
Early Renoprotective Effect of Ruxolitinib in a Rat Model of Diabetic Nephropathy

 ,
, _Talaat.jpg) , and
, and
Abstract
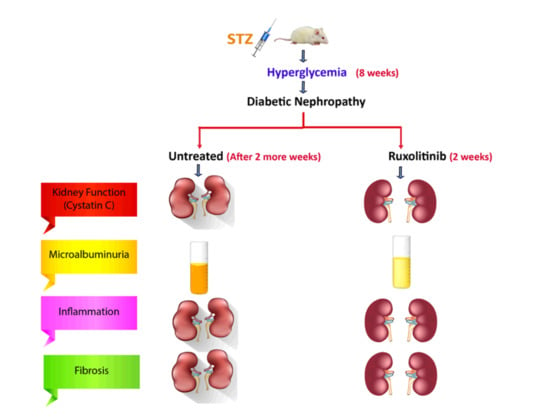
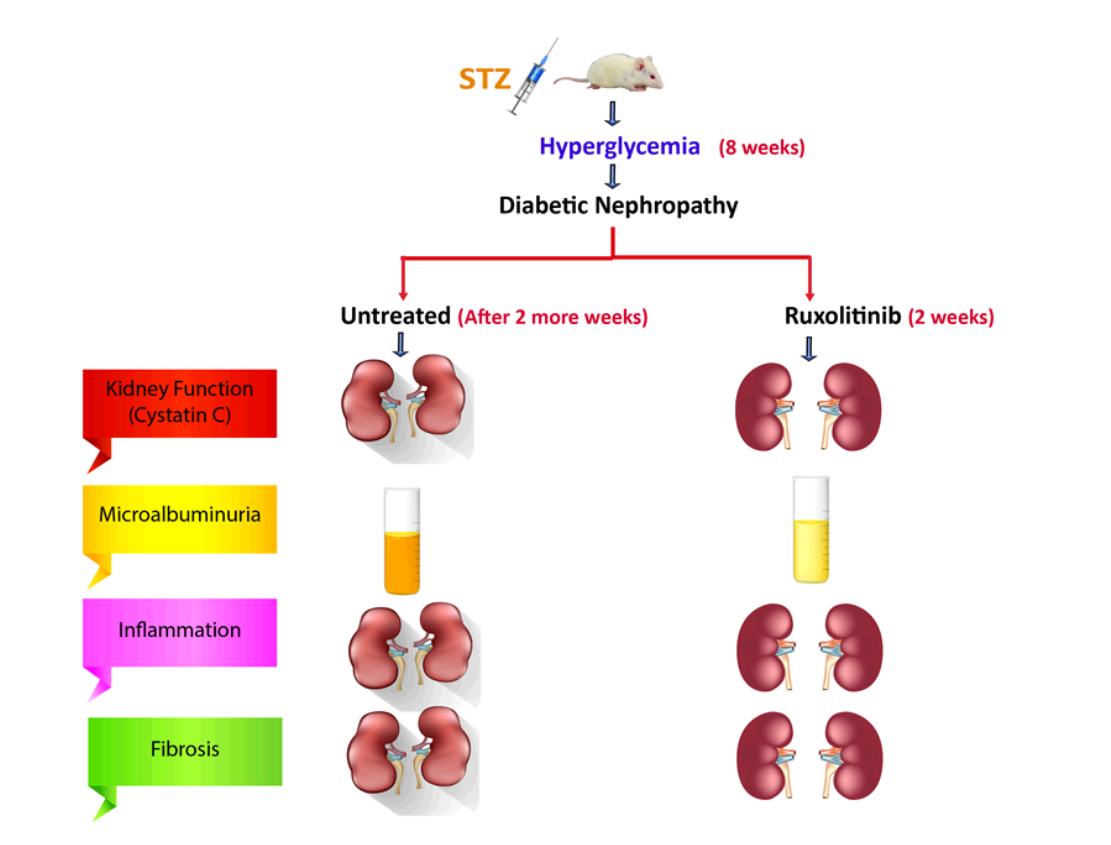
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
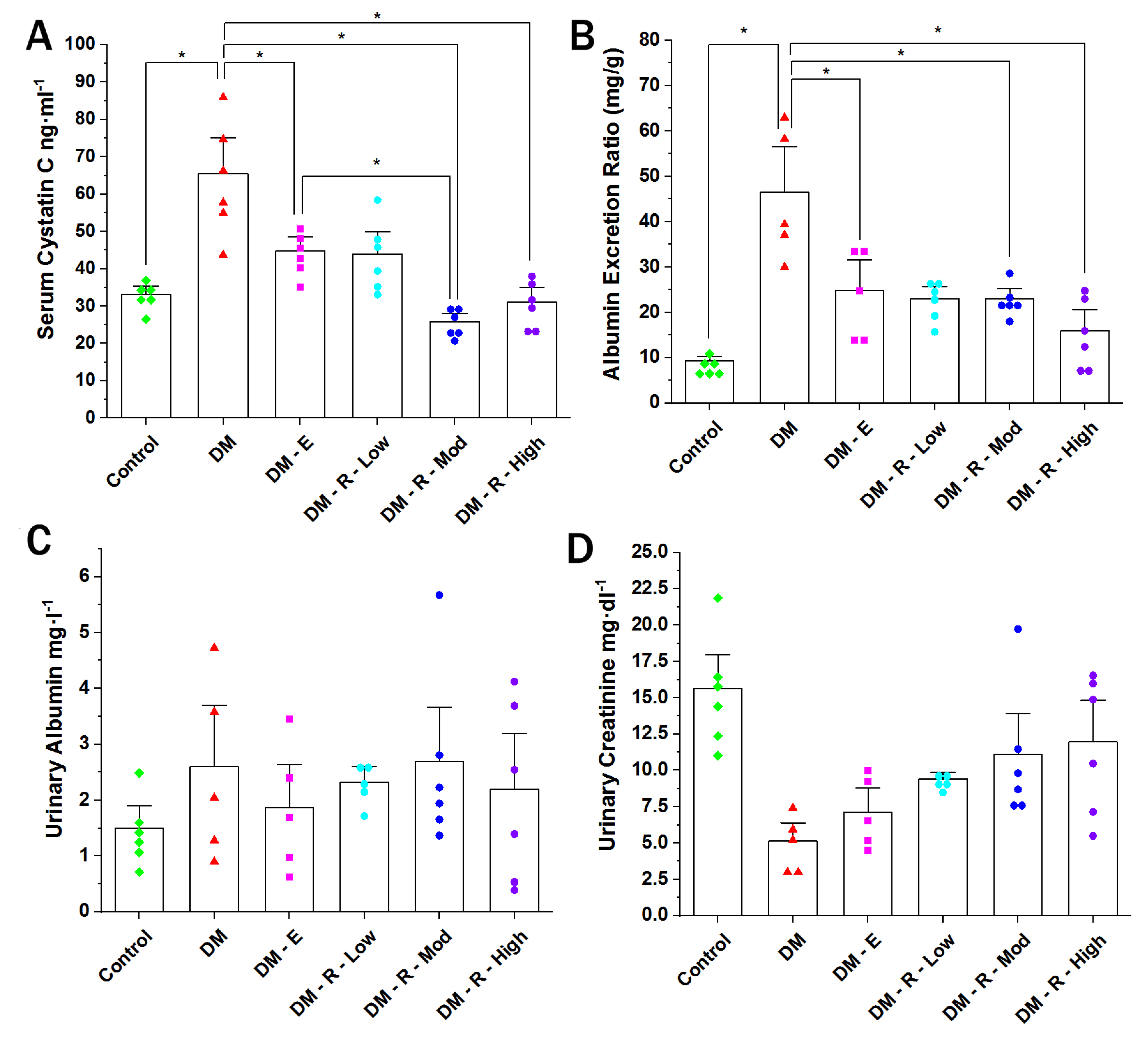
2.1. Effect of Ruxolitinib on Serum Cystatin, Albumin Excretion Ratio, Urinary Albumin, and Urinary Creatinine Levels
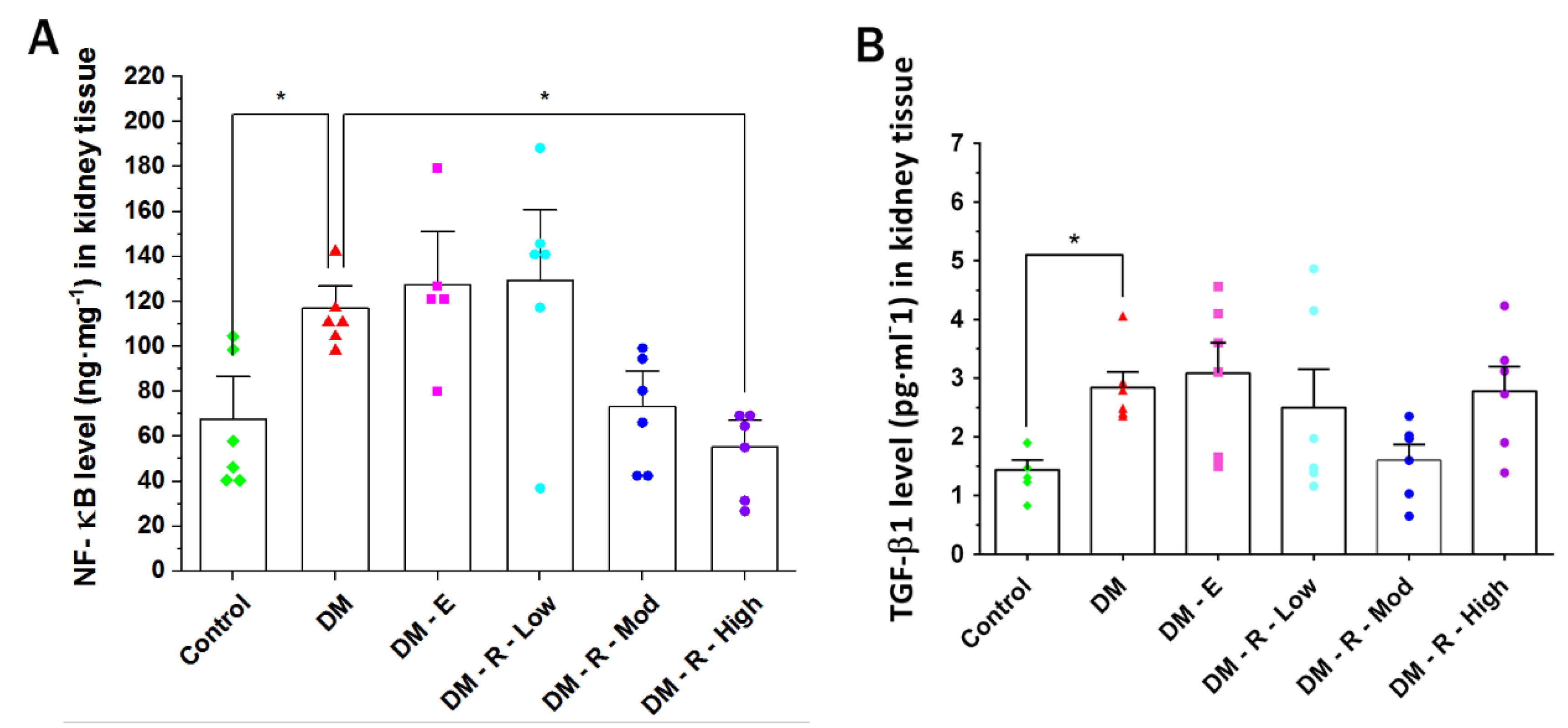
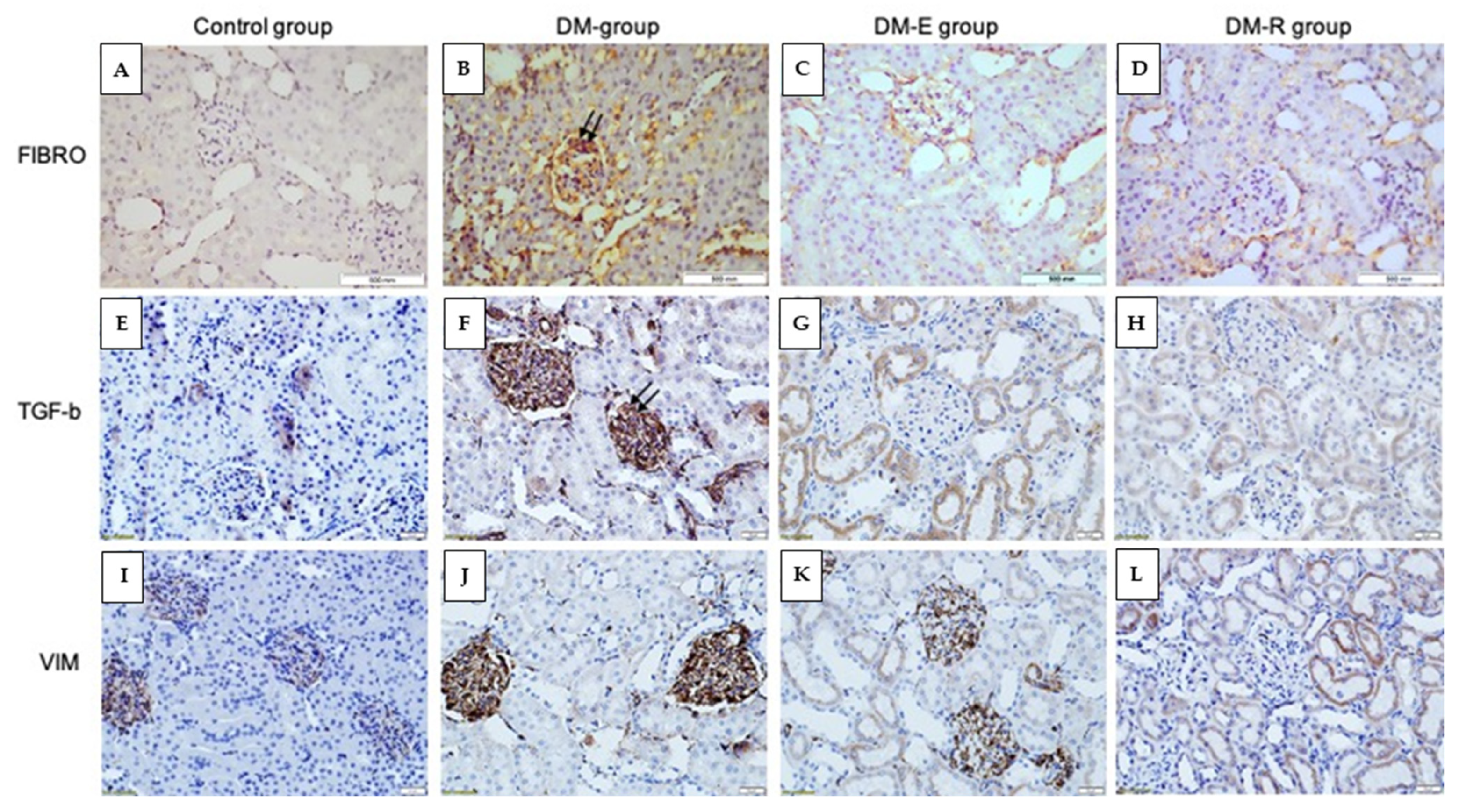
2.2. Effects of Ruxolitinib on Kidney NF-κB and TGF-β1 Levels
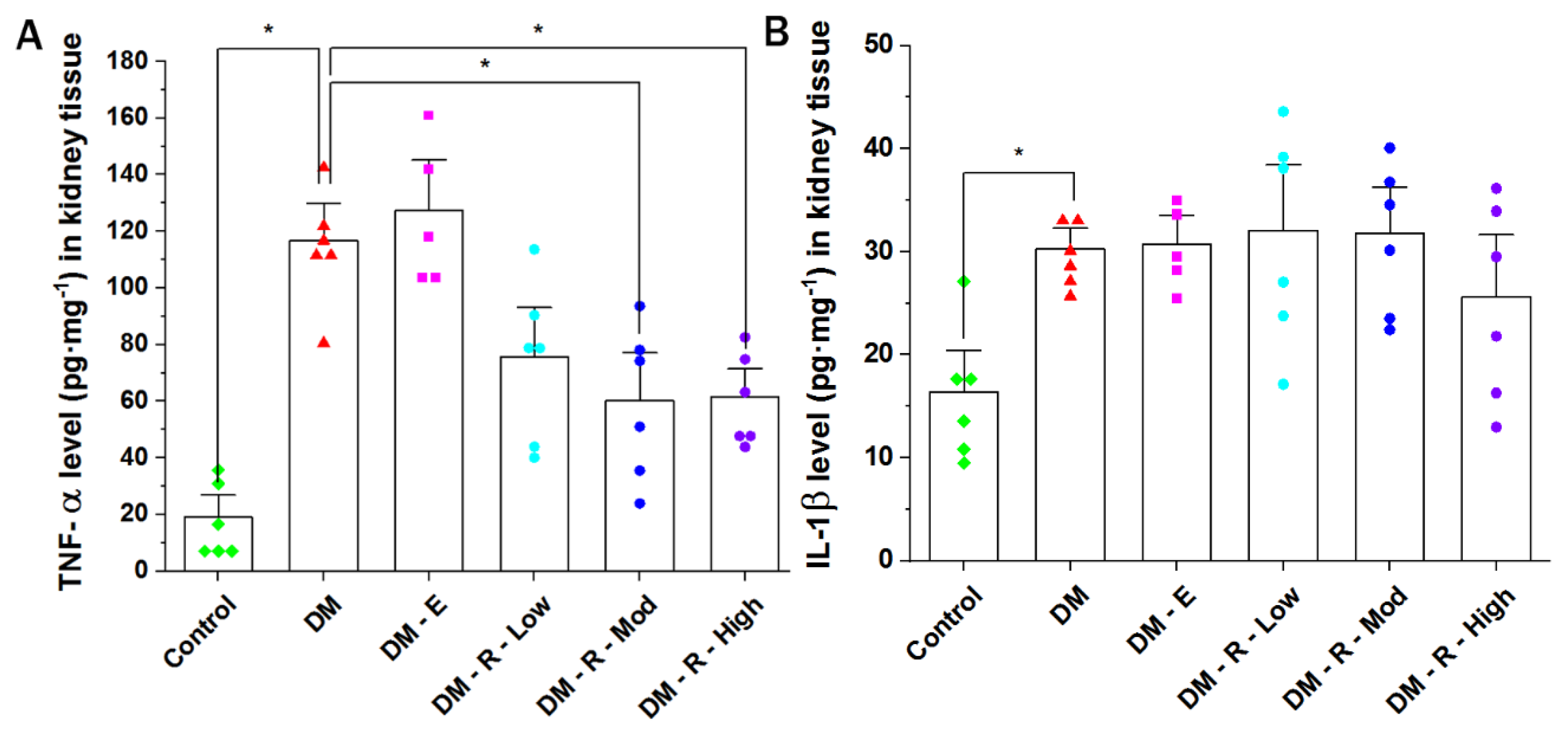
2.3. Effect of Ruxolitinib on Kidney TNF-α and IL-1β Levels
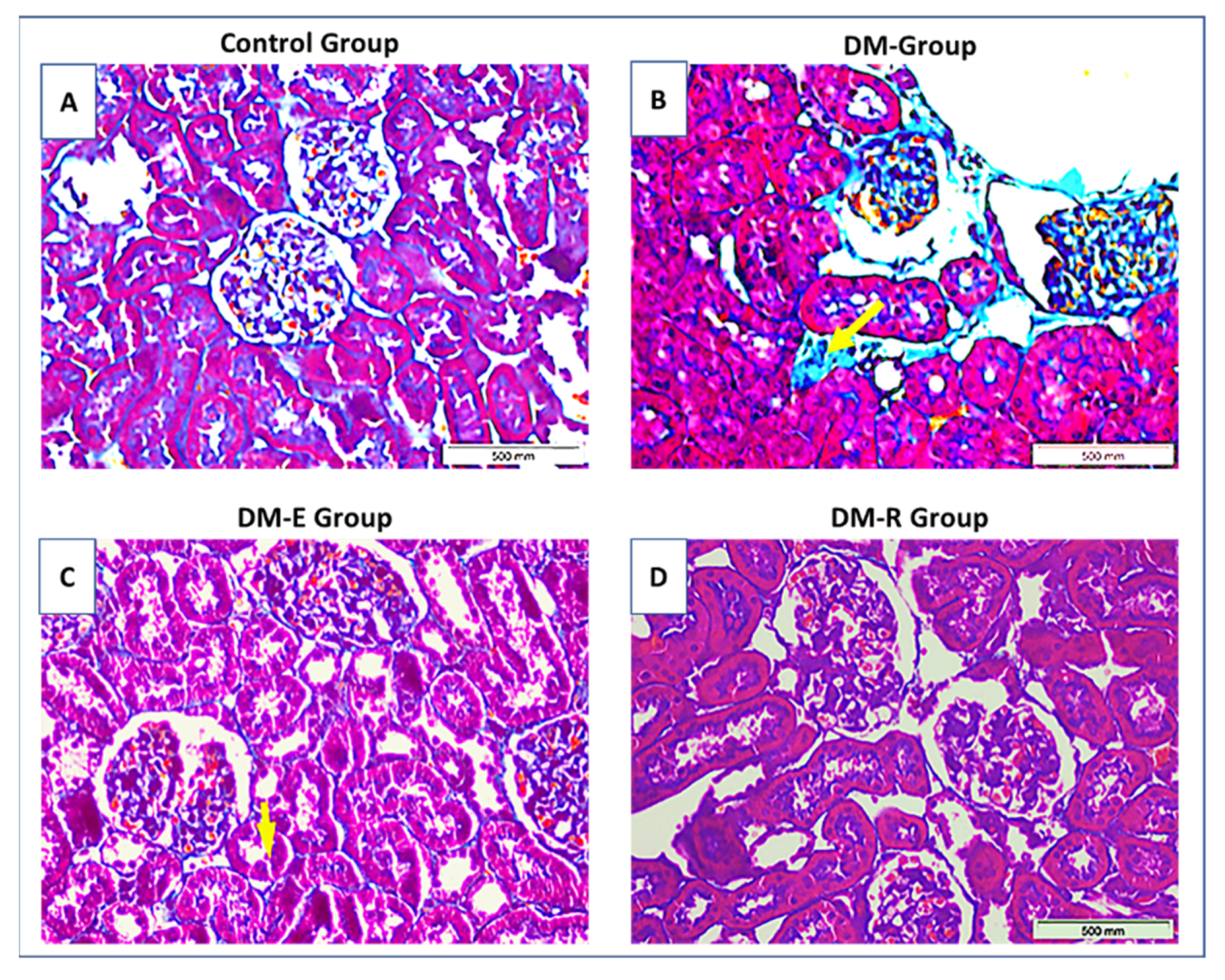
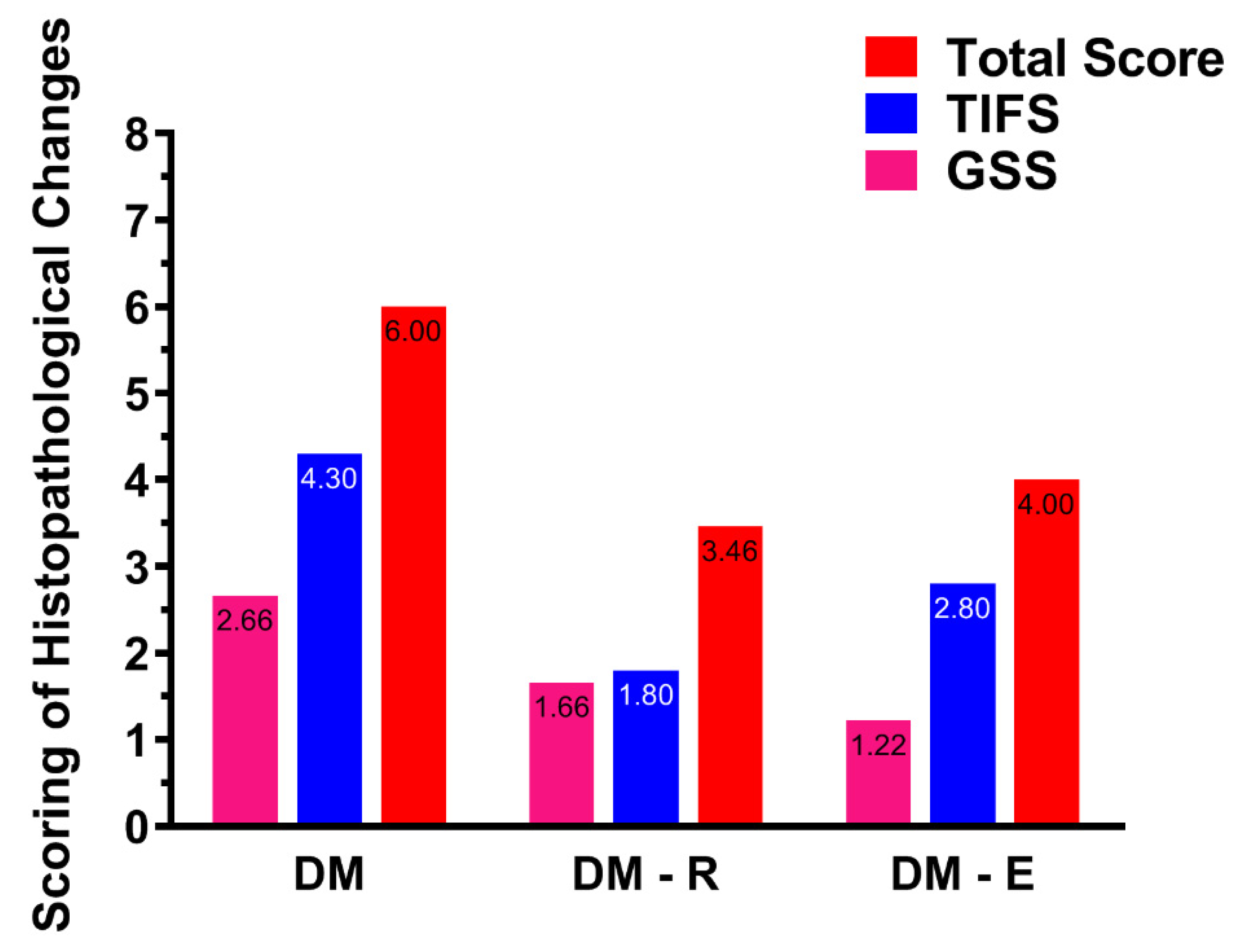
2.4. Effects of Ruxolitinib on Renal Histological Findings in Streptozotocin-Induced Diabetic Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Model Induction
4.3. Animal Groups and Treatment Protocol
4.4. Drugs and Chemicals
4.5. Collecting Biospecimens
4.6. Biochemical Assessment
4.7. Histopathological Evaluation of Renal Changes
4.8. Immunohistochemistry
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, H.-T.; Xu, X.; Lim, P.S.; Hung, K.-Y. Worldwide Epidemiology of Diabetes-Related End-Stage Renal Disease, 2000–2015. Diabetes Care 2021, 44, 89–97. [Google Scholar] [CrossRef]
- Saran, R.; Robinson, B.; Abbott, K.C.; Bragg-Gresham, J.; Chen, X.; Gipson, D.; Gu, H.; Hirth, R.A.; Hutton, D.; Jin, Y.; et al. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2020, 75, A6–A7. [Google Scholar] [CrossRef]
- Sharma, D.; Bhattacharya, P.; Kalia, K.; Tiwari, V. Diabetic nephropathy: New insights into established therapeutic paradigms and novel molecular targets. Diabetes Res. Clin. Pr. 2017, 128, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Komers, R. Pathophysiology of the Diabetic Kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [CrossRef]
- Bonner, R.; Albajrami, O.; Hudspeth, J.; Upadhyay, A. Diabetic Kidney Disease. Prim. Care Clin. Off. Pr. 2020, 47, 645–659. [Google Scholar] [CrossRef]
- Nishiyama, A.; Seth, D.M.; Navar, L.G. Renal Interstitial Fluid Concentrations of Angiotensins I and II in Anesthetized Rats. Hypertension 2002, 39, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gembillo, G.; Cernaro, V.; Salvo, A.; Siligato, R.; Laudani, A.; Buemi, M.; Santoro, D. Role of Vitamin D Status in Diabetic Patients with Renal Disease. Medicina 2019, 55, 273. [Google Scholar] [CrossRef] [Green Version]
- Ziyadeh, F.N.; Wolf, G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr. Diabetes Rev. 2008, 4, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Barma, S.; Konwar, N.; Dewanjee, S.; Manna, P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur. J. Pharmacol. 2016, 791, 8–24. [Google Scholar] [CrossRef]
- Qi, W.; Twigg, S.; Chen, X.; Polhill, T.S.; Poronnik, P.; Gilbert, R.E.; Pollock, C.A. Integrated actions of transforming growth factor-β1 and connective tissue growth factor in renal fibrosis. Am. J. Physiol. Renal. Physiol. 2005, 288, F800–F809. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, I.; Wolf, G. Mechanisms of Interstitial Fibrosis in Diabetic Nephropathy; Springer: Cham, Switzerland, 2018; pp. 227–251. [Google Scholar]
- Fuchs, S.; Xiao, H.D.; Cole, J.M.; Adams, J.W.; Frenzel, K.; Michaud, A.; Zhao, H.; Keshelava, G.; Capecchi, M.R.; Corvol, P.; et al. Role of the N-terminal Catalytic Domain of Angiotensin-converting Enzyme Investigated by Targeted Inactivation in Mice. J. Biol. Chem. 2004, 279, 15946–15953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, C.A.; Kumar, N.; Nakagawa, P.; Worou, M.E.; Liao, T.-D.; Peterson, E.L.; Carretero, O.A. Renal release of N-acetyl-seryl-aspartyl-lysyl-proline is part of an antifibrotic peptidergic system in the kidney. Am. J. Physiol. Physiol. 2019, 316, F195–F203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Y.; Chun, B.; Potthoff, S.A.; Kazi, N.; Brolin, T.J.; Orhan, D.; Yang, H.-C.; Ma, L.-J.; Kon, V.; Myöhänen, T.; et al. Thymosin β4 and its degradation product, Ac-SDKP, are novel reparative factors in renal fibrosis. Kidney Int. 2013, 84, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S.P.; Koya, D. Linagliptin-Mediated DPP-4 Inhibition Ameliorates Kidney Fibrosis in Streptozotocin-Induced Diabetic Mice by Inhibiting Endothelial-to-Mesenchymal Transition in a Therapeutic Regimen. Diabetes 2014, 63, 2120–2131. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.P.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Metabolic reprogramming by N -acetyl-seryl-aspartyl-lysyl-proline protects against diabetic kidney disease. Br. J. Pharmacol. 2020, 177, 3691–3711. [Google Scholar] [CrossRef]
- Remuzzi, G.; Macia, M.; Ruggenenti, P. Prevention and Treatment of Diabetic Renal Disease in Type 2 Diabetes: The BENEDICT Study. J. Am. Soc. Nephrol. 2006, 17, S90–S97. [Google Scholar] [CrossRef]
- Bakris, G.L.; Weir, M.R.; Shanifar, S.; Zhang, Z.; Douglas, J.; Van Dijk, D.J.; Brenner, B.M. Effects of Blood Pressure Level on Progression of Diabetic NephropathyResults From the RENAAL Study. Arch. Intern. Med. 2003, 163, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Barnett, A.H.; Bain, S.C.; Bouter, P.; Karlberg, B.; Madsbad, S.; Jervell, J.; Mustonen, J. Angiotensin-Receptor Blockade versus Converting–Enzyme Inhibition in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2004, 351, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Bjorck, S.; Mulec, H.; Johnsen, S.A.; Norden, G.; Aurell, M. Renal protective effect of enalapril in diabetic nephropathy. BMJ 1992, 304, 339–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perico, N.; Ruggenenti, P.; Remuzzi, G. ACE and SGLT2 inhibitors: The future for non-diabetic and diabetic proteinuric renal disease. Curr. Opin. Pharmacol. 2017, 33, 34–40. [Google Scholar] [CrossRef]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef] [Green Version]
- Hattori, S. Sitagliptin reduces albuminuria in patients with type 2 diabetes [Rapid Communication]. Endocr. J. 2011, 58, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Ke, Q.; Fang, Y.; Wen, P.; Chen, H.; Yuan, Q.; Luo, J.; Zhang, Y.; Sun, Q.; Lv, Y.; et al. Sodium–glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020, 11, 390. [Google Scholar] [CrossRef]
- Nagai, T.; Kanasaki, M.; Srivastava, S.P.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Shi, S.; Kanasaki, K.; Koya, D. N-acetyl-seryl-aspartyl-lysyl-proline Inhibits Diabetes-Associated Kidney Fibrosis and Endothelial-Mesenchymal Transition. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Jiang, L.; Xu, J.; Bai, F.; Zhou, Y.; Yuan, Q.; Luo, J.; Zen, K.; Yang, J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Physiol. 2017, 313, F561–F575. [Google Scholar] [CrossRef] [Green Version]
- Afkarian, M.; Zelnick, L.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; De Boer, I.H. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988–2014. JAMA 2016, 316, 602–610. [Google Scholar] [CrossRef]
- Thomas, M.C.; Groop, P.-H. New approaches to the treatment of nephropathy in diabetes. Expert Opin. Investig. Drugs 2011, 20, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Pak, W.L.W.; Tanaka, T.; Tang, S.C.W.; Nangaku, M. Update on diagnosis, pathophysiology, and management of diabetic kidney disease. Nephrology 2021, 26, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Jiang, P.; Zhou, L.; Sun, X.; Bi, J.; Cui, L.; Nie, X.; Luo, R.; Zhao, X.; Liu, Y. Additive Effect of Qidan Dihuang Grain, a Traditional Chinese Medicine, and Angiotensin Receptor Blockers on Albuminuria Levels in Patients with Diabetic Nephropathy: A Randomized, Parallel-Controlled Trial. Evid. Based Complement. Altern. Med. 2016, 2016, 1064924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivian, E.; Mannebach, C. Therapeutic approaches to slowing the progression of diabetic nephropathy—is less best? Drugs Context 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT—Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; et al. Enhanced Expression of Janus Kinase-Signal Transducer and Activator of Transcription Pathway Members in Human Diabetic Nephropathy. Diabetes 2008, 58, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosius, F.C.; Tuttle, K.; Kretzler, M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia 2016, 59, 1624–1627. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Opazo-Ríos, L.; Guerrero-Hue, M.; García-Caballero, C.; Vázquez-Carballo, C.; Mas, S.; Sanz, A.B.; Herencia, C.; Mezzano, S.; et al. Pathogenic Pathways and Therapeutic Approaches Targeting Inflammation in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 3798. [Google Scholar] [CrossRef]
- Sun, M.-Y.; Wang, S.-J.; Li, X.-Q.; Shen, Y.-L.; Lu, J.-R.; Tian, X.-H.; Rahman, K.; Zhang, L.-J.; Nian, H.; Zhang, H. CXCL6 Promotes Renal Interstitial Fibrosis in Diabetic Nephropathy by Activating JAK/STAT3 Signaling Pathway. Front. Pharmacol. 2019, 10, 224. [Google Scholar] [CrossRef]
- Hashimoto, R.; Kakigi, R.; Miyamoto, Y.; Nakamura, K.; Itoh, S.; Daida, H.; Okada, T.; Katoh, Y. JAK-STAT-dependent regulation of scavenger receptors in LPS-activated murine macrophages. Eur. J. Pharmacol. 2020, 871, 172940. [Google Scholar] [CrossRef]
- Riazi, S.; Maric, C.; Ecelbarger, C. 17-β Estradiol attenuates streptozotocin-induced diabetes and regulates the expression of renal sodium transporters. Kidney Int. 2006, 69, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, M.C.; Brunner, H.R.; Foster, C.; Huo, Y. Angiotensin II type 1 receptor antagonists in animal models of vascular, cardiac, metabolic and renal disease. Pharmacol. Ther. 2016, 164, 1–81. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.; Thomas, D.; Harris, P.J.; Skinner, S.L. The role of endogenous angiotensin II in the regulation of renal haemodynamics and proximal fluid reabsorption in the rat. J. Physiol. 1992, 453, 1–13. [Google Scholar] [CrossRef]
- Brodsky, S.; Gurbanov, K.; Abassi, Z.; Hoffman, A.; Ruffolojr, R.R.; Feuerstein, G.Z.; Winaver, J. Effects of Eprosartan on Renal Function and Cardiac Hypertrophy in Rats with Experimental Heart Failure. Hypertension 1998, 32, 746–752. [Google Scholar] [CrossRef] [Green Version]
- La Jeon, Y.; Kim, M.H.; Lee, W.-I.; Kang, S.Y. Cystatin C as an Early Marker of Diabetic Nephropathy in Patients with Type 2 Diabetes. Clin. Lab. 2013, 59, 1221–1229. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Luis-Rodríguez, D.; Martín-Núñez, E.; Tagua, V.G.; Hernández-Carballo, C.; Ferri, C.; Rodríguez-Rodríguez, A.E.; Mora-Fernández, C.; Navarro-González, J.F. Inflammatory Targets in Diabetic Nephropathy. J. Clin. Med. 2020, 9, 458. [Google Scholar] [CrossRef] [Green Version]
- Sanachai, K.; Mahalapbutr, P.; Choowongkomon, K.; Poo-Arporn, R.P.; Wolschann, P.; Rungrotmongkol, T. Insights into the Binding Recognition and Susceptibility of Tofacitinib toward Janus Kinases. ACS Omega 2020, 5, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Pichler, R.; Afkarian, M.; Dieter, B.P.; Tuttle, K. Immunity and inflammation in diabetic kidney disease: Translating mechanisms to biomarkers and treatment targets. Am. J. Physiol. Physiol. 2017, 312, F716–F731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Prim. 2015, 1, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Kim, S.M.; Eisner, C.; Oppermann, M.; Huang, Y.; Mizel, D.; Li, L.; Chen, M.; Lopez, M.L.S.; Weinstein, L.S.; et al. Stimulation of Renin Secretion by Angiotensin II Blockade is Gsα-Dependent. J. Am. Soc. Nephrol. 2010, 21, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Border, W.A.; Noble, N.A. Interactions of Transforming Growth Factor-β and Angiotensin II in Renal Fibrosis. Hypertension 1998, 31, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semino, C.; Carta, S.; Gattorno, M.; Sitia, R.; Rubartelli, A. Progressive waves of IL-1β release by primary human monocytes via sequential activation of vesicular and gasdermin D-mediated secretory pathways. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Gao, T.; Cooper, T.K.; Reeves, W.B.; Awad, A.S. Macrophages directly mediate diabetic renal injury. Am. J. Physiol. Physiol. 2013, 305, F1719–F1727. [Google Scholar] [CrossRef] [PubMed]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Ohshiro, Y.; Ma, R.C.; Yasuda, Y.; Hiraoka-Yamamoto, J.; Clermont, A.C.; Isshiki, K.; Yagi, K.; Arikawa, E.; Kern, T.S.; King, G.L. Reduction of Diabetes-Induced Oxidative Stress, Fibrotic Cytokine Expression, and Renal Dysfunction in Protein Kinase CBeta-Null Mice. Diabetes 2006, 55, 3112–3120. [Google Scholar] [CrossRef] [Green Version]
- Mima, A.; Kitada, M.; Geraldes, P.; Li, Q.; Matsumoto, M.; Mizutani, K.; Qi, W.; Li, C.; Leitges, M.; Rask-Madsen, C.; et al. Glomerular VEGF resistance induced by PKCδ/SHP-1 activation and contribution to diabetic nephropathy. FASEB J. 2012, 26, 2963–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Liu, W.; Lan, T.; Xie, X.; Peng, J.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Berberine Reduces Fibronectin Expression by Suppressing the S1P-S1P2 Receptor Pathway in Experimental Diabetic Nephropathy Models. PLoS ONE 2012, 7, e43874. [Google Scholar] [CrossRef]
- Vansthertem, D.; Gossiaux, A.; Declèves, A.-E.; Caron, N.; Nonclercq, D.; Legrand, A.; Toubeau, G. Expression of Nestin, Vimentin, and NCAM by Renal Interstitial Cells after Ischemic Tubular Injury. J. Biomed. Biotechnol. 2010, 2010, 193259. [Google Scholar] [CrossRef]
- Wang, Z.; Divanyan, A.; Jourd’Heuil, F.L.; Goldman, R.D.; Ridge, K.M.; Jourd’Heuil, D.; Lopez-Soler, R.I. Vimentin expression is required for the development of EMT-related renal fibrosis following unilateral ureteral obstruction in mice. Am. J. Physiol. Physiol. 2018, 315, F769–F780. [Google Scholar] [CrossRef] [PubMed]
- Tervaert, T.W.C.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; De Heer, E.; et al. Pathologic Classification of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Kanasaki, K.; Taduri, G.; Koya, D. Diabetic nephropathy: The role of inflammation in fibroblast activation and kidney fibrosis. Front. Endocrinol. 2013, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Kuno, Y.; Iyoda, M.; Shibata, T.; Hirai, Y.; Akizawa, T. Sildenafil, a phosphodiesterase type 5 inhibitor, attenuates diabetic nephropathy in non-insulin-dependent Otsuka Long-Evans Tokushima Fatty rats. Br. J. Pharmacol. 2011, 162, 1389–1400. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.P.; Hedayat, A.F.; Kanasaki, K.; Goodwin, J.E. microRNA Crosstalk Influences Epithelial-to-Mesenchymal, Endothelial-to-Mesenchymal, and Macrophage-to-Mesenchymal Transitions in the Kidney. Front. Pharmacol. 2019, 10, 904. [Google Scholar] [CrossRef]
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.; Echeverría, C.; Kalergis, A.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Sanz, L.; Bernal, S.; Recio, C.; Lazaro, I.; Oguiza, A.; Melgar, A.; Jimenez-Castilla, L.; Egido, J.; Gomez-Guerrero, C. SOCS1-targeted therapy ameliorates renal and vascular oxidative stress in diabetes via STAT1 and PI3K inhibition. Lab. Investig. 2018, 98, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Park, A.S.D.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome Analysis of Human Diabetic Kidney Disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Payne, C.; Zhang, X.; Shahri, N.; Williams, W.; Cannady, E. AB0492 Evaluation of Potential Drug-Drug Interactions with Baricitinib. Ann. Rheum. Dis. 2015, 74, 1063. [Google Scholar] [CrossRef]
- Amrhein, J.; Drynda, S.; Schlatt, L.; Karst, U.; Lohmann, C.H.; Ciarimboli, G.; Bertrand, J. Tofacitinib and Baricitinib Are Taken up by Different Uptake Mechanisms Determining the Efficacy of Both Drugs in RA. Int. J. Mol. Sci. 2020, 21, 6632. [Google Scholar] [CrossRef]
- Schwartz, D.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Kalender, B.; Öztürk, M.; Tunçdemir, M.; Uysal, Ö.; Dağıstanlı, F.K.; Yeğenağa, I.; Erek, E. Renoprotective effects of valsartan and enalapril in STZ-induced diabetes in rats. Acta Histochem. 2002, 104, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Gu, J.; Liang, X.; Mao, X.; Wang, Z.; Huang, W. Low dose ruxolitinib plus HLH-94 protocol: A potential choice for secondary HLH. Semin. Hematol. 2020, 57, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Sirhan, S.; Busque, L.; Foltz, L.; Grewal, K.; Hamm, C.; Laferriere, N.; Laneuville, P.; Leber, B.; Liew, E.; Olney, H.J.; et al. Evolving Therapeutic Options for Polycythemia Vera: Perspectives of the Canadian Myeloproliferative Neoplasms Group. Clin. Lymphoma Myeloma Leuk. 2015, 15, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, K.M.; Gopal, G.S.; Chalam, C.R.; Madan, K.; Kumar, V.K.; Prakash, G.J.; Annapurna, A. The influence of sulindac on diabetic cardiomyopathy: A non-invasive evaluation by Doppler echocardiography in streptozotocin-induced diabetic rats. Vasc. Pharmacol. 2005, 43, 91–100. [Google Scholar] [CrossRef]
- Kestenbaum, B.; De Boer, I.H. Urine Albumin-to-Creatinine Ratio: What’s in a Number? J. Am. Soc. Nephrol. 2010, 21, 1243–1244. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.L.; He, J.K.; Xie, Y.; Han, R.; Qin, Z.H.; Zhu, L.J. Therapeutic potential of Naja naja atra venom in a rat model of diabetic nephropathy. Biomed. Environ. Sci. 2012, 25, 630–638. [Google Scholar] [CrossRef]
- Iyoda, M.; Shibata, T.; Hirai, Y.; Kuno, Y.; Akizawa, T. Nilotinib Attenuates Renal Injury and Prolongs Survival in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 1486–1496. [Google Scholar] [CrossRef]
- Liu, I.-M.; Tzeng, T.-F.; Liou, S.-S.; Chang, C.J. The amelioration of streptozotocin diabetes-induced renal damage by Wu-Ling-San (Hoelen Five Herb Formula), a traditional Chinese prescription. J. Ethnopharmacol. 2009, 124, 211–218. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Kady, M.M.; Naggar, R.A.; Guimei, M.; Talaat, I.M.; Shaker, O.G.; Saber-Ayad, M. Early Renoprotective Effect of Ruxolitinib in a Rat Model of Diabetic Nephropathy. Pharmaceuticals 2021, 14, 608. https://doi.org/10.3390/ph14070608
El-Kady MM, Naggar RA, Guimei M, Talaat IM, Shaker OG, Saber-Ayad M. Early Renoprotective Effect of Ruxolitinib in a Rat Model of Diabetic Nephropathy. Pharmaceuticals. 2021; 14(7):608. https://doi.org/10.3390/ph14070608
Chicago/Turabian StyleEl-Kady, Mohamed M., Reham A. Naggar, Maha Guimei, Iman M. Talaat, Olfat G. Shaker, and Maha Saber-Ayad. 2021. "Early Renoprotective Effect of Ruxolitinib in a Rat Model of Diabetic Nephropathy" Pharmaceuticals 14, no. 7: 608. https://doi.org/10.3390/ph14070608
APA StyleEl-Kady, M. M., Naggar, R. A., Guimei, M., Talaat, I. M., Shaker, O. G., & Saber-Ayad, M. (2021). Early Renoprotective Effect of Ruxolitinib in a Rat Model of Diabetic Nephropathy. Pharmaceuticals, 14(7), 608. https://doi.org/10.3390/ph14070608