; Ray, C.G.; Sherris, J.C. Sherris Medical Microbiology: An Introduction to Infectious Diseases; McGraw-Hill: New York, NY, USA, 2004. [Google Scholar]
- Buglyó, P.; Parajdi-Losonczi, P.L.; Bényei, A.C.; Lihi, N.; Bíró, L.; Farkas, E. Versatility of coordination modes in complexes of monohydroxamic acids with half-sandwich type ruthenium, rhodium, osmium and iridium cations. Chem. Sel. 2017, 2, 8127–8136. [Google Scholar] [CrossRef]
- Mészáros, J.P.; Poljarevic, J.M.; Gál, G.T.; May, N.V.; Spengler, G.; Enyedy, É.A. Comparative solution and structural studies of half-sandwich rhodium and ruthenium complexes bearing curcumin and acetylacetone. J. Inorg. Biochem. 2019, 195, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Bíró, L.; Farkas, E.; Buglyó, P. Complex formation between Ru(η6-p-cym)(H2O)3]2+ and (O,O) donor ligands with biological relevance in aqueous solution. Dalton Trans. 2010, 39, 10272–10278. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Sija, É.; Jakusch, T.; Hartinger, C.G.; Kandioller, W.; Keppler, B.K.; Kiss, T. Solution equilibria of anticancer ruthenium(II)-(η6-p-cymene)-hydroxy(thio)pyr(id)one complexes: Impact of sulfur vs. oxygen donor systems on the speciation and bioactivity. J. Inorg. Biochem. 2013, 127, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dömötör, O.; Pape, V.F.S.; May, N.V.; Szakács, G.; Enyedy, É.A. Comparative solution equilibrium studies of antitumor ruthenium(η6-p-cymene) and rhodium(η5-C5Me5) complexes of 8-hydroxyquinolines. Dalton Trans. 2017, 46, 4382–4396. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, P.J.; Poljarevic, J.M.; Szatmári, I.; Csuvik, O.; Fülöp, F.; Szoboszlai, M.; Spengler, G.; Enyedy, É.A. An 8-hydroxyquinoline-proline hybrid with multidrug resistance reversal activity and solution chemistry of its half-sandwich organometallic Ru and Rh complexes. Dalton Trans. 2020, 49, 7977–7992. [Google Scholar] [CrossRef]
- Sija, É.; Hartinger, C.G.; Keppler, B.K.; Kiss, T.; Enyedy, É.A. Solution equilibrium studies of anticancer ruthenium(II)-η6-p-cymene complexes of pyridinecarboxylic acids. Polyhedron 2014, 67, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Jakusch, T.; Gajda-Schrantz, K.; Adachi, Y.; Sakurai, H.; Kiss, T.; Horváth, L. Solution equilibrium characterization of insulin-mimetic Zn(II) complexes. J. Inorg. Biochem. 2006, 100, 1521–1526. [Google Scholar] [CrossRef]
- Marković, K.; Milačič, R.; Marković, S.; Kladnik, J.; Turel, I.; Ščančar, J. Binding kinetics of ruthenium pyrithione chemotherapeutic candidates to human serum proteins studied by HPLC-ICP-MS. Molecules 2020, 25, 1512. [Google Scholar] [CrossRef] [Green Version]
- Poljarević, J.M.; Tamás, G.G.; May, N.V.; Spengler, G.; Dömötör, O.; Savić, A.R.; Grgurić-Šipka, S.; Enyedy, É.A. Comparative solution equilibrium and structural studies of half-sandwich ruthenium(II)(η6-toluene) complexes of picolinate derivatives. J. Inorg. Biochem. 2018, 181, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Elsadek, B.; Kratz, F. Impact of albumin on drug delivery—New applications on the horizon. J. Control. Release 2012, 157, 4–28. [Google Scholar] [CrossRef]
- Dömötör, O.; Enyedy, É.A. Binding mechanisms of half-sandwich Rh(III) and Ru(II) arene complexes on human serum albumin: A comparative study. J. Biol. Inorg. Chem. 2019, 24, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Activation mechanisms for organometallic anticancer complexes. Top Organomet. Chem. 2010, 32, 21–56. [Google Scholar]
- Briš, A.; Jašík, J.; Turel, I.; Roithová, J. Anti-cancer organoruthenium(II) complexes and their interactions with cysteine and its analogues. A mass-spectrometric study. Dalton Trans. 2019, 48, 2626–2634. [Google Scholar] [CrossRef]
- Sheng, Y.; Hou, Z.; Cui, S.; Cao, K.; Yuan, S.; Sun, M.; Kljun, J.; Huang, G.; Turel, I.; Liu, Y. Covalent versus non-covalent binding of ruthenium η6-p-cymene complexes to zinc-finger protein NCp7. Chem. Eur. J. 2019, 25, 12789–12794. [Google Scholar] [CrossRef]
- Hassan, M.; Azzazy, E.; Christenson, H.R. All About Albumin: Biochemistry, Genetics, and Medical Applications; Peters, T., Jr., Ed.; Academic Press: San Diego, CA, USA, 1996; p. 432. [Google Scholar]
- Dömötör, O.; Hartinger, C.G.; Bytzek, A.K.; Kiss, T.; Keppler, B.K.; Enyedy, E.A. Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. J. Biol. Inorg. Chem. 2013, 18, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Daigle, D.J.; Decuir, T.J.; Robertson, J.B.; Darensbourg, D.J. 1,3,5-Triaz-7-phosphatricyclo[3.3.1.13,7]decane and derivatives. In Inorganic Syntheses; Darensbourg, M.Y., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1998; Volume 32, pp. 40–45. [Google Scholar]
- Beaven, G.H.; Chen, S.-H.; D’albis, A.; Gratzer, W.B. A Spectroscopic study of the haemin-human-serum-albumin system. Eur. J. Biochem. 1974, 42, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- SCQuery, The IUPAC Stability Constants Database, Academic Software, version 5.5; Royal Society of Chemistry: London, UK, 1993.
- Zékány, L.; Nagypál, I. Computational Methods for the Determination of Stability Constants; Leggett, D.L., Ed.; Plenum Press: New York, NY, USA, 1985; pp. 291–353. [Google Scholar]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A novel design of artificial membrane for improving the PAMPA model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef]
- Yu, H.; Wang, Q.; Sun, Y.; Shen, M.; Li, H.; Duan, Y. A new PAMPA model proposed on the basis of a synthetic phospholipid membrane. PLoS ONE 2015, 10, e0116502. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006. [Google Scholar]
- CLSI. Susceptibility testing process. In Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 10th ed.; Christopher, P.J., Polgar, E.P., Eds.; Clinical and Laboratory Standards Institute: Wayne, MI, USA, 2015; Volume 32, pp. 15–19. [Google Scholar]
- Sabet, S.F.; Simmons, J.; Caldwell, H.D. Enhancement of Chlamydia trachomatis infectious progeny by cultivation of HeLa 229 cells treated with DEAE-dextran and cycloheximide. J. Clin. Microbiol. 1984, 20, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, A.; Endrész, V.; Urbán, S.; Lantos, I.; Deák, J.; Burián, K.; Önder, K.; Ayaydin, F.; Balázs, P.; Virok, D.P. Application of DNA chip scanning technology for automatic detection of Chlamydia trachomatis and Chlamydia pneumoniae inclusions. Antimicrob. Agents Chemother. 2014, 58, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Blaho, J.A.; Morton, E.R.; Yedowitz, J.C. Herpes simplex virus: Propagation, quantification, and storage. In Current Protocols in Microbiology; Coico, R., McBride, A., Quarles, J.M., Stevenson, B., Taylor, R.K., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 14E.1.1–14E.1.23. [Google Scholar]
- Mucsi, I.; Molnár, J.; Motohashi, N. Combination of benzo[a]phenothiazines with acyclovir against herpes simplex virus. Int. J. Antimicrob. Agents 2001, 18, 67–72. [Google Scholar] [CrossRef]
- Eszik, I.; Lantos, I.; Önder, K.; Somogyvári, F.; Burián, K.; Endrész, V.; Virok, D.P. High dynamic range detection of Chlamydia trachomatis growth by direct quantitative PCR of the infected cells. J. Microbiol. Methods 2016, 120, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virók, D.P.; Eszik, I.; Mosolygó, T.; Önder, K.; Endrész, V.; Burián, K. A direct quantitative PCR-based measurement of herpes simplex virus susceptibility to antiviral drugs and neutralizing antibodies. J. Virol. Methods 2017, 242, 46–52. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | S.I. | ||||
|---|---|---|---|---|---|
| Colo 205 | Colo 320 | MRC-5 | MRC-5/Colo 205 | MRC-5/Colo 320 | |
| 1 | 14.04 ± 0.62 | 3.3 ± 1.3 | 2.17 ± 0.22 | 0.15 | 0.66 |
| 2 | >100 | >100 | >100 | – | – |
| 3 | 17.3 ± 3.1 | 10.7 ±1.5 | 7.6 ± 1.6 | 0.44 | 0.71 |
| 4 | >100 | >100 | 81.0 ± 6.2 | <0.81 | <0.81 |
| 5 | 21.0 ± 2.4 | 13.74 ± 0.85 | 2.95 ± 0.71 | 0.14 | 0.21 |
| 6 | >100 | >100 | >100 | – | – |
| 7 | 52.4 ± 2.7 | 29.1 ± 2.0 | 26.5 ± 3.2 | 0.51 | 0.91 |
| 8 | 80.6 ± 1.1 | 47.8 ± 7.3 | 17.6 ± 1.5 | 0.22 | 0.37 |
| RAPTA-C | >100 | >100 | >100 | – | – |
| cisplatin | 29.8 ± 1.2 | 5.58 ± 0.70 | 0.88 ± 0.09 | 0.03 | 0.16 |
| MIC (μM) | S. aureus1 | E. faecalis 2 | E. coli 3 | K. pneumoniae4 |
|---|---|---|---|---|
| Gram-Positive | Gram-Negative | |||
| 1 | 50 | 100 | >100 | >100 |
| 2 | >100 | >100 | >100 | >100 |
| 3 | 12.5 | 12.5 | >100 | >100 |
| 4 | >100 | >100 | >100 | >100 |
| 5 | 25 | 12.5 | >100 | >100 |
| 6 | 100 | >100 | >100 | >100 |
| 7 | 50 | 50 | >100 | >100 |
| 8 | 50 | 100 | >100 | >100 |
| RAPTA-C | >100 | >100 | >100 | >100 |
| cisplatin | >100 | >100 | >100 | >100 |
| Compound | pKa | Method | c |
|---|---|---|---|
| HPYR | 4.52 ± 0.04 1 | pH-potentiometry | 1.3 mM |
| HHiQT | 4.63 ± 0.08 | UV-vis | 27 μM |
| 1 | 10.37 ± 0.06 | pH-potentiometry | 1.30 mM |
| 10.34 ± 0.03 | UV-vis | 250 μM | |
| 3 | 10.29 ± 0.09 | pH-potentiometry 2 | 0.6 mM |
| 10.25 ± 0.03 | UV-vis | 250 μM |
| c(KCl) | 1 | 2 | 3 | |
|---|---|---|---|---|
| logD7.4 | 4 mM | −0.43 ± 0.09 | n.d. | +0.92 ± 0.06 |
| logD7.4 | 24 mM | −0.10 ± 0.05 | n.d. | +1.22 ± 0.04 |
| logD7.4 | 100 mM | +0.31 ± 0.03 | n.d. | +1.37 ± 0.06 |
| Peff (cm/s) | 100 mM | 1.13 × 10−6 | 2.88 × 10−6 | 3.35 × 10−6 |
| recovery | 1.3% | 15% | 8.1% |
| logKQ’ | logKWF’ | logKDG’ | ||
|---|---|---|---|---|
| 1 | 5.81 ± 0.03 | (titration) | 6.16 ± 0.03 | 5.80 ± 0.03 |
| 2 | <4 | (titration) | – | – |
| 3 | 6.18 ± 0.03 | (titration) | 5.98 ± 0.03 | 5.61 ± 0.03 |
| 4 | 4.46 ± 0.03 5.39 ± 0.03 | (titration) (48 h) | – | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pivarcsik, T.; Tóth, G.; Szemerédi, N.; Bogdanov, A.; Spengler, G.; Kljun, J.; Kladnik, J.; Turel, I.; Enyedy, É.A. Comparison of Solution Chemical Properties and Biological Activity of Ruthenium Complexes of Selected β-Diketone, 8-Hydroxyquinoline and Pyrithione Ligands. Pharmaceuticals 2021, 14, 518. https://doi.org/10.3390/ph14060518
Pivarcsik T, Tóth G, Szemerédi N, Bogdanov A, Spengler G, Kljun J, Kladnik J, Turel I, Enyedy ÉA. Comparison of Solution Chemical Properties and Biological Activity of Ruthenium Complexes of Selected β-Diketone, 8-Hydroxyquinoline and Pyrithione Ligands. Pharmaceuticals. 2021; 14(6):518. https://doi.org/10.3390/ph14060518
Chicago/Turabian StylePivarcsik, Tamás, Gábor Tóth, Nikoletta Szemerédi, Anita Bogdanov, Gabriella Spengler, Jakob Kljun, Jerneja Kladnik, Iztok Turel, and Éva A. Enyedy. 2021. "Comparison of Solution Chemical Properties and Biological Activity of Ruthenium Complexes of Selected β-Diketone, 8-Hydroxyquinoline and Pyrithione Ligands" Pharmaceuticals 14, no. 6: 518. https://doi.org/10.3390/ph14060518
APA StylePivarcsik, T., Tóth, G., Szemerédi, N., Bogdanov, A., Spengler, G., Kljun, J., Kladnik, J., Turel, I., & Enyedy, É. A. (2021). Comparison of Solution Chemical Properties and Biological Activity of Ruthenium Complexes of Selected β-Diketone, 8-Hydroxyquinoline and Pyrithione Ligands. Pharmaceuticals, 14(6), 518. https://doi.org/10.3390/ph14060518