
Synthesis, Characterization and DNA-Binding Affinity of a New Zinc(II) Bis(5-methoxy-indol-3-yl)propane-1,3-dione Complex

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Spectroscopic and Structural Studies
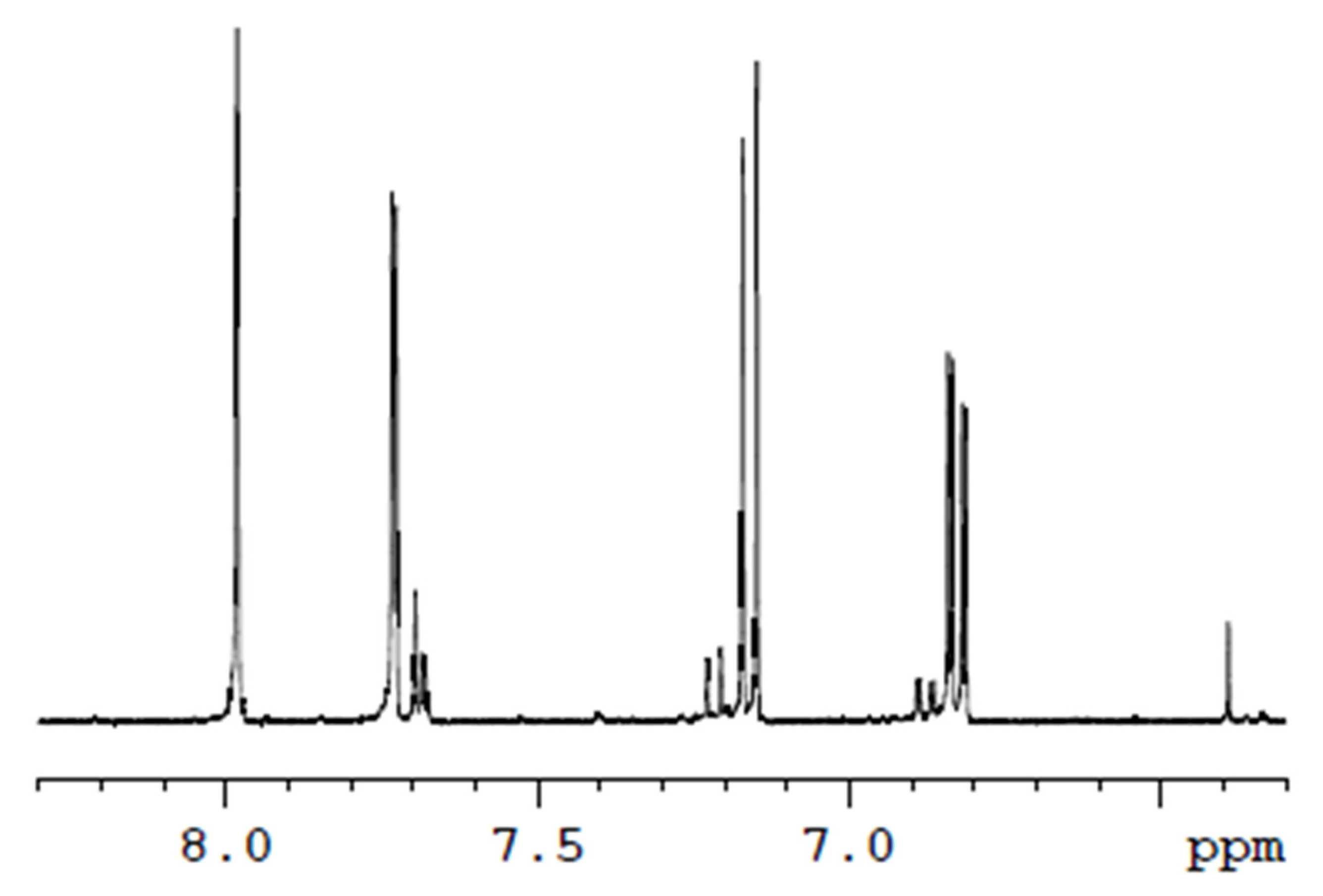
2.2.1. NMR Spectroscopy
2.2.2. IR Spectroscopy
2.2.3. Electronic-State Transition Spectroscopy
UV-Vis Absorption in Organic Solvents
Steady-State Fluorescence in Organic Solvents
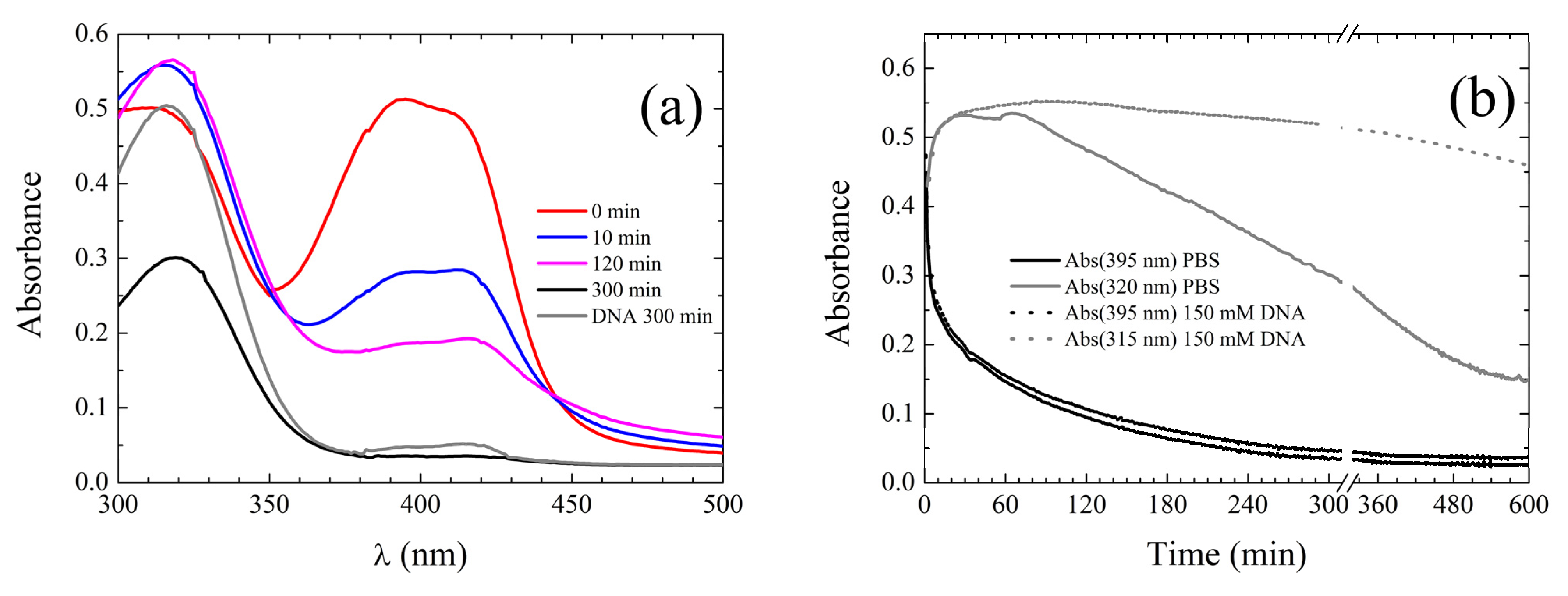
Spectroscopic Properties in Aqueous Environment
Binding to DNA
2.2.4. Powder X-ray Diffraction Studies
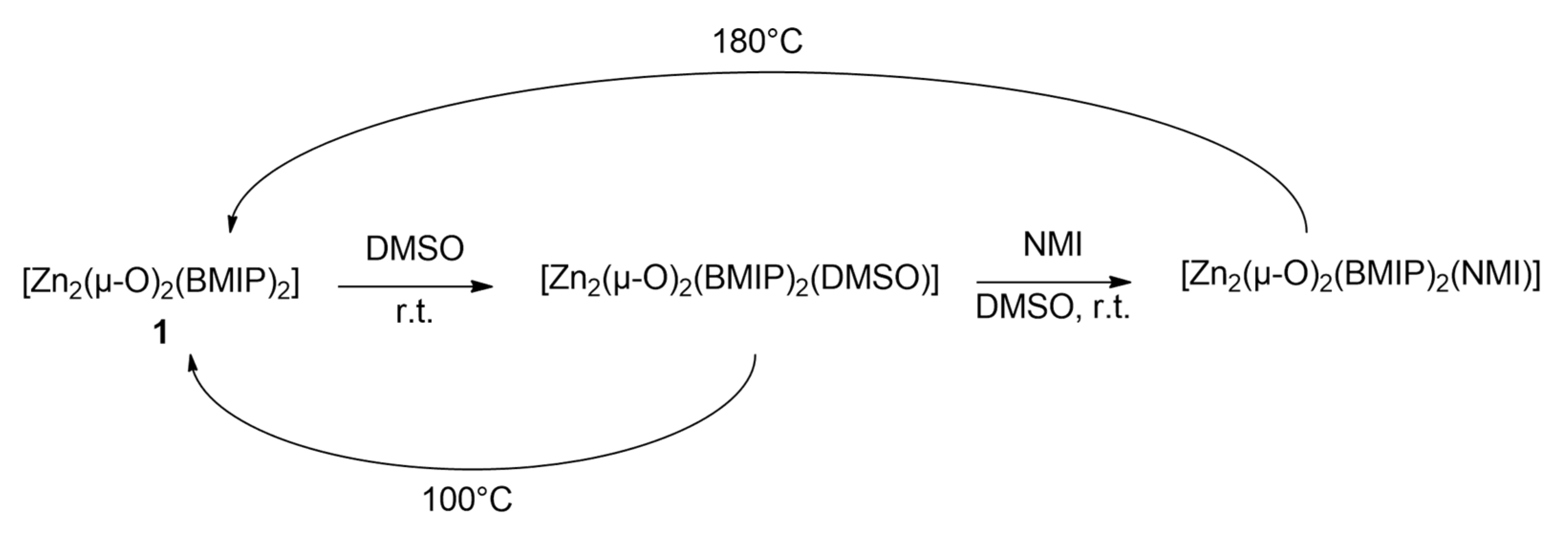
2.3. Coordination with DMSO and NMI
3. Materials and Methods
3.1. Chemicals and Samples Preparation
3.2. Spectroscopy Experiments
3.3. Powder X-ray Diffraction
3.4. Synthesis of 1-methyl-5-methoxyindole
3.5. Synthesis of BMIP
3.6. Synthesis of [Zn2(µ-O)2(BMIP)2]
3.7. Synthesis of [Zn2(µ-O)2(BMIP)2(DMSO)]—Direct Method
3.8. Synthesis of [Zn2(µ-O)2(BMIP)2(DMSO)]—Indirect Method
3.9. Synthesis of [Zn2(µ-O)2(BMIP)2(NMI)]
3.10. Synthesis of [Zn2(µ-O)2(BMIP)2(NMI)]—Ligand Exchange
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 208, 698–699. [Google Scholar] [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Hanif, M.; Hartinger, C.G. Anticancer metallodrugs: Where is the next cisplatin? Future Med. Chem. 2018, 10, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Contel, M. Unconventional anticancer metallodrugs and strategies to improve their pharmacological profile. Inorganics 2019, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Komeda, S.; Casini, A. Next-generation anticancer metallodrugs. Curr. Top. Med. Chem. 2012, 12, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Kljun, J.; Turel, I. β-Diketones as scaffolds for anticancer drug design-from organic building blocks to natural products and metallodrug components. Eur. J. Inorg. Chem. 2017, 2017, 1655–1666. [Google Scholar] [CrossRef] [Green Version]
- Pettinari, R.; Marchetti, F.; Di Nicola, C.; Pettinari, C. Half-sandwich metal complexes with β-Diketone-like ligands and their anticancer activity. Eur. J. Inorg. Chem. 2018, 2018, 3521–3536. [Google Scholar] [CrossRef] [Green Version]
- Shakeri, A.; Panahi, Y.; Johnston, T.P.; Sahebkar, A. Biological properties of metal complexes of curcumin. BioFactors 2019, 45, 304–317. [Google Scholar] [CrossRef]
- Wilson, J.J.; Lippard, S.J. In vitro anticancer activity of cis-diammineplatinum(II) complexes with β-Diketonate leaving group ligands. J. Med. Chem. 2012, 55, 5326–5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, T.; Keppler, K.B.; Heim, M.E.; Niebch, G.; Dietzfelbinger, H.; Rastetter, J.; Hanauske, A.R. Clinical phase I and pharmacokinetic trial of the new titanium complex budotitane. Investig. New Drugs 1995, 13, 327–332. [Google Scholar] [CrossRef]
- Seršen, S.; Kljun, J.; Kryeziu, K.; Panchuk, R.; Alte, B.; Körner, W.; Heffeter, P.; Berger, W.; Turel, I. Structure-related mode-of-action differences of anticancer organoruthenium complexes with β-Diketonates. J. Med. Chem. 2015, 58, 3984–3996. [Google Scholar] [CrossRef]
- Sheikh, J.; Juneja, H.; Ingle, V.; Ali, P.; Ben Hadda, T. Synthesis and in vitro biology of Co(II), Ni(II), Cu(II) and Zinc(II) complexes of functionalized beta-diketone bearing energy buried potential antibacterial and antiviral O,O pharmacophore sites. J. Saudi Chem. Soc. 2013, 17, 269–276. [Google Scholar] [CrossRef] [Green Version]
- dos Santos Portas, A.; Miguel, D.C.; Yokoyama-Yasunaka, J.K.; Uliana, S.R.B.; Espósito, B.P. Increasing the activity of copper(II) complexes against Leishmania through lipophilicity and pro-oxidant ability. J. Biol. Inorg. Chem. 2012, 17, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Gómez, M.E.; Campero-Peredo, C.; García-Conde, D.; Mosqueira-Santillán, M.J.; Serment-Guerrero, J.; Ruiz-Azuara, L. DNA-binding mode of antitumoral copper compounds (Casiopeinas®) and analysis of its biological meaning. Polyhedron 2015, 102, 530–538. [Google Scholar] [CrossRef]
- Deepthi, T.V.; Venugopalan, P. Synthesis, characterization and biological studies on NiII and CuII complexes of two novel α,β-unsaturated 1,3-diketones related to curcuminoids. Inorg. Chim. Acta 2016, 450, 243–250. [Google Scholar] [CrossRef]
- Sousa, L.M.; Souza, W.A.; Paixão, D.A.; Fazzi, R.B.; Tezuka, D.Y.; Lopes, C.D.; Carneiro, Z.A.; Moreira, M.B.; Pivatto, M.; Netto, A.V.G.; et al. DNA binding, cleavage, apoptosis and cytotoxicity studies of three heteroleptic nickel complexes bearing β-diketones. Inorg. Chim. Acta 2020, 511, 119824. [Google Scholar] [CrossRef]
- Inamdar, P.R.; Sheela, A. Exploration of DNA binding mode, chemical nuclease, cytotoxic and apoptotic potentials of diketone based oxovanadium(IV) complexes. Int. J. Biol. Macromol. 2015, 76, 269–278. [Google Scholar] [CrossRef]
- Kolenko, V.; Teper, E.; Kutikov, A.; Uzzo, R. Zinc and zinc transporters in prostate carcinogenesis. Nat. Rev. Urol. 2013, 10, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapiero, H.; Tew, K.D. Trace elements in human physiology and pathology: Zinc and metallothioneins. Biomed. Pharmacother. 2003, 57, 399–411. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Lees, G.J. Zinc Metabolism in the Brain: Relevance to Human Neurodegenerative Disorders. J. Neurobiol. Dis. 1997, 4, 137–169. [Google Scholar] [CrossRef] [Green Version]
- Krebs, N.F.; Hambidge, K.M. Zinc metabolism and homeostasis: The application of tracer techniques to human zinc physiology. BioMetals 2001, 14, 397–412. [Google Scholar] [CrossRef]
- Anjum, R.; Palanimuthu, D.; Kalinowski, D.S.; Lewis, W.; Park, K.C.; Kovacevic, Z.; Khan, I.U.; Richardson, D.R. Synthesis, characterization, and in vitro anticancer activity of copper and zinc bis (thiosemicarbazone) complexes. Inorg. Chem. 2019, 58, 13709–13723. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shen, R.; Wu, J.; Tang, N. Antitumor activity and DNA-binding investigations of the Zn (II) and Cu (II) complexes with isoeuxanthone. Chem. Pharm. Bull. 2009, 57, 814–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.C.; Wei, J.H.; Chen, Z.F.; Liu, M.; Gu, Y.Q.; Bin Huang, K.; Li, Z.Q.; Liang, H. The Antitumor Activity of Zinc(II) and Copper(II) Complexes with 5,7-Dihalo-Substituted-8-Quinolinoline. Eur. J. Med. Chem. 2013, 69, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Tarushi, A.; Psomas, G.; Raptopoulou, C.P.; Kessissoglou, D.P. Zinc complexes of the antibacterial drug oxolinic acid: Structure and DNA-binding properties. J. Inorg. Biochem. 2009, 103, 898–905. [Google Scholar] [CrossRef]
- Maheswari, P.; Barends, S.; Özalp-Yaman, S.; De Hoog, P.; Casellas, H.; Teat, S.; Massera, C.; Lutz, M.; Spek, A.; Van Wezel, G.P.; et al. Unique ligand-based oxidative DNA cleavage by zinc (II) complexes of Hpyramol and Hpyrimol. Chem. A Eur. J. 2007, 13, 5213–5222. [Google Scholar] [CrossRef] [Green Version]
- Dołega, A.; Farmas, A.; Baranowska, K.; Herman, A. Novel zinc complexes with acetyloacetonate, imidazole and thiolate ligands: Crystal structure of a zinc complex of relevance to farnesyl transferase. Inorg. Chem. Commun. 2009, 12, 823–827. [Google Scholar] [CrossRef]
- Malekshah, R.E.; Salehi, M.; Kubicki, M.; Khaleghian, A. Crystal structure, molecular docking, and biological activity of the zinc complexes with 2-thenoyltrifluoroacetone and N-donor heterocyclic ligands. J. Mol. Struct. 2017, 1150, 155–165. [Google Scholar] [CrossRef]
- Mendiguchia, B.S.; Aiello, I.; Crispini, A. Zn (II) and Cu (II) complexes containing bioactive O, O-chelated ligands: Homoleptic and heteroleptic metal-based biomolecules. Dalt. Trans. 2015, 44, 9321–9334. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.-Q.; Liang, C.-J.; Zhou, Z.; Qin, P.Q.; Wei, Z.-Z.; Tan, M.-X.; Liang, H. Mitochondria-localizing curcumin-cryptolepine Zn (II) complexes and their antitumor activity. Bioorg. Med. Chem. 2021, 30, 115948. [Google Scholar] [CrossRef]
- Banerjee, S.; Chakravarty, A.R. Metal complexes of curcumin for cellular imaging, targeting, and photoinduced anticancer activity. Acc. Chem. Res. 2015, 48, 2075–2083. [Google Scholar] [CrossRef]
- Bosi, A.; Banfi, D.; Bistoletti, M.; Giaroni, C.; Baj, A. Tryptophan metabolites along the microbiota-gut-brain axis: An interkingdom communication system influencing the gut in health and disease. Int. J. Tryptophan Res. 2020, 13, 1–25. [Google Scholar] [CrossRef]
- Sharma, V.; Kumar, P.; Pathak, D. Biological importance of the indole nucleus in recent years: A comprehensive review. J. Heterocycl. Chem. 2010, 47, 491–502. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical importance of indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef]
- Han, Y.; Dong, W.; Guo, Q.; Li, X.; Huang, L. The importance of indole and azaindole scaffold in the development of antitumor agents. Eur. J. Med. Chem. 2020, 203, 112506. [Google Scholar] [CrossRef]
- Chen, F.Y.; Li, X.; Zhu, H.P.; Huang, W. Regulation of the Ras-Related Signaling Pathway by Small Molecules Containing an Indole Core Scaffold: A Potential Antitumor Therapy. Front. Pharmacol. 2020, 11, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Bao, G.; Wang, L.; Li, W.; Xu, B.; Du, B.; Lu, J.; Zhai, X.; Gong, P. Design, synthesis, biological evaluation and preliminary mechanism study of novel benzothiazole derivatives bearing indole-based moiety as potent antitumor agents. Eur. J. Med. Chem. 2015, 96, 173–186. [Google Scholar] [CrossRef] [Green Version]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.H.; Cirrincione, G. Synthesis and antitumor activity of 2, 5-bis (3′-indolyl)-furans and 3, 5-bis (3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorganic Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef] [PubMed]
- Ieronimo, G.; Palmisano, G.; Maspero, A.; Marzorati, A.; Scapinello, L.; Masciocchi, N.; Cravotto, G.; Barge, A.; Simonetti, M.; Ameta, K.L.; et al. A novel synthesis of N-hydroxy-3-aroylindoles and 3-aroylindoles. Org. Biomol. Chem. 2018, 16, 6853–6859. [Google Scholar] [CrossRef] [PubMed]
- Scapinello, L.; Maspero, A.; Tollari, S.; Palmisano, G.; Nicholas, K.M.; Penoni, A. A Direct, Regioselective and Atom-Economical Synthesis of 3-Aroyl-N-hydroxy-5-nitroindoles by Cycloaddition of 4-Nitronitrosobenzene with Alkynones. J. Vis. Exp. JoVE 2020, 155, e60201. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, G.; Penoni, A.; Sisti, M.; Tibiletti, F.; Tollari, S.; Nicholas, K.M. Synthesis of indole derivatives with biological activity by reactions between unsaturated hydrocarbons and N-aromatic precursors. Curr. Org. Chem. 2010, 14, 2409–2441. [Google Scholar] [CrossRef]
- Cramer, R.E.; Chudyk, M.A. Acetylacetone complex of zinc perchlorate. Inorg. Chem. 1973, 12, 1193–1195. [Google Scholar] [CrossRef]
- Nakamura, Y.; Isobe, K.; Morita, H.; Yamazaki, S.; Kawaguchi, S. Metal complexes containing acetylacetone as a neutral ligand. Inorg. Chem. 1972, 11, 1573–1578. [Google Scholar] [CrossRef]
- Kawaguchi, S. Variety in the coordination modes of β-dicarbonyl compounds in metal complexes. Coord. Chem. Rev. 1986, 70, 51–84. [Google Scholar] [CrossRef]
- Prochowicz, D.; Sokołowski, K.; Lewiński, J. Zinc hydroxides and oxides supported by organic ligands: Synthesis and structural diversity. Coord. Chem. Rev. 2014, 270–271, 112–126. [Google Scholar] [CrossRef]
- Ruqia, B.; Nam, K.M.; Lee, H.; Lee, G.; Choi, S.I. Facile synthesis of highly crystalline ZnO nanorods with controlled aspect ratios and their optical properties. CrystEngComm 2017, 19, 1454–1458. [Google Scholar] [CrossRef]
- Adati, R.D.; Lima, S.A.M.; Davolos, M.R.; Jafelicci, M. A new β-diketone complex with high color purity. J. Alloy. Compd. 2006, 418, 222–225. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 3rd ed.; Wiley Interscience: New York, NY, USA, 1969. [Google Scholar]
- Nardo, L.; Maspero, A.; Selva, M.; Bondani, M.; Palmisano, G.; Ferrari, E.; Saladini, M. Excited-state dynamics of bis-dehydroxycourcumin carboxylic acid, a water-soluble derivative of the photosensitizer curcumin. J. Phys. Chem. A 2012, 116, 9321–9330. [Google Scholar] [CrossRef] [PubMed]
- Nardo, L.; Maspero, A.; Penoni, A.; Palmisano, G.; Ferrari, E.; Saladini, M. Excited state dynamics of bis-dehydroxycurcumin tert-butyl ester, a diketo-shifted derivative of the photosensitizer curcumin. PLoS ONE 2017, 12, e0175225. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, E.; Pignedoli, F.; Imbrano, C.; Marverti, G.; Basile, V.; Venturi, E.; Saladini, M. Newly synthesized curcumin derivatives: Crosstalk between chemico-physical properties and biological activity. J. Med. Chem. 2011, 54, 8066–8077. [Google Scholar] [CrossRef]
- Chignell, C.F.; Bilskj, P.; Reszka, K.J.; Motten, A.G.; Sik, R.H.; Dahl, T.A. Spectral and photochemical properties of curcumin. Photochem. Photobiol. 1994, 59, 295–302. [Google Scholar] [CrossRef]
- Nardo, L.; Paderno, R.; Andreoni, A.; Masson, M.; Haukvik, T.; Tonnesen, H.H. Role of H-bond formation in the photoreactivity of curcumin. Spectroscopy 2008, 22, 187–198. [Google Scholar] [CrossRef]
- Nardo, L.; Andreoni, A.; Bondani, M.; Masson, M.; Tonnesen, H.H. Studies on curcumin and curcuminoids. XXXIV. Photophysical properties of a symmetrical, non-substituted curcumin analogue. J. Photochem. Photobiol. B Biol. 2009, 97, 77–86. [Google Scholar] [CrossRef]
- Nardo, L.; Andreoni, A.; Masson, M.; Tonnesen, H.H. Photophysical properties of bis-demethoxy-curcumin. J. Fluoresc. 2011, 21, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardo, L.; Andreoni, A.; Bondani, M.; Masson, M.; Haukvik, T.; Tonnesen, H.H. Photophysical properties of dimethoxycurcumin and bis-dehydroxycurcumin. J. Fluoresc. 2012, 22, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Lamperti, M.; Maspero, A.; Tønnesen, H.H.; Bondani, M.; Nardo, L. Elucidation of the relationships between H-bonding patterns and excited state dynamics in Cyclovalone. Molecules 2014, 19, 13282–13304. [Google Scholar] [CrossRef] [Green Version]
- Nardo, L.; Bondani, M.; Andreoni, A. Discrimination of the binding-mode of DNA-ligands by single photon timing. Spectroscopy 2009, 23, 11–28. [Google Scholar] [CrossRef]
- Giani, A.M.; Lamperti, M.; Maspero, A.; Cimino, A.; Negri, R.; Giovenzana, G.B.; Palmisano, G.; Nardo, L. Fluorescence studies on 2-(het)aryl perimidine derivatives. J. Lumin. 2016, 179, 384–392. [Google Scholar] [CrossRef]
- Franks, M.; Gadzhieva, A.; Ghandhi, L.; Murrell, D.; Blake, A.J.; Davies, E.S.; Lewis, W.; Moro, F.; McMaster, J.; Schröder, M. Five coordinate M (II)-diphenolate [M= Zn (II), Ni (II), and Cu (II)] Schiff base complexes exhibiting metal-and ligand-based redox chemistry. Inorg. Chem. 2013, 52, 660–670. [Google Scholar] [CrossRef]
- Song, X.W.; Gao, X.J.; Liu, H.X.; Chen, H.; Chen, C.N. Synthesis and characterization of a supramolecular assembly based on a pyridyl-functionalized [FeFe]-hydrogenase mimic and zinc tetraphenylporphyrin. Inorg. Chem. Commun. 2016, 70, 1–3. [Google Scholar] [CrossRef]
- Che, W.; Yu, T.; Jin, D.; Ren, X.; Zhu, D.; Su, Z.; Bryce, M.R. A simple oxazoline as fluorescent sensor for Zn2+ in aqueous media. Inorg. Chem. Commun. 2016, 69, 89–93. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Ng, Y.M.; Tiong, P.J.; Abu Talip, R.A.; Jasin, N.; Jong, V.Y.M.; Tay, M.G. Five-Coordinate Zinc(II) Complex: Synthesis, Characterization, Molecular Structure, and Antibacterial Activities of Bis-[(E)-2-hydroxy-N’-{1-(4-methoxyphenyl)ethylidene}benzohydrazido]dimethylsulfoxidezinc(II) Complex. Int. J. Inorg. Chem. 2017, 2017, 7520640. [Google Scholar] [CrossRef] [Green Version]
- Evans, I.P.; Spencer, A.; Wilkinson, G. Dichlorotetrakis(dimethyl sulphoxide)ruthenium(II) and its use as a source material for some new ruthenium(II) complexes. J. Chem. Soc. Dalton Trans. 1973, 2, 204–209. [Google Scholar] [CrossRef]
- Berlman, I.B. Handbook of Fluorescence Spectra of Aromatic Molecules; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Rochette, E.; Desrosiers, V.; Soltani, Y.; Fontaine, F.G. Isodesmic C-H Borylation: Perspectives and Proof of Concept of Transfer Borylation Catalysis. J. Am. Chem. Soc. 2019, 141, 12305–12311. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | νC–H | νC–H | νC=O | νC–O–C | νC=N NMI | νC–N NMI | Νρ(Me) DMSO | ν(S=O) DMSO | νZn–O | νZn–O DMSO |
|---|---|---|---|---|---|---|---|---|---|---|
| ligand | 2939 | 1620 | 1210 | |||||||
| 1 | 3020 | 2950 | 1520 | 1260 | 507 | |||||
| 2 | 3020 | 2950 | 1520 | 1260 | 1031 | 961 | 510 | 435 | ||
| 3 | 3020 | 2950 | 1520 | 1260 | 1421 | 1285 | 518 |
| Solvent | λabs,1 (nm) | λabs,2 (nm) | f1 | fDiketo,NMR | <ε> (M−1cm−1) |
|---|---|---|---|---|---|
| DCM | 313 (35,000) | 392 (32,900) | 0.79 | 0.85 | 34,600 |
| DMF | 302 | 392 | 0.57 | 44,100 | |
| DMSO | 309 (41,500) | 393 (43,000) | 0.45 | 0.46 | 42,400 |
| EtOH | 311 | 397 | 0.76 | -- |
| Solvent | λemi,enol (nm) | λemi,diketo (nm) † | λexc,enol (nm) | λexc,diketo (nm) | Φfluo,enol |
|---|---|---|---|---|---|
| DCM | 426 | 403 | 326 (398) | 315 | 0.06 |
| DMF | 424 | 405 | 400 (325) | 280 (309) | 0.08 |
| DMSO | 428 | 403 | 400, 408 (322) | 293, 304 | 0.12 |
| EtOH | 447 | 413 | 401 (325) | 292 | 0.20 |
| Solvent | Kdiss (M) | n (ligands/bp) | F0 (a.u.) | δF (μM−1) |
|---|---|---|---|---|
| DMSO | 0.69 ± 0.32 | 0.26 ± 0.05 | 2.07 ± 0.03 | −0.098 ± 0.004 |
| DMF | 0.87 ± 0.39 | 0.29 ± 0.05 | 1.19 ± 0.02 | −0.057 ± 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scapinello, L.; Vesco, G.; Nardo, L.; Maspero, A.; Vavassori, F.; Galli, S.; Penoni, A. Synthesis, Characterization and DNA-Binding Affinity of a New Zinc(II) Bis(5-methoxy-indol-3-yl)propane-1,3-dione Complex. Pharmaceuticals 2021, 14, 760. https://doi.org/10.3390/ph14080760
Scapinello L, Vesco G, Nardo L, Maspero A, Vavassori F, Galli S, Penoni A. Synthesis, Characterization and DNA-Binding Affinity of a New Zinc(II) Bis(5-methoxy-indol-3-yl)propane-1,3-dione Complex. Pharmaceuticals. 2021; 14(8):760. https://doi.org/10.3390/ph14080760
Chicago/Turabian StyleScapinello, Luca, Guglielmo Vesco, Luca Nardo, Angelo Maspero, Federico Vavassori, Simona Galli, and Andrea Penoni. 2021. "Synthesis, Characterization and DNA-Binding Affinity of a New Zinc(II) Bis(5-methoxy-indol-3-yl)propane-1,3-dione Complex" Pharmaceuticals 14, no. 8: 760. https://doi.org/10.3390/ph14080760
APA StyleScapinello, L., Vesco, G., Nardo, L., Maspero, A., Vavassori, F., Galli, S., & Penoni, A. (2021). Synthesis, Characterization and DNA-Binding Affinity of a New Zinc(II) Bis(5-methoxy-indol-3-yl)propane-1,3-dione Complex. Pharmaceuticals, 14(8), 760. https://doi.org/10.3390/ph14080760