
In Silico Approaches: A Way to Unveil Novel Therapeutic Drugs for Cervical Cancer Management

 ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
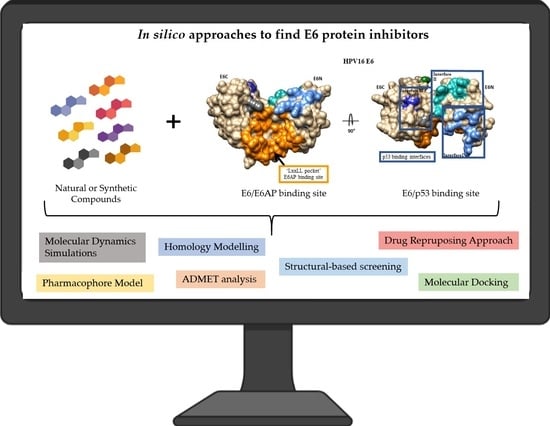
:
1. Introduction
2. HPV, CC Development, and Clinical Treatment
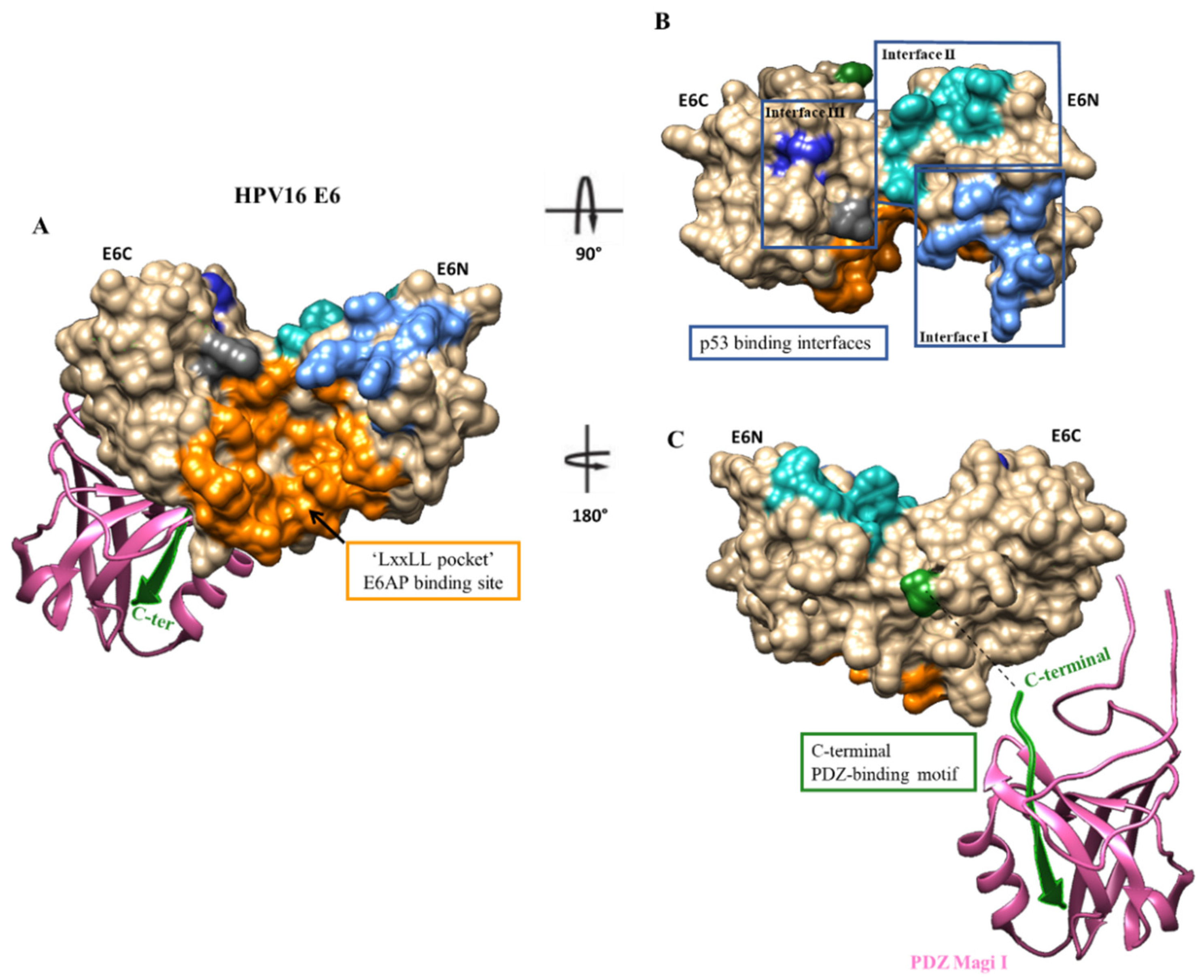
3. E6 Protein
3.1. E6-E6AP Complex Inhibitors
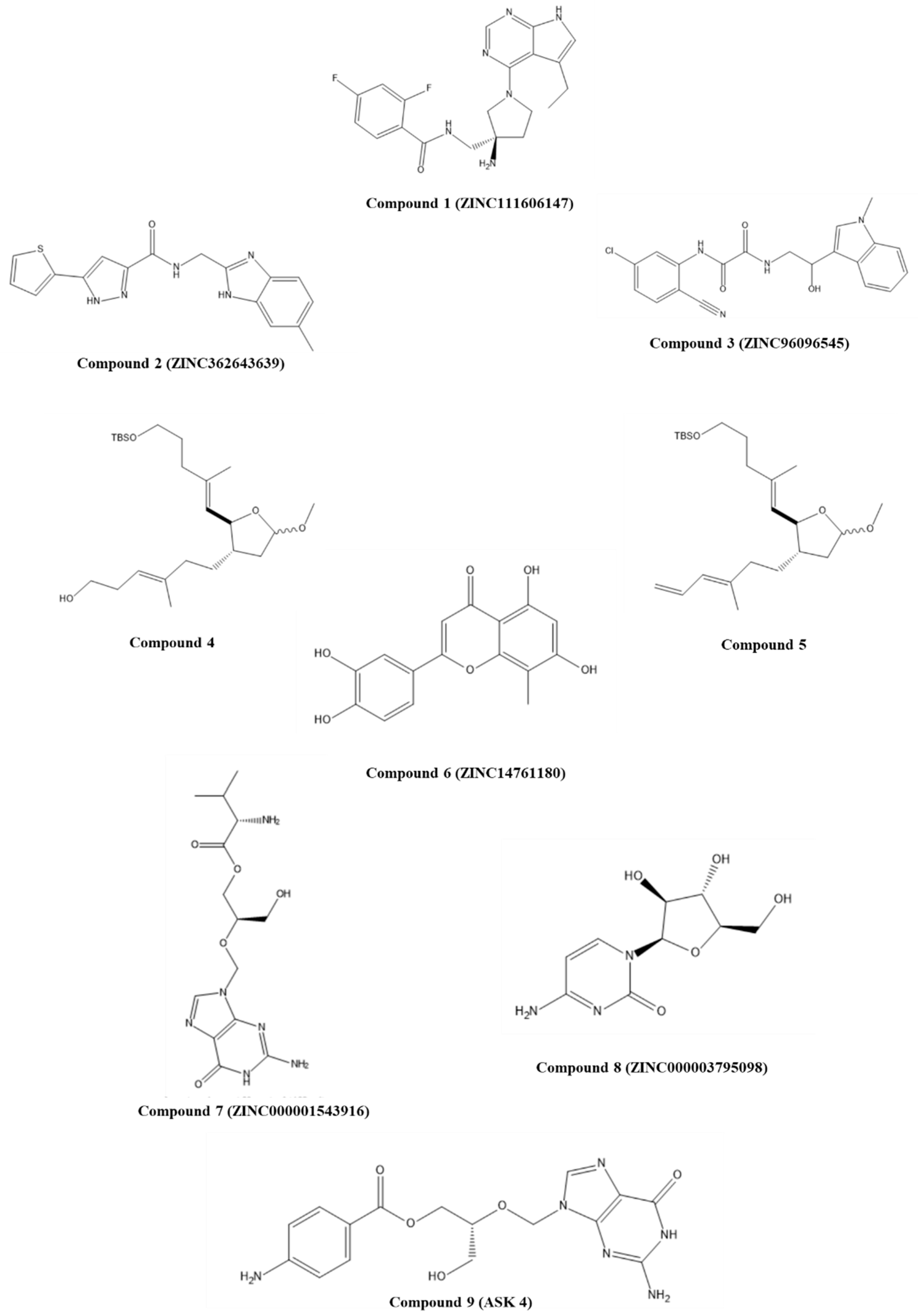
3.1.1. Synthetic Compounds
3.1.2. Natural Compounds
3.2. E6-p53 Complex Inhibitors
3.2.1. Natural Compounds
3.2.2. Synthetic Compounds
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GLOBOCAN 2020: New Global Cancer Data. Available online: https://www.uicc.org/news/globocan-2020-new-global-cancer-data (accessed on 25 May 2021).
- Bober, P.; Alexovič, M.; Tomková, Z.; Kilík, R.; Sabo, J. RHOA and mDia1 promotes apoptosis of breast cancer cells via a high dose of doxorubicin treatment. Open Life Sci. 2019, 14, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Queiroz, J.A.; Sousa, F.; Sousa, A. Cervical cancer and HPV infection: Ongoing therapeutic research to counteract the action of E6 and E7 oncoproteins. Drug Discov. Today 2019, 24, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2019, 10, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prigge, E.S.; von Knebel Doeberitz, M.; Reuschenbach, M. Clinical relevance and implications of HPV-induced neoplasia in different anatomical locations. Mutat. Res. Rev. Mutat. Res. 2017, 772, 51–66. [Google Scholar] [CrossRef]
- Cordeiro, M.N.; Lima, R.D.C.P.D.; Paolini, F.; Melo, A.R.S.; Campos, A.P.F.; Venuti, A.; De Freitas, A.C. Current research into novel therapeutic vaccines against cervical cancer. Expert Rev. Anticancer Ther. 2018, 18, 365–376. [Google Scholar] [CrossRef]
- Barra, F.; Lorusso, D.; Leone Roberti Maggiore, U.; Ditto, A.; Bogani, G.; Raspagliesi, F.; Ferrero, S. Investigational drugs for the treatment of cervical cancer. Expert Opin. Investig. Drugs 2017, 26, 389–402. [Google Scholar] [CrossRef]
- Kumar, A.; Rathi, E.; Hariharapura, R.C.; Kini, S.G. Is viral E6 oncoprotein a viable target? A critical analysis in the context of cervical cancer. Med. Res. Rev. 2020, 40, 2019–2048. [Google Scholar] [CrossRef]
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Yella, J.K.; Yaddanapudi, S.; Wang, Y.; Jegga, A.G. Changing Trends in Computational Drug Repositioning. Pharmaceuticals 2018, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.T. Computational approaches in target identification and drug discovery. Comput. Struct Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Martínez, F.D.; López-López, E.; Eurídice Juárez-Mercado, K.; Medina-Franco, J.L. Computational Drug Design Methods—Current and Future Perspectives. In In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 19–44. [Google Scholar] [CrossRef]
- Bernetti, M.; Bertazzo, M.; Masetti, M. Data-Driven Molecular Dynamics: A Multifaceted Challenge. Pharmaceuticals 2020, 13, 253. [Google Scholar] [CrossRef]
- Cruz-Vicente, P.; Passarinha, L.A.; Silvestre, S.; Gallardo, E. Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches. Molecules 2021, 26, 2193. [Google Scholar] [CrossRef]
- McBride, A.A. Oncogenic human papillomaviruses. Phil. Trans. R. Soc. B 2017, 372, 20160273. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, M.E.; Chiocca, S. Human papillomavirus as a driver of head and neck cancers. Br. J. Cancer 2020, 122, 306–314. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2016, 25, 2–23. [Google Scholar] [CrossRef] [Green Version]
- De Freitas, N.L.; Deberaldini, M.G.; Gomes, D.; Pavan, A.R.; Sousa, Â.; Dos Santos, J.L.; Soares, C.P. Histone Deacetylase Inhibitors as Therapeutic Interventions on Cervical Cancer Induced by Human Papillomavirus. Front. Cell Dev. Biol. 2021, 8, 1–22. [Google Scholar] [CrossRef]
- Farthing, A.J.; Vousden, K.H. Functions of human papillomavirus E6 and E7 oncoproteins. Trends Microbiol. 1994, 2, 170–173. [Google Scholar] [CrossRef]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef]
- Martinez-Ramirez, I.; Carrillo-Garcia, A.; Contreras-Paredes, A.; Ortiz-Sanchez, E.; Cruz-Gregorio, A.; Lizano, M. Regulation of Cellular Metabolism by High-Risk Human Papillomaviruses. Int. J. Mol. Sci. 2018, 19, 1839. [Google Scholar] [CrossRef] [Green Version]
- Viarisio, D.; Gissmann, L.; Tommasino, M. Human papillomaviruses and carcinogenesis: Well-established and novel models. Curr. Opin. Virol. 2017, 26, 56–62. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Diagnosis and treatment of invasive cervical cancer. In Comprehensive Cervical Cancer Control: A Guide to Essential Practice, 2nd ed.; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Liontos, M.; Kyriazoglou, A.; Dimitriadis, I.; Dimopoulos M-Athanasios, A.B. Systemic therapy in cervical cancer: 30 years in review. Crit. Rev. Oncol. Hematol. 2019, 137, 9–17. [Google Scholar] [CrossRef]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Nomine, Y.; Ristriani, T.; Laurent, C.; Lefèvre, J.O.; Weiss, É.; Travé, G. A strategy for optimizing the monodispersity of fusion proteins: Application to purification of recombinant HPV E6 oncoprotein. Protein Eng. 2001, 14, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Zanier, K.; Sidi, A.o.M.h.o.; Boulade-Ladame, C.; Rybin, V.; Chappelle, A.; Atkinson, A.; Kieffer, B.; Travé, G. Solution structure analysis of the HPV16 E6 oncoprotein reveals a self-association mechanism required for E6-mediated degradation of p53. Structure 2012, 20, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Pol, S.B.V.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [Green Version]
- Grm, H.S.; Banks, L. Degradation of hDlg and MAGIs by human papillomavirus E6 is E6-AP-independent. J. Gen. Virol. 2004, 85, 2815–2819. [Google Scholar] [CrossRef]
- Charbonnier, S.; Nominé, Y.; Ramírez, J.; Luck, K.; Chapelle, A.; Stote, R.H.; Travé, G.; Kieffer, B.; Atkinson, R.A. The Structural and Dynamic Response of MAGI-1 PDZ1 with Noncanonical Domain Boundaries to the Binding of Human Papillomavirus E6. J. Mol. Biol. 2011, 406, 745–763. [Google Scholar] [CrossRef] [PubMed]
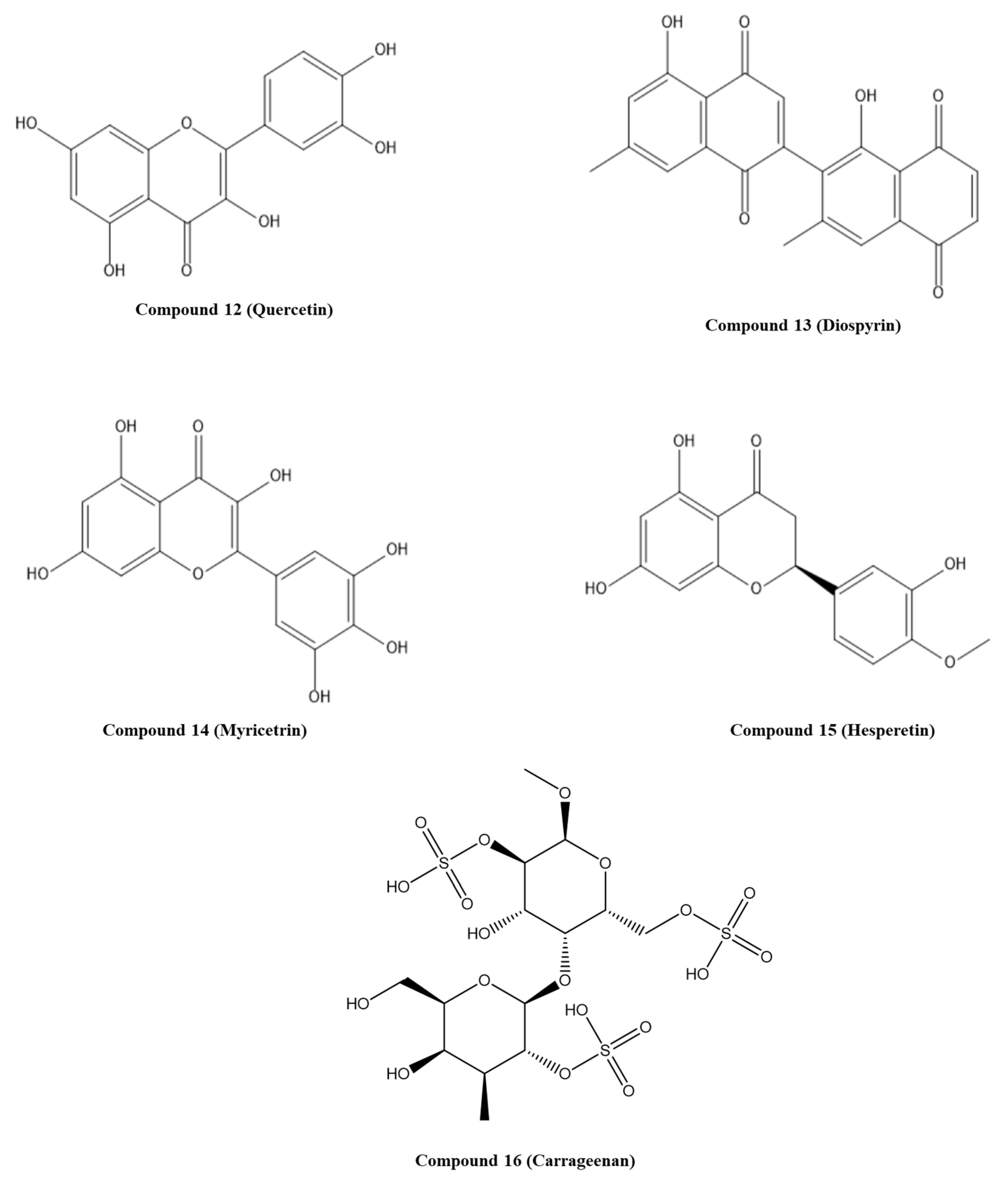
- Cherry, J.J.; Rietz, A.; Malinkevich, A.; Liu, Y.; Xie, M.; Bartolowits, M.; Davisson, V.J.; Baleja, J.D.; Androphy, E.J. Structure based identification and characterization of flavonoids that disrupt human papillomavirus-16 E6 function. PLoS ONE 2013, 8, e84506. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Stutz, C.; Kintscher, S.; Reinz, E.; Sehr, P.; Bulkescher, J.; Hoppe-Seyler, K.; Trave, G.; Hoppe-Seyler, F. The E6AP binding pocket of the HPV16 E6 oncoprotein provides a docking site for a small inhibitory peptide unrelated to E6AP, indicating druggability of E6. PLoS ONE 2014, 9, e112514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci-Lopez, J.; Vidal-Limon, A.; Zunniga, M.; Jimenez, V.A.; Alderete, J.B.; Brizuela, C.A.; Aguila, S. Molecular modeling simulation studies reveal new potential inhibitors against HPV E6 protein. PLoS ONE 2019, 14, e0213028. [Google Scholar] [CrossRef]
- Senthilkumar, R.; Brusentsev, Y.; Paul, P.; Marimuthu, P.; Cheng, F.; Eklund, P.C.; Eriksson, J.E. Synthesis and Evaluation of Anisomelic acid-like Compounds for the Treatment of HPV-Mediated Carcinomas. Sci. Rep. 2019, 9, 20295. [Google Scholar] [CrossRef] [Green Version]
- Rietz, A.; Petrov, D.P.; Bartolowits, M.; DeSmet, M.; Davisson, V.J.; Androphy, E.J. Molecular Probing of the HPV-16 E6 Protein Alpha Helix Binding Groove with Small Molecule Inhibitors. PLoS ONE 2016, 11, e0149845. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Rathi, E.; Kini, S.G. E-pharmacophore modelling, virtual screening, molecular dynamics simulations and in-silico ADME analysis for identification of potential E6 inhibitors against cervical cancer. J. Mol. Struct. 2019, 1189, 299–306. [Google Scholar] [CrossRef]
- Kumar, A.; Rathi, E.; Kini, S.G. Drug repurposing approach for the identification and designing of potential E6 inhibitors against cervical cancer: An in silico investigation. Struct. Chem. 2019, 31, 141–153. [Google Scholar] [CrossRef]
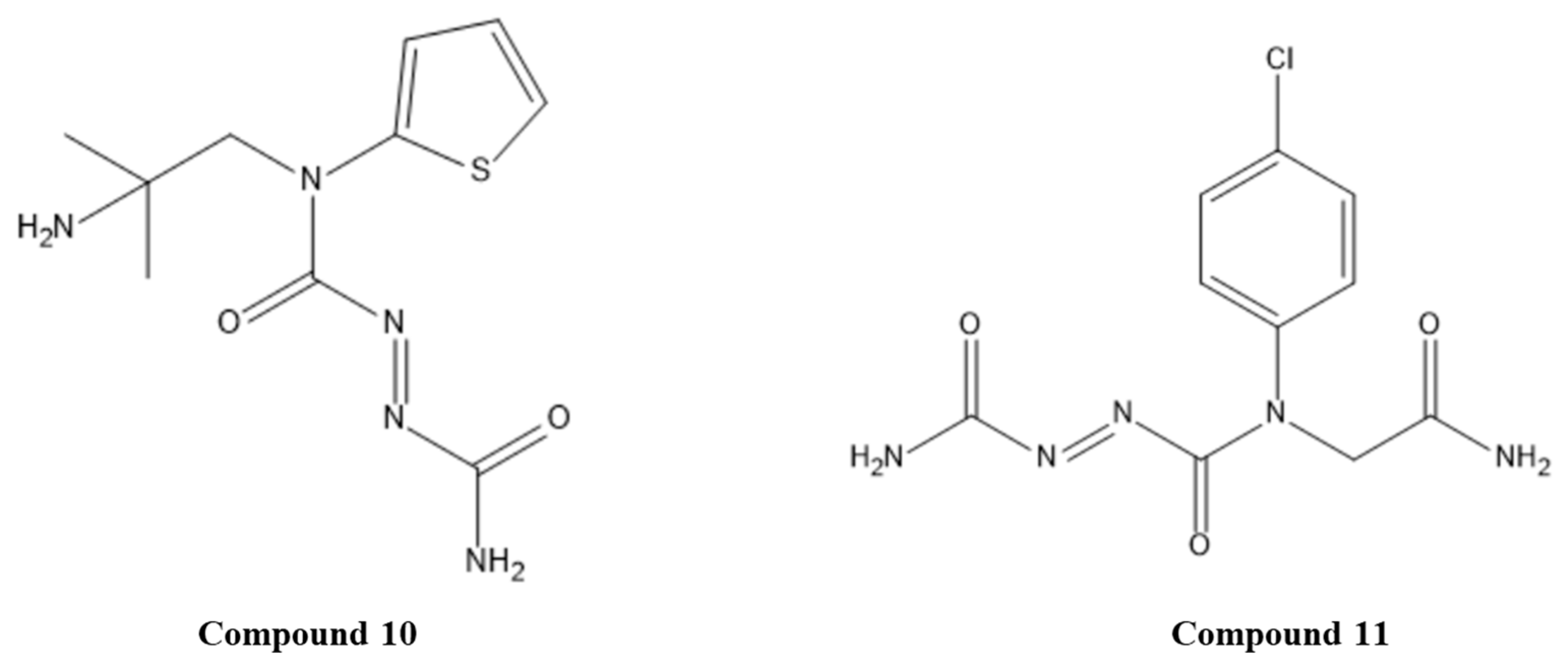
- Choudhury, A.D.; Choudhury, M.D.; Chetia, P.; Chowdhury, A.; Talukdar, A.D. An In Silico Appraisal of Azoic and Disulphide Derivatives for Anticancer Activity Against HPV E6 Oncoprotein to Medicate Cervical Cancer. Comb. Chem. High. Throughput Screen. 2014, 17, 38–46. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E. Plant-Derived Compounds in Cancer Therapy: Traditions of Past and Drugs of Future. In Anticancer Plants: Properties and Application; Akhtar, M., Swamy, M., Eds.; Springer: Singapore, 2018. [Google Scholar] [CrossRef]
- Franconi, R.; Massa, S.; Paolini, F.; Vici, P.; Venuti, A. Plant-Derived Natural Compounds in Genetic Vaccination and Therapy for HPV-Associated Cancers. Cancers 2020, 12, 3101. [Google Scholar] [CrossRef]
- Clemente-Soto, A.F.; Salas-Vidal, E.; Milan-Pacheco, C.; Sanchez-Carranza, J.N.; Peralta-Zaragoza, O.; Gonzalez-Maya, L. Quercetin induces G2 phase arrest and apoptosis with the activation of p53 in an E6 expressionindependent manner in HPVpositive human cervical cancerderived cells. Mol. Med. Rep. 2019, 19, 2097–2106. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Rathi, E.; Kini, S.G. Identification of E6 Inhibitors Employing Energetically Optimized Structure-Based Pharmacophore Modelling, Ligand Docking and Molecular Dynamics Simulations Studies. ChemistrySelect 2019, 4, 10701–10708. [Google Scholar] [CrossRef]
- Kolluru, S.; Momoh, R.; Lin, L.; Mallareddy, J.R.; Krstenansky, J.L. Identification of potential binding pocket on viral oncoprotein HPV16 E6: A promising anti-cancer target for small molecule drug discovery. BMC Mol. Cell Biol. 2019, 20, 30. [Google Scholar] [CrossRef]
- Prakash, S.; Elavarasan, N.; Subashini, K.; Kanaga, S.; Dhandapani, R.; Sivanandam, M.; Kumaradhas, P.; Thirunavukkarasu, C.; Sujatha, V. Isolation of hesperetin—A flavonoid from Cordia sebestena flower extract through antioxidant assay guided method and its antibacterial, anticancer effect on cervical cancer via in vitro and in silico molecular docking studies. J. Mol. Struct. 2020, 1207. [Google Scholar] [CrossRef]
- Rajasekhar, P.; Dhrub Kumar, Y.; Prasad, G.; Srinivasulu, K. In Silico Analysis of HPV E6 as Drug Target with Natural Antioxidants. Int. J. Pharma Bio Sci. 2020, 10. [Google Scholar] [CrossRef]
- Medina-Alarcón, K.P.; Voltan, A.R.; Fonseca-Santos, B.; Moro, I.J.; Souza, F.D.O.; Chorilli, M.; Soares, C.P.; dos Santos, A.G.; Giannini, M.J.M.; Fusco-Almeida, A.M. Highlights in nanocarriers for the treatment against cervical cancer. Mater. Sci. Eng. C 2017, 80, 748–759. [Google Scholar] [CrossRef] [Green Version]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Anwar, M.A.; Park, S.; Jafri, S.S.; Choi, S. In silico mechanistic analysis of IRF3 inactivation and high-risk HPV E6 species-dependent drug response. Sci. Rep. 2015, 5, 13446. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, B.; Joe, A. Oncogene Addiction. Cancer Res. 2008, 68. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Jena, L.; Mohod, K.; Daf, S.; Varma, A.K. Virtual Screening for Potential Inhibitors of High-Risk Human Papillomavirus 16 E6 Protein. Interdiscip. Sci. 2015, 7, 136–142. [Google Scholar] [CrossRef]
- Atabaki, V.; Pourahmad, J.; Hosseinabadi, T. Phytochemical compounds from Jurinea macrocephala subsp.elbursensis and their cytotoxicity evaluation. S. Afr. J. Bot. 2021, 137, 399–405. [Google Scholar] [CrossRef]
- Mamgain, S.; Sharma, P.; Pathak, R.K.; Baunthiyal, M. Computer aided screening of natural compounds targeting the E6 protein of HPV using molecular docking. Bioinformation 2015, 11, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Nabati, F.; Moradi, M.; Mohabatkar, H. In silico analyzing the molecular interactions of plant-derived inhibitors against E6AP, p53, and c-Myc binding sites of HPV type 16 E6 oncoprotein. Mol. Biol. Res. Commun. 2020, 9, 71–82. [Google Scholar] [CrossRef]
- Kumar, S.; Jena, L.; Sahoo, M.; Kakde, M.; Daf, S.; Varma, A.K. In Silico Docking to Explicate Interface between Plant-Originated Inhibitors and E6 Oncogenic Protein of Highly Threatening Human Papillomavirus 18. Genom. Inf. 2015, 13, 60–67. [Google Scholar] [CrossRef] [Green Version]
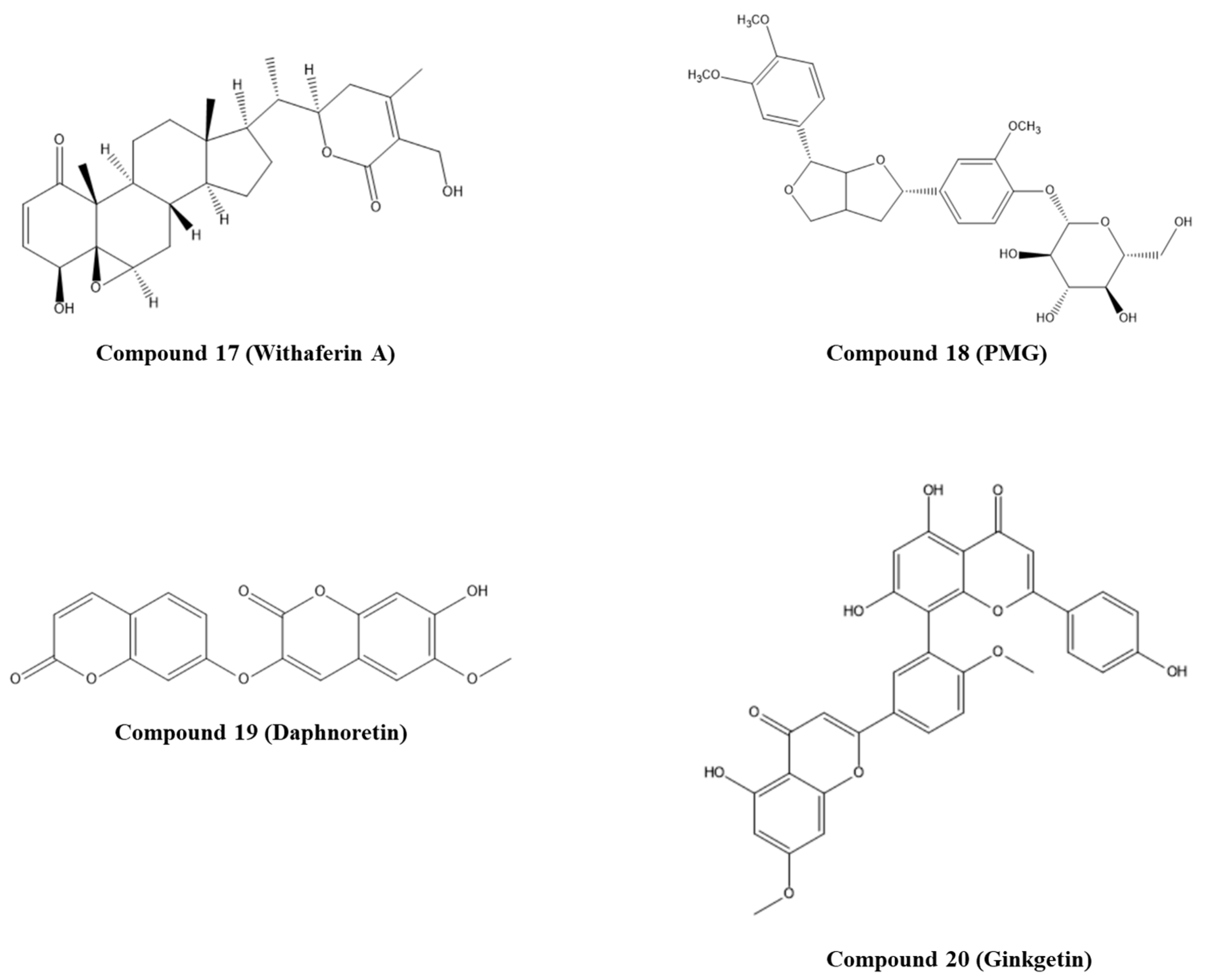
- Celegato, M.; Messa, L.; Goracci, L.; Mercorelli, B.; Bertagnin, C.; Spyrakis, F.; Suarez, I.; Cousido-Siah, A.; Trave, G.; Banks, L.; et al. A novel small-molecule inhibitor of the human papillomavirus E6-p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Lett. 2020, 470, 115–125. [Google Scholar] [CrossRef]
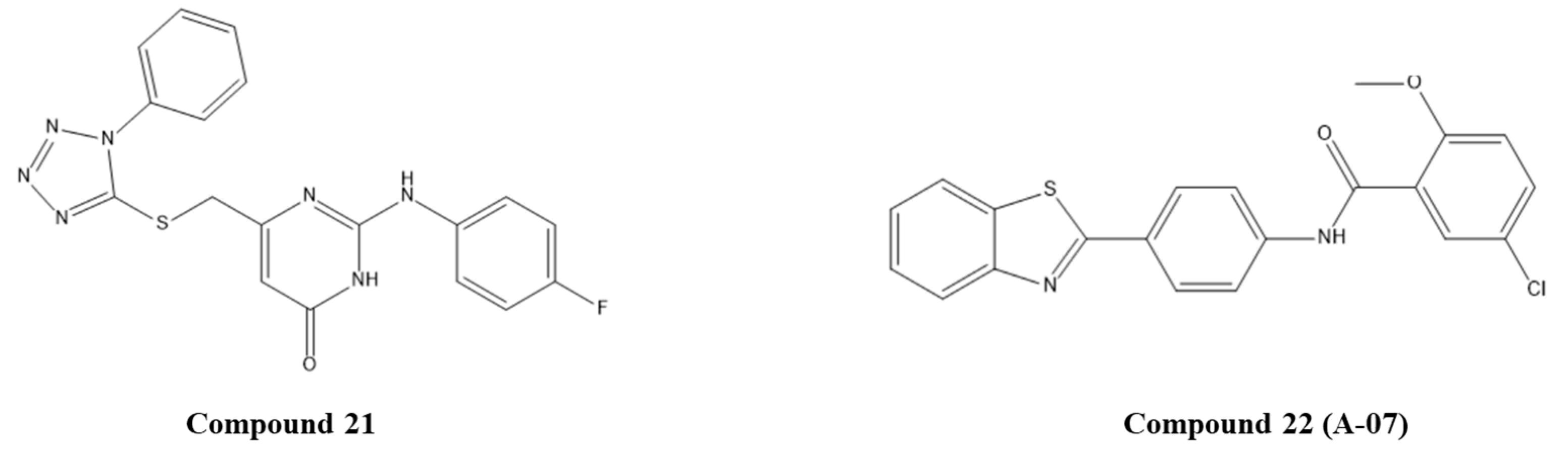
- Modi, A.; Singh, M.; Gutti, G.; Shanker, O.R.; Singh, V.K.; Singh, S.; Singh, S.K.; Pradhan, S.; Narayan, G. Benzothiazole derivative bearing amide moiety induces p53-mediated apoptosis in HPV16 positive cervical cancer cells. Investig. New Drugs 2020, 38, 934–945. [Google Scholar] [CrossRef]
- Duenas-Gonzalez, A.; Gonzalez-Fierro, A. Pharmacodynamics of current and emerging treatments for cervical cancer. Expert Opin. Drug Metab. Toxicol. 2019, 15, 671–682. [Google Scholar] [CrossRef]
- Kunckler, M.; Schumacher, F.; Kenfack, B.; Catarino, R.; Viviano, M.; Tincho, E.; Tebeu, P.M.; Temogne, L.; Vassilakos, P.; Petignat, P. Cervical cancer screening in a low-resource setting: A pilot study on an HPV-based screen-and-treat approach. Cancer Med. 2017, 6, 1752–1761. [Google Scholar] [CrossRef]
- Kamath Mulki, A.; Withers, M. Human Papilloma Virus self-sampling performance in low- and middle-income countries. BMC Women’s Health 2021, 21, 12. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Substage | Description | Recommended Treatment |
|---|---|---|---|
| I | IA1 | The disease is only found in the cervix | Conization or cone biopsy; simple hysterectomy |
| IA2 | Conization; simple hysterectomy with pelvic lymphadenectomy; radical hysterectomy | ||
| IB1 | Radical hysterectomy or radical trachelectomy with lymphadenectomy | ||
| IB2 | |||
| II | IIA1 | Outside the cervix, cancer has progressed to the upper vaginal wall or tissue next to the cervix (parametrium), but not to the pelvic sidewall(s) | Radical hysterectomy or radical trachelectomy with lymphadenectomy |
| IIA2 | Radiotherapy (RT) with or without chemotherapy | ||
| IIB | Cisplatin-based chemotherapy and concurrent radiation (CRT) | ||
| III | IIIA | Cancer has advanced to the lower region of the vagina and through the parametrium until the pelvic sidewall(s) | Cisplatin-based chemotherapy and concurrent radiation |
| IIIB | |||
| IV | IVA | Cancer has advanced to adjacent organs or distant tissue, such as lungs and distant lymph nodes | Palliative radiotherapy or palliative systemic cytotoxic chemotherapy |
| IVB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, D.; Silvestre, S.; Duarte, A.P.; Venuti, A.; Soares, C.P.; Passarinha, L.; Sousa, Â. In Silico Approaches: A Way to Unveil Novel Therapeutic Drugs for Cervical Cancer Management. Pharmaceuticals 2021, 14, 741. https://doi.org/10.3390/ph14080741
Gomes D, Silvestre S, Duarte AP, Venuti A, Soares CP, Passarinha L, Sousa Â. In Silico Approaches: A Way to Unveil Novel Therapeutic Drugs for Cervical Cancer Management. Pharmaceuticals. 2021; 14(8):741. https://doi.org/10.3390/ph14080741
Chicago/Turabian StyleGomes, Diana, Samuel Silvestre, Ana Paula Duarte, Aldo Venuti, Christiane P. Soares, Luís Passarinha, and Ângela Sousa. 2021. "In Silico Approaches: A Way to Unveil Novel Therapeutic Drugs for Cervical Cancer Management" Pharmaceuticals 14, no. 8: 741. https://doi.org/10.3390/ph14080741
APA StyleGomes, D., Silvestre, S., Duarte, A. P., Venuti, A., Soares, C. P., Passarinha, L., & Sousa, Â. (2021). In Silico Approaches: A Way to Unveil Novel Therapeutic Drugs for Cervical Cancer Management. Pharmaceuticals, 14(8), 741. https://doi.org/10.3390/ph14080741