
Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities

 , , and
, , and
Abstract
:1. Introduction
2. Epithelial to Mesenchymal Transition
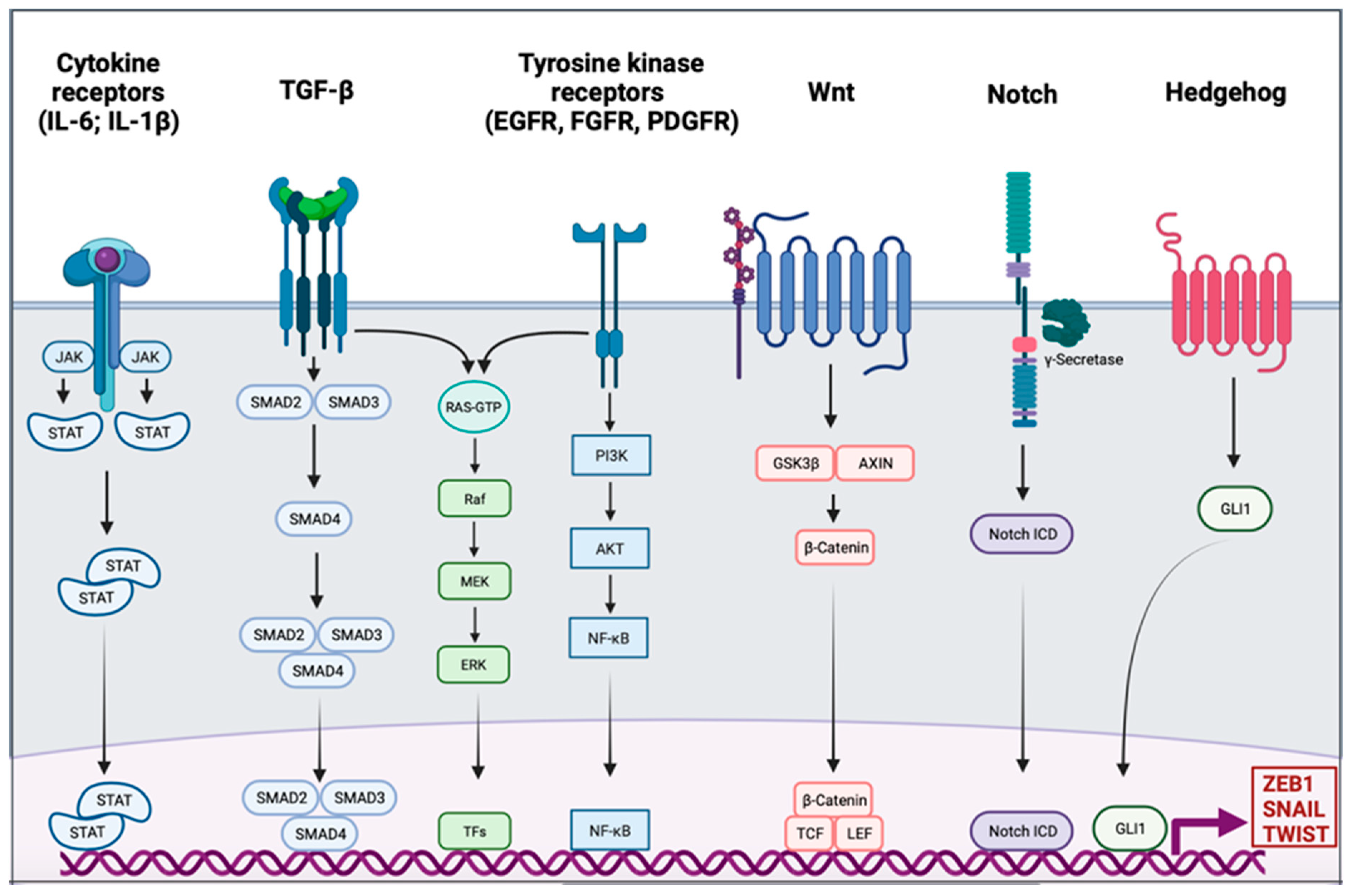
EMT Signaling
3. EMT in PDAC
3.1. Activation of EMT in PDAC
3.2. Role of EMT in PDAC Metastasis
4. Clinical Implication of EMT in PDAC
4.1. EMT in Prognosis of Patients with PDAC
4.2. EMT and Chemoresistance
4.3. EMT as a Biomarker in the Choice of Treatment and to Predict Chemoresistance
5. EMT as a New Target?
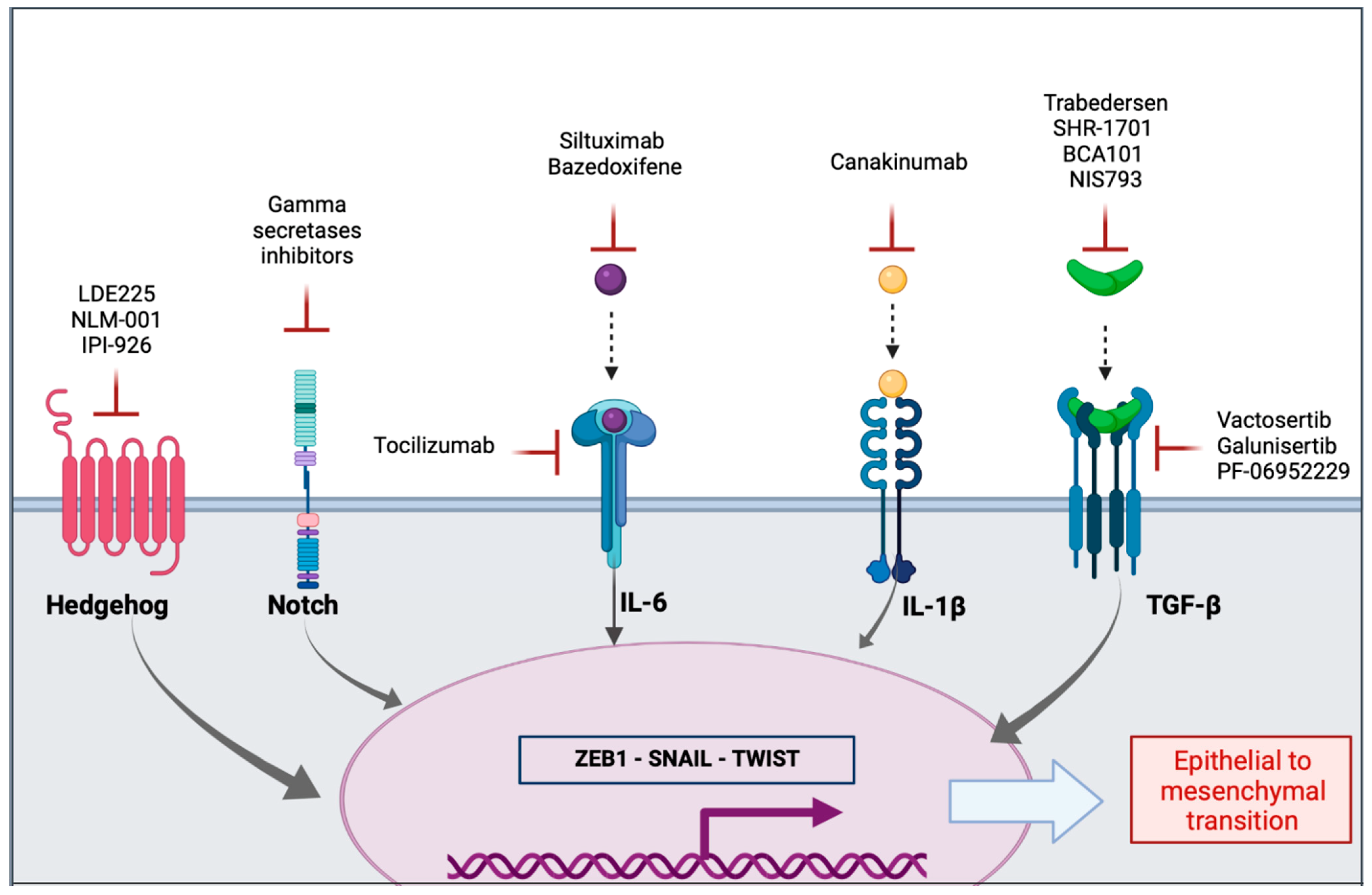
5.1. Inhibition of Extracellular Mediators and Their Receptors
5.1.1. Transforming Growth Factor β
5.1.2. Interleukins
5.1.3. Sonic Hedgehog Signaling Pathway
5.1.4. Notch Signaling Pathway
5.2. MicroRNAs
5.3. Nanomedicine to Target EMT
6. Conclusions
7. Methods
7.1. Search Strategy
7.2. Figures
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Laethem, J.L.V.; Conroy, T.; et al. Cancer of the Pancreas: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up†. Ann. Oncol. 2015, 26, v56–v68. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.-L.; Malka, D.; Ducreux, M.; Conroy, T. An Update on Treatment Options for Pancreatic Adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, Cell Plasticity and Metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Lutfi, W.; Talamonti, M.S.; Kantor, O.; Wang, C.-H.; Liederbach, E.; Stocker, S.J.; Bentrem, D.J.; Roggin, K.K.; Winchester, D.J.; Marsh, R.; et al. Perioperative Chemotherapy Is Associated with a Survival Advantage in Early Stage Adenocarcinoma of the Pancreatic Head. Surgery 2016, 160, 714–724. [Google Scholar] [CrossRef]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the Outcomes of Pancreatic Cancer Surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci 2020, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends in Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiques, O.; Fanshawe, B.; Crosas-Molist, E.; Rodriguez-Hernandez, I.; Volpe, A.; Cantelli, G.; Boehme, L.; Orgaz, J.L.; Mardakheh, F.K.; Sanz-Moreno, V.; et al. A Preclinical Pipeline to Evaluate Migrastatics as Therapeutic Agents in Metastatic Melanoma. Br. J. Cancer 2021. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-Beta-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- McCormack, N.; O’Dea, S. Regulation of Epithelial to Mesenchymal Transition by Bone Morphogenetic Proteins. Cell Signal. 2013, 25, 2856–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, I.; Miele, L. Deadly Crosstalk: Notch Signaling at the Intersection of EMT and Cancer Stem Cells. Cancer Lett. 2013, 341, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt Signalling Pathways in Cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling Mechanisms of the Epithelial-Mesenchymal Transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The Transcription Factor Snail Is a Repressor of E-Cadherin Gene Expression in Epithelial Tumour Cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial-Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Snail: More than EMT. Cell Adh. Migr. 2010, 4, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 Is a Transcriptional Repressor of E-Cadherin and Regulates Epithelial Plasticity in Breast Cancer Cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [Green Version]
- Vandewalle, C.; Comijn, J.; De Craene, B.; Vermassen, P.; Bruyneel, E.; Andersen, H.; Tulchinsky, E.; Van Roy, F.; Berx, G. SIP1/ZEB2 Induces EMT by Repressing Genes of Different Epithelial Cell–Cell Junctions. Nucleic Acids Res. 2005, 33, 6566–6578. [Google Scholar] [CrossRef]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noël, A.; van Roy, F.; Berx, G.; Foidart, J.-M.; Gilles, C. Regulation of Vimentin by SIP1 in Human Epithelial Breast Tumor Cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Casas, E.; Kim, J.; Bendesky, A.; Ohno-Machado, L.; Wolfe, C.J.; Yang, J. Snail2 Is an Essential Mediator of Twist1-Induced Epithelial Mesenchymal Transition and Metastasis. Cancer Res. 2011, 71, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of Epithelial-Mesenchymal Transition through Epigenetic and Post-Translational Modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-Mesenchymal Transition: A New Target in Anticancer Drug Discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulle, A.; Lim, K.-H. Beyond Just a Tight Fortress: Contribution of Stroma to Epithelial-Mesenchymal Transition in Pancreatic Cancer. Signal. Transduct. Target. Ther. 2020, 5, 249. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Dong, L.; Zhao, B.; Lu, J.; Zhao, Y. E-cadherin Is Downregulated by Microenvironmental Changes in Pancreatic Cancer and Induces EMT. Oncol. Rep. 2018, 40, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Porciuncula, A.; Hajdu, C.; David, G. The Dual Role of Senescence in Pancreatic Ductal Adenocarcinoma. Adv. Cancer Res. 2016, 131, 1–20. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [Green Version]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardare, J.; Witz, A.; Merlin, J.-L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach-Carson, M.C.; Levine, H. Tumor Budding: The Name Is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal Transition Is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. EMT Is Not Required for Lung Metastasis but Contributes to Chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-Activator Zeb1 Is a Key Factor for Cell Plasticity and Promotes Metastasis in Pancreatic Cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-Activator ZEB1 Promotes Tumorigenicity by Repressing Stemness-Inhibiting MicroRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual Microdissection Identifies Distinct Tumor- and Stroma-Specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Rashid, N.U.; Peng, X.L.; Jin, C.; Moffitt, R.A.; Volmar, K.E.; Belt, B.A.; Panni, R.Z.; Nywening, T.M.; Herrera, S.G.; Moore, K.J.; et al. Purity Independent Subtyping of Tumors (PurIST), A Clinically Robust, Single-Sample Classifier for Tumor Subtyping in Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Yamazaki, K.; Hibi, T.; Aiura, K.; Kitagawa, Y.; Sakamoto, M. Solitary Cell Infiltration Is a Novel Indicator of Poor Prognosis and Epithelial-Mesenchymal Transition in Pancreatic Cancer. Hum. Pathol 2010, 41, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-Cell Transcriptome Analysis of Tumor and Stromal Compartments of Pancreatic Ductal Adenocarcinoma Primary Tumors and Metastatic Lesions. Genome Med. 2020, 12, 80. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Wang, Z.-R.; Chen, C.-L.; Di, L.; Bi, Z.-F.; Li, Z.-H.; Liu, Y.-M. Molecular Detection of Epithelial-Mesenchymal Transition Markers in Circulating Tumor Cells from Pancreatic Cancer Patients: Potential Role in Clinical Practice. World J. Gastroenterol. 2019, 25, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Ma, Y.; Zhao, X.; Tian, X.; Sun, Y.; Yang, Y.; Zhao, X. Spatial Heterogeneity in Epithelial to Mesenchymal Transition Properties of Circulating Tumor Cells Associated with Distant Recurrence in Pancreatic Cancer Patients. Ann. Transl Med. 2020, 8, 676. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in Survival and Clinical Benefit with Gemcitabine as First-Line Therapy for Patients with Advanced Pancreas Cancer: A Randomized Trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and Characterization of Gemcitabine-Resistant Pancreatic Tumor Cells. Ann. Surg Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of Epithelial-Mesenchymal Transition Phenotype of Gemcitabine-Resistant Pancreatic Cancer Cells Is Linked with Activation of the Notch Signaling Pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.-H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription Phenotypes of Pancreatic Cancer Are Driven by Genomic Events during Tumor Evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Martinelli, P.; Cañamero, M.; del Pozo, N.; Madriles, F.; Zapata, A.; Real, F.X. Gata6 Is Required for Complete Acinar Differentiation and Maintenance of the Exocrine Pancreas in Adult Mice. Gut 2013, 62, 1481–1488. [Google Scholar] [CrossRef]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. GATA6 Regulates EMT and Tumour Dissemination, and Is a Marker of Response to Adjuvant Chemotherapy in Pancreatic Cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Li, K.; Lou, J.; Wu, Y.; Peng, C. An EMT-Related Gene Signature for Predicting Response to Adjuvant Chemotherapy in Pancreatic Ductal Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus Gemcitabine vs. Gemcitabine for First-Line Treatment of Patients with Unresectable Pancreatic Cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Takahashi, H.; Kondo, S.; Lahn, M.M.F.; Ogasawara, K.; Benhadji, K.A.; Fujii, H.; Ueno, H. Phase 1b Study of Galunisertib in Combination with Gemcitabine in Japanese Patients with Metastatic or Locally Advanced Pancreatic Cancer. Cancer Chemother. Pharmacol. 2017, 79, 1169–1177. [Google Scholar] [CrossRef]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel Therapies Emerging in Oncology to Target the TGF-β Pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Holmer, R.; Goumas, F.A.; Waetzig, G.H.; Rose-John, S.; Kalthoff, H. Interleukin-6: A Villain in the Drama of Pancreatic Cancer Development and Progression. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 371–380. [Google Scholar] [CrossRef]
- Guan, J.; Zhang, H.; Wen, Z.; Gu, Y.; Cheng, Y.; Sun, Y.; Zhang, T.; Jia, C.; Lu, Z.; Chen, J. Retinoic Acid Inhibits Pancreatic Cancer Cell Migration and EMT through the Downregulation of IL-6 in Cancer Associated Fibroblast Cells. Cancer Lett. 2014, 345, 132–139. [Google Scholar] [CrossRef]
- Li, H.; Xiao, H.; Lin, L.; Jou, D.; Kumari, V.; Lin, J.; Li, C. Drug Design Targeting Protein-Protein Interactions (PPIs) Using Multiple Ligand Simultaneous Docking (MLSD) and Drug Repositioning: Discovery of Raloxifene and Bazedoxifene as Novel Inhibitors of IL-6/GP130 Interface. J. Med. Chem. 2014, 57, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 Axis Is Associated with Cancer Stem Cell-like Properties and Correlates with Clinical Prognosis in Human Pancreatic Cancer Cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef] [PubMed]
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J. Simultaneous Blocking of IL-6 and IL-8 Is Sufficient to Fully Inhibit CAF-Induced Human Melanoma Cell Invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Ai, K.; Du, Y.; Chen, G. Sonic Hedgehog Expression Correlates with Distant Metastasis in Pancreatic Adenocarcinoma. Pancreas 2011, 40, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Bahra, M.; Kamphues, C.; Boas-Knoop, S.; Lippert, S.; Esendik, U.; Schüller, U.; Hartmann, W.; Waha, A.; Neuhaus, P.; Heppner, F.; et al. Combination of Hedgehog Signaling Blockage and Chemotherapy Leads to Tumor Reduction in Pancreatic Adenocarcinomas. Pancreas 2012, 41, 222–229. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 Study of Vismodegib, a Hedgehog Inhibitor, Combined with Gemcitabine and Nab-Paclitaxel in Patients with Untreated Metastatic Pancreatic Adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Laheru, D.; Maitra, A.; Arcaroli, J.; Rudek, M.A.; Dasari, A.; Blatchford, P.J.; Quackenbush, K.; Messersmith, W. A Phase II Study of the Gamma Secretase Inhibitor RO4929097 in Patients with Previously Treated Metastatic Pancreatic Adenocarcinoma. Investig. New Drugs 2014, 32, 739–745. [Google Scholar] [CrossRef]
- Richter, S.; Bedard, P.L.; Chen, E.X.; Clarke, B.A.; Tran, B.; Hotte, S.J.; Stathis, A.; Hirte, H.W.; Razak, A.R.A.; Reedijk, M.; et al. A Phase I Study of the Oral Gamma Secretase Inhibitor R04929097 in Combination with Gemcitabine in Patients with Advanced Solid Tumors (PHL-078/CTEP 8575). Investig. New Drugs 2014, 32, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Cook, N.; Basu, B.; Smith, D.-M.; Gopinathan, A.; Evans, J.; Steward, W.P.; Palmer, D.; Propper, D.; Venugopal, B.; Hategan, M.; et al. A Phase I Trial of the γ-Secretase Inhibitor MK-0752 in Combination with Gemcitabine in Patients with Pancreatic Ductal Adenocarcinoma. Br. J. Cancer 2018, 118, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Dvořánková, B.; Szabo, P.; Lacina, L.; Kodet, O.; Matoušková, E.; Smetana, K. Fibroblasts Prepared from Different Types of Malignant Tumors Stimulate Expression of Luminal Marker Keratin 8 in the EM-G3 Breast Cancer Cell Line. Histochem. Cell Biol. 2012, 137, 679–685. [Google Scholar] [CrossRef]
- Li, Y.; VandenBoom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-Regulation of MiR-200 and Let-7 by Natural Agents Leads to the Reversal of Epithelial-to-Mesenchymal Transition in Gemcitabine-Resistant Pancreatic Cancer Cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef] [Green Version]
- Chitkara, D.; Mittal, A.; Mahato, R.I. MiRNAs in Pancreatic Cancer: Therapeutic Potential, Delivery Challenges and Strategies. Adv. Drug Deliv. Rev. 2015, 81, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Li, J.; Zhang, Z.; Huang, C.; Lian, G.; Yang, K.; Chen, S.; Lin, Y.; Wang, L.; et al. Co-Delivery of MicroRNA-21 Antisense Oligonucleotides and Gemcitabine Using Nanomedicine for Pancreatic Cancer Therapy. Cancer Sci. 2017, 108, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Saha, M.; Ghosh, S.S. Nanoparticle Mediated Alteration of EMT Dynamics: An Approach to Modulate Cancer Therapeutics. Mater. Adv. 2020, 1, 2614–2630. [Google Scholar] [CrossRef]
- Huai, Y.; Zhang, Y.; Xiong, X.; Das, S.; Bhattacharya, R.; Mukherjee, P. Gold Nanoparticles Sensitize Pancreatic Cancer Cells to Gemcitabine. Cell Stress 2019, 3, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Cordani, M.; Strippoli, R.; Somoza, Á. Nanomaterials as Inhibitors of Epithelial Mesenchymal Transition in Cancer Treatment. Cancers 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra Boinpelly, V.; Verma, R.K.; Srivastav, S.; Srivastava, R.K.; Shankar, S. A-Mangostin-encapsulated PLGA Nanoparticles Inhibit Colorectal Cancer Growth by Inhibiting Notch Pathway. J. Cell Mol. Med. 2020, 24, 11343–11354. [Google Scholar] [CrossRef]
- Verma, R.K.; Yu, W.; Singh, S.P.; Shankar, S.; Srivastava, R.K. Anthothecol-Encapsulated PLGA Nanoparticles Inhibit Pancreatic Cancer Stem Cell Growth by Modulating Sonic Hedgehog Pathway. Nanomedicine 2015, 11, 2061–2070. [Google Scholar] [CrossRef]
- Daman, Z.; Faghihi, H.; Montazeri, H. Salinomycin Nanoparticles Interfere with Tumor Cell Growth and the Tumor Microenvironment in an Orthotopic Model of Pancreatic Cancer. Drug Dev. Ind. Pharmacy 2018, 44, 1434–1442. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Name | Combination | Functional Class | Study Population | Phase | Study NCT Registry Number |
|---|---|---|---|---|---|
| Vactosertib (TEW-7197) | FOLFOX | Inhibitor of the serine/threonine kinase TGF-βR1 | Metastatic PDAC who have failed first-line gemcitabine and nab-paclitaxel | Ib | NCT03666832 |
| Nanoliposomal irinotecan with 5-FU and leucovorin | Metastatic PDAC | II | NCT04258072 | ||
| Galunisertib (LY2157299) | Durvalumab | TGF-βR1 kinase inhibitor | Recurrent or refractory metastatic pancreatic cancer | Ib | NCT02734160 |
| Gemcitabine | Inoperable or metastatic pancreatic cancer | Ib | NCT02154646 | ||
| Gemcitabine | Advanced or metastatic unresectable pancreatic cancer | Ib/II | NCT01373164 | ||
| Trabedersen (AP 12009) | _____ | Antisense oligonucleotide specific for the mRNA TGF-β2 | Advanced tumors known to overproduce TGF-β2 (Pancreatic neoplasm) | I | NCT00844064 |
| SHR-1701 | Gemcitabine and albumine paclitaxel | Bifunctional fusion protein targeting PD-L1 and TGF-β | Advanced/Metastatic pancreatic cancer in first line treatment | Ib /II | NCT04624217 |
| PF-06952229 | _____ | TFG-β receptor I inhibitor | Advanced solid tumors (Pancreatic neoplasms) | I | NCT03685591 |
| BCA101 | Alone or with pembrolizumab | EGFR/TGF-β fusion mAb | Patients with EGFR-driven advanced solid tumors (Pancreas cancer with KRAS G12D mutation) | I | NCT04429542 |
| NIS793 | Spartalizumab | Anti-TGF-β mAb | Advanced malignancies (Pancreatic cancer) | I | NCT02947165 |
| With and without spartalizumab in combination with gemcitabine and nab-paclitaxel | First-line in metastatic PDAC | II | NCT04390763 | ||
| Tocilizumab | Gemcitabine and nab-paclitaxel | Anti -IL-6 Receptor mAb | Unresectable pancreatic carcinoma | II | NCT02767557 |
| Ipilimumab, nivolumab and radiation | Advanced pancreatic cancer | II | NCT04258150 | ||
| Nab-paclitaxel, gemcitabine, oxaliplatine, leucovorin, fluorouracil, atezolizumab, cobimetinib, PEGPH20, BL-8040, selicrelumab, bevacizumab, RO6874281, AB928, tiragolumab | Metastatic PDAC | I/II | NCT03193190 | ||
| Siltuximab | _____ | Anti-IL-6 mAb | Solid tumors (Pancreatic neoplasms) | I/II | NCT00841191 |
| Spartalizumab | Metastatic pancreatic cancer | Ib/II | NCT04191421 | ||
| Bazedoxifene | Gemcitabine and nab-paclitaxel | Selective estrogen receptor modulator—Inhibitor of IL-6/glycoprotein 130 | Metastatic pancreatic cancer | - | NCT04812808 |
| Canakinumab (ACZ885) | Spartalizumab, gemcitabine and nab-paclitaxel | Anti-IL-1-β mAb | Metastatic pancreatic cancer | Ib | NCT04581343 |
| LDE225 | Gemcitabine and nab-paclitaxel | Hedgehog inhibitor | Locally advanced or metastasized pancreatic cancer | I/II | NCT02358161 |
| Fluorouracil, leucovorin, oxaliplatin, irinotecan | Untreated advanced pancreatic cancer | Ib | NCT01485744 | ||
| NLM-001 | Gemcitabine and nab-paclitaxel, zalifrelimab | Advanced pancreatic cancer | Ib/IIa | NCT04827953 | |
| IPI-926 | Gemcitabine | Metastatic pancreatic cancer | Ib/II | NCT01130142 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dardare, J.; Witz, A.; Merlin, J.-L.; Bochnakian, A.; Toussaint, P.; Gilson, P.; Harlé, A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals 2021, 14, 740. https://doi.org/10.3390/ph14080740
Dardare J, Witz A, Merlin J-L, Bochnakian A, Toussaint P, Gilson P, Harlé A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals. 2021; 14(8):740. https://doi.org/10.3390/ph14080740
Chicago/Turabian StyleDardare, Julie, Andréa Witz, Jean-Louis Merlin, Agathe Bochnakian, Paul Toussaint, Pauline Gilson, and Alexandre Harlé. 2021. "Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities" Pharmaceuticals 14, no. 8: 740. https://doi.org/10.3390/ph14080740
APA StyleDardare, J., Witz, A., Merlin, J. -L., Bochnakian, A., Toussaint, P., Gilson, P., & Harlé, A. (2021). Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals, 14(8), 740. https://doi.org/10.3390/ph14080740