
Human Hair Keratin Composite Scaffold: Characterisation and Biocompatibility Study on NIH 3T3 Fibroblast Cells

 ,
,  ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
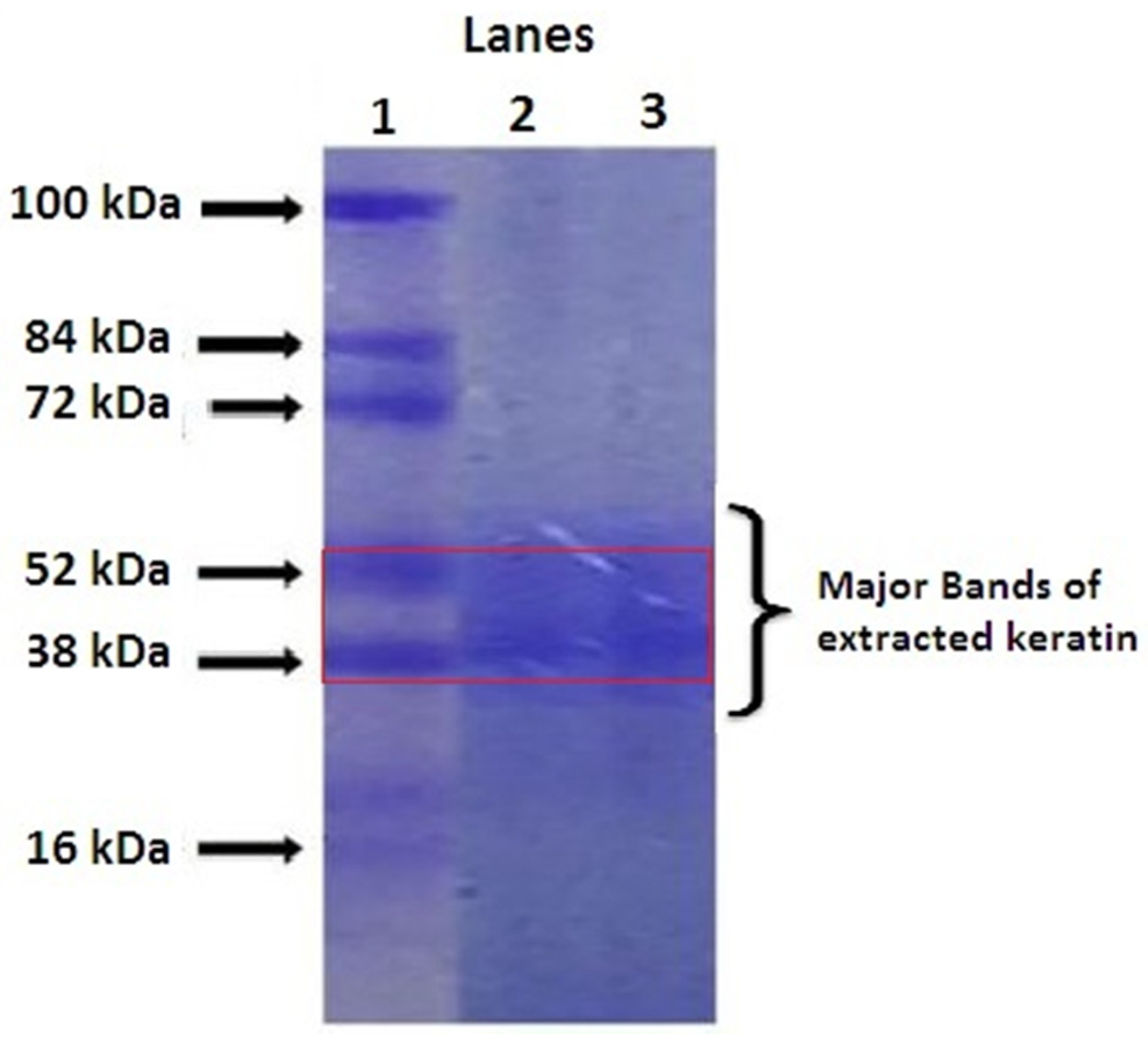
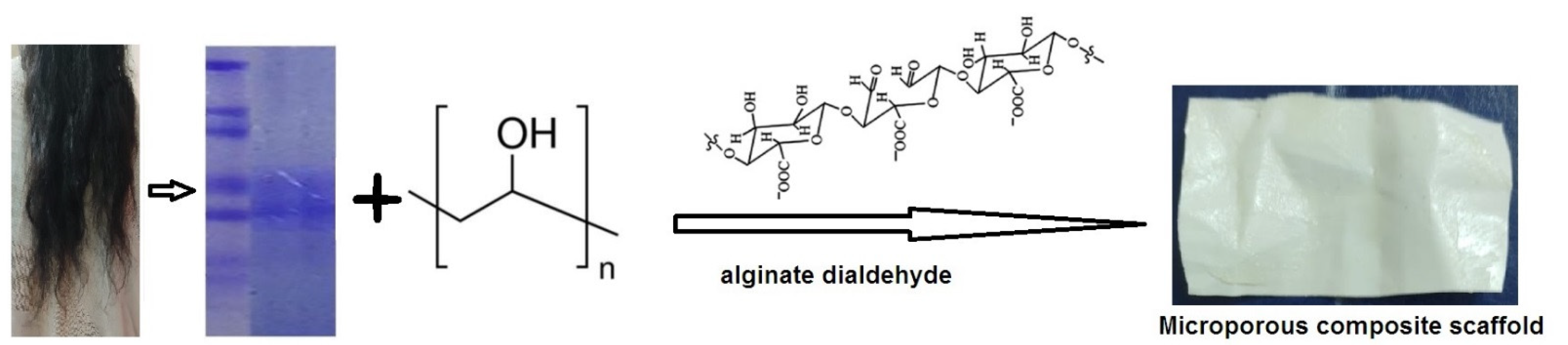
2.1. Quantification of Extracted Keratin by SDS-PAGE
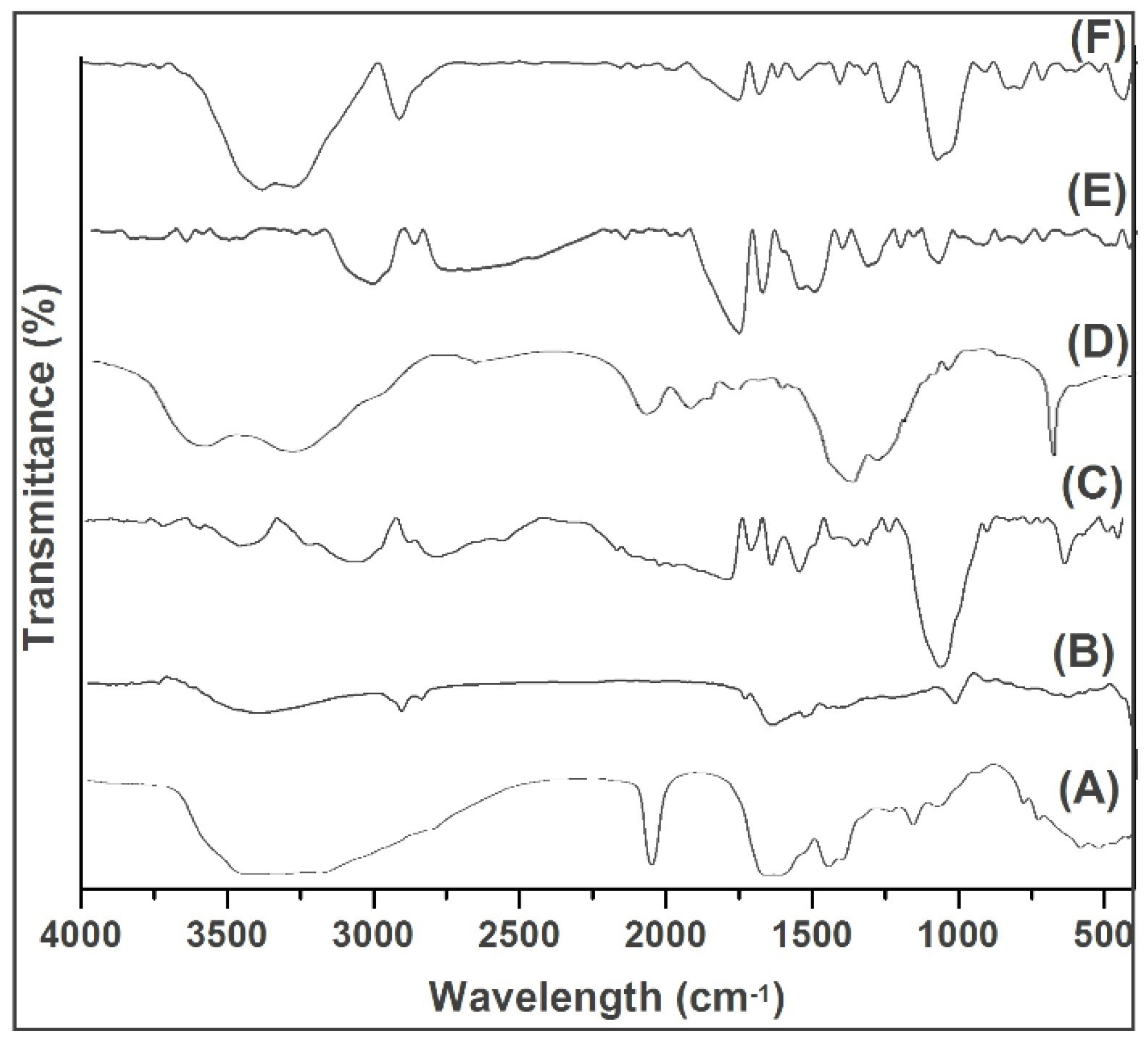
2.2. FTIR
2.3. DSC
2.4. TGA
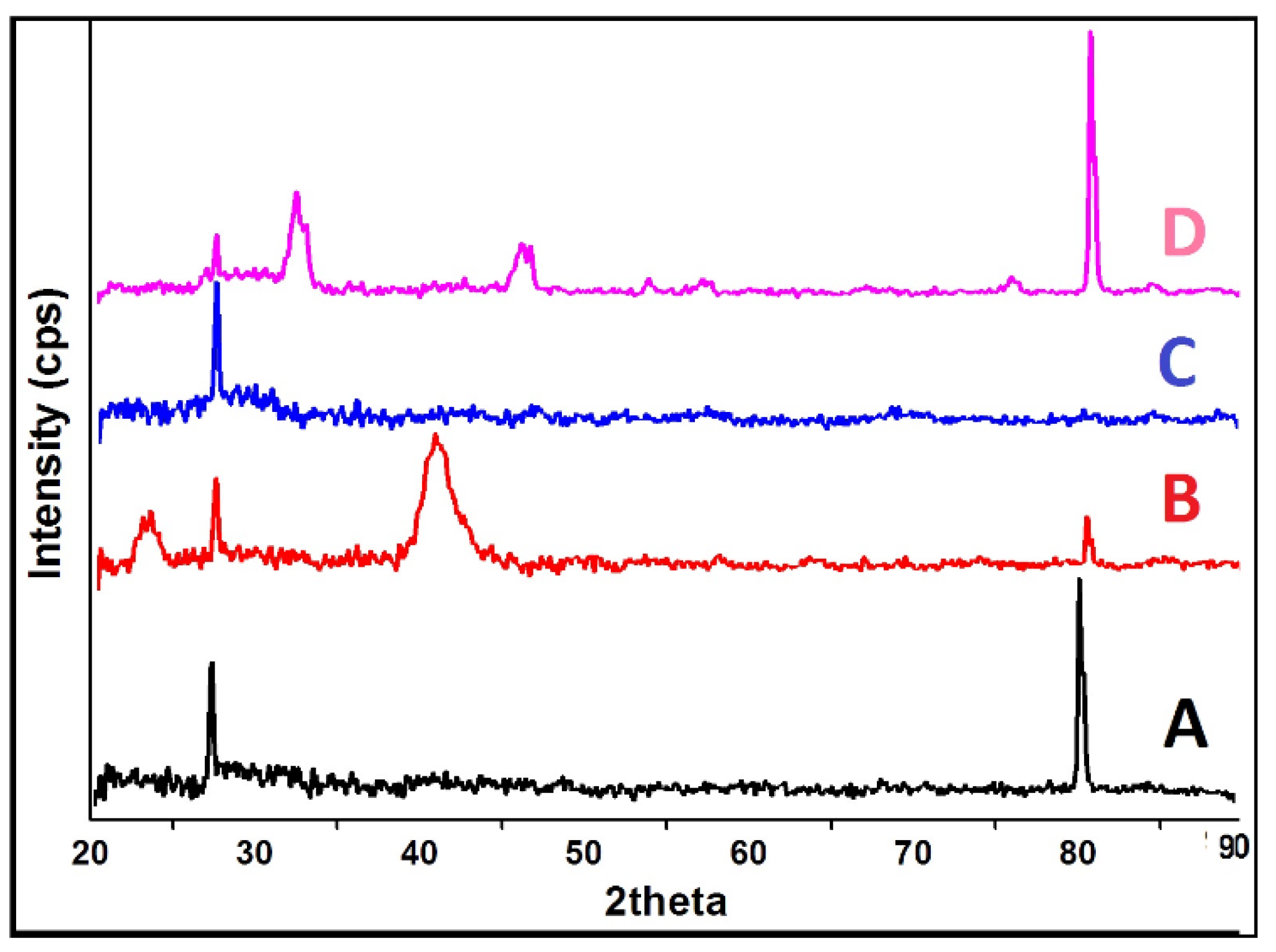
2.5. XRD
2.6. Swelling, Water Absorption and Porosity
2.7. SEM
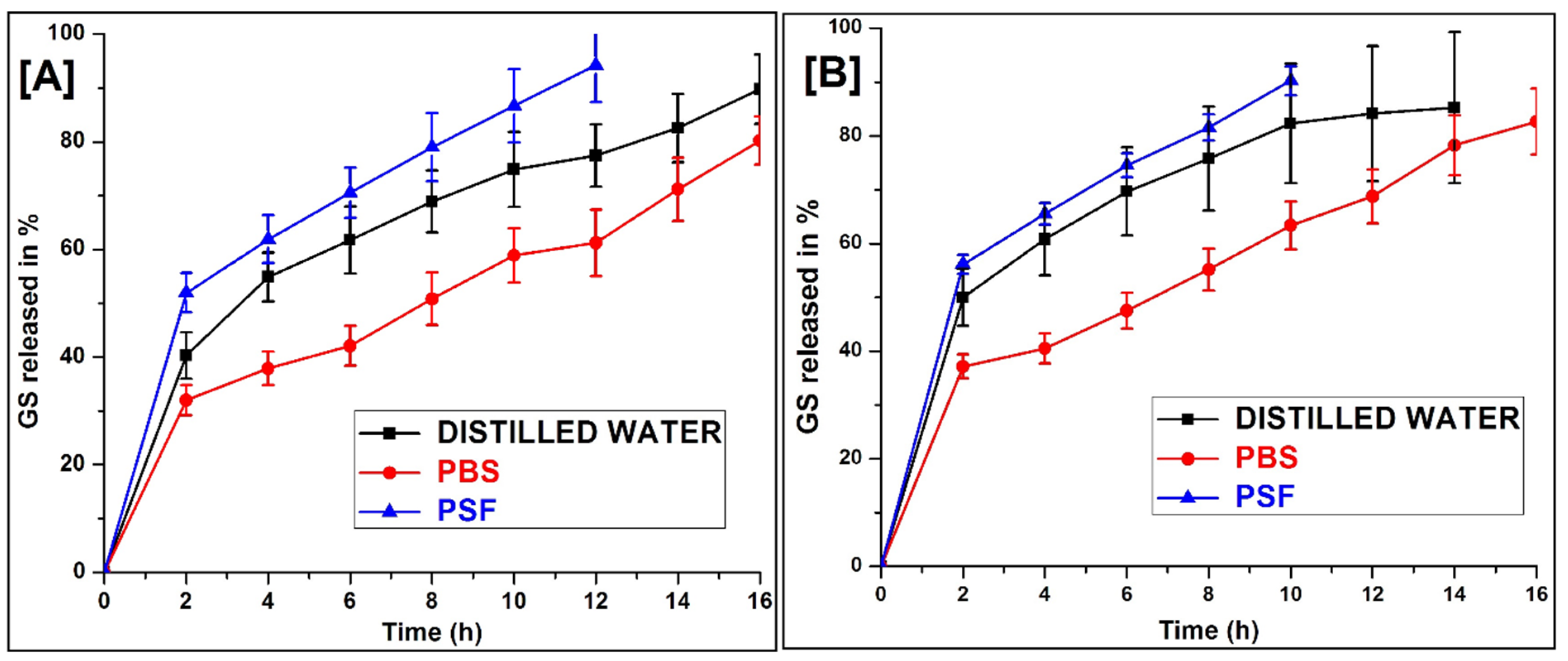
2.8. In Vitro Release
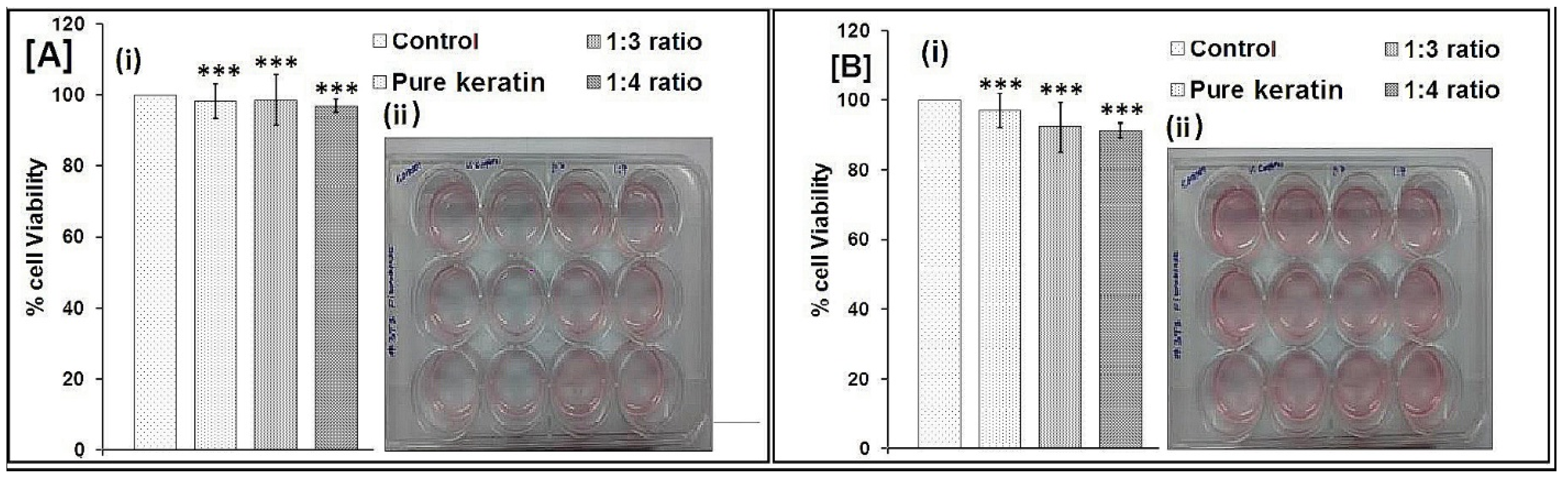
2.9. Biocompatibility Testing by MTT Assay
3. Materials and Methods
3.1. Materials
3.2. Extraction of Keratin
3.2.1. Preparation of Buffer Solution for Keratin Extraction
3.2.2. Extraction of Keratin by Shindai Method
3.3. Preparation of GS Loaded Scaffold
3.4. Characterisation of Scaffolds
3.4.1. SDS Polyacrylamide Gel Electrophoresis (SDS-PAGE)
3.4.2. Fourier Transform Infrared (FTIR) Spectral Analysis
3.4.3. Thermal Properties
3.4.4. Crystallization Analysis
3.5. Swelling, Water Absorption and Porosity
3.6. Morphology
3.7. In-Vitro Drug Release Studies
3.8. Biocompatibility Test
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhivya, S.; Padma, V.V.; Santhini, E. Wound dressings—A review. Biomedicine 2015, 5, 22. [Google Scholar] [CrossRef]
- Das, S.; Baker, A.B. Biomaterials and Nanotherapeutics for Enhancing Skin Wound Healing. Front. Bioeng. Biotechnol. 2016, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, L.E.; Gerecht, S. Engineered Biopolymeric Scaffolds for Chronic Wound Healing. Front. Physiol. 2016, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.; Yu, D. Human Hair: Scaffold Materials for Regenerative Medicine. Adv. Exp. Med. Biol. 2020, 1249, 223–229. [Google Scholar] [CrossRef]
- Bo, T.H.; Yu, W.F.; Wei, D.; Ying, Z.; Jing, D.; Xin, C.D.; Fu, Y.K.; Jun, Y.; Liu, Y.; Qing, X.Y. Fabrication and Evaluation of Porous Keratin/chitosan (KCS) Scaffolds for Effectively Accelerating Wound Healing. Biomed Environ. Sci. 2015, 28, 178–189. [Google Scholar] [CrossRef]
- Mogosanu, G.D.; Grumezescu, A.M.; Chifiriuc, M.C. Keratin-based biomaterials for biomedical applications. Curr. Drug Targets 2014, 15, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.A.; Amorim, M.T.P.; Felgueiras, H.P. Poly(Vinyl Alcohol)-Based Nanofibrous Electrospun Scaffolds for Tissue Engineering Applications. Polymers 2020, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Webster, T.J.; Sinha, A. Evolution of PVA gels prepared without crosslinking agentsas a cell adhesive surface. J. Mater. Sci. Mater. Med. 2011, 22, 1763. [Google Scholar] [CrossRef]
- Fathi, A.; Khanmohammadi, M.; Goodarzi, A.; Foroutani, L.; Mobarakeh, Z.T.; Saremi, J.; Arabpour, Z.; Ai, J. Fabrication of chitosan-polyvinyl alcohol and silk electrospun fiber seeded with differentiated keratinocyte for skin tissue regeneration in animal wound model. J. Biol. Eng. 2020, 14, 27. [Google Scholar] [CrossRef]
- Jang, H.Y.; Shin, J.Y.; Oh, S.H.; Byun, J.-H.; Lee, J.H. PCL/HA Hybrid Microspheres for Effective Osteogenic Differentiation and Bone Regeneration. ACS Biomater. Sci. Eng. 2020, 6, 5172–5180. [Google Scholar] [CrossRef] [PubMed]
- Kirubanandan, S.; Babu, U.D. Macroporous keratin scaffold—A novel biomaterial for biomedical applications. Int. J. Pharm. Chem. Sci. 2012, 1, 1447. [Google Scholar]
- Toivola, D.M.; Zhou, Q.; English, L.S.; Omary, B. Type II Keratins Are Phosphorylated on a Unique Motif during Stress and Mitosis in Tissues and Cultured Cells. Mol. Biol. Cell 2002, 13, 1857–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, T.; Singh, O.; Arora, S.; Murthy, R.S.R. Scaffold: A Novel Carrier for Cell and Drug Delivery. Crit. Rev. Ther. Drug Carr. Syst. 2012, 29, 1–63. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, A.; Cavaco-Paulo, A. The use of keratin in biomedical applications. Curr. Drug Targets 2013, 14, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Sow, W.T.; Devi, D.; Ng, K.W.; Mandal, B.B.; Cho, N.J. Silk fibroin–keratin based 3D scaffolds as a dermal substitute for skin tissue engineering. Integr. Biol. 2014, 7, 53–63. [Google Scholar] [CrossRef]
- Wang, X.; Shi, Z.; Zhao, Q.; Yun, Y. Study on the Structure and Properties of Biofunctional Keratin from Rabbit Hair. Materials 2021, 14, 379. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.M.N. Physically Cross-Linked Gels of PVA with Natural Polymers as Matrices for Manuka Honey Release in Wound-Care Applications. Materials 2019, 12, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichl, S.; Borrelli, M.; Geerling, G. Keratin Films for Ocular Surface Reconstruction. Biomaterials 2011, 32, 3375–3386. [Google Scholar] [CrossRef]
- Xing, Z.; Yuan, J.; Chae, W.; Kang, I.; Kim, S. Keratin Nanofibers as Biometerials. IPCBEE 2011, 2, 120. [Google Scholar]
- Arslan, Y.E.; Efe, B.; Arslan, T.S. A novel method for constructing an acellular 3D biomatrix from bovine spinal cord for neural tissue engineering applications. Biotechnol. Prog. 2019, 35, e2814. [Google Scholar] [CrossRef]
- Selmin, F.; Cilurzoa, F.; Aluigi, A.; Franze, S.; Minghettia, P. Regenerated keratin membrane to match thein vitrodrug diffusion through human epidermis. Result Pharma Sci. 2012, 2, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Dou, Y.; Zhang, B.; He, M.; Yin, G.; Cui, Y.; Savina, I.N. Keratin/Polyvinyl Alcohol Blend Films Cross-Linked by Dialdehyde Starch and Their Potential Application for Drug Release. Polymers 2015, 7, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, P.; Madhan, B.; Shanmugam, G. Extraction and characterization of keratin from bovine hoof: A potential material for biomedical applications. SpringerPlus 2014, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sow, W.T.; Lui, Y.S.; Ng, K.W. Electrospun human keratin matrices as templates for tissue regeneration. Nanomedicine 2013, 8, 531–541. [Google Scholar] [CrossRef]
- Barua, E.; Deoghare, A.; Chatterjee, S.; Mate, V.R. Characterization of Mechanical and Micro-Architectural Properties of Porous Hydroxyapatite Bone Scaffold Using Green MicroAlgae as Binder. Arab. J. Sci. Eng. 2019, 44, 7707–7722. [Google Scholar] [CrossRef]
- Mabrouk, M.; Mostafa, A.; Oudadesse, H.; Mahmoud, A.; El-Gohary, M. Effect of ciprofloxacin incorporation in PVA and PVA bioactive glass composite scaffolds. Ceram. Int. 2014, 40, 4833–4845. [Google Scholar] [CrossRef] [Green Version]
- Golafshan, N.; Rezahasani, R.; Esfahani, M.T.; Kharaziha, M.; Khorasani, S. Nanohybrid hydrogels of laponite: PVA-Alginate as a potential wound healing material. Carbohydr. Polym. 2017, 176, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Mohanty, S.; Nayak, S. Effect of Different Solvents in Solvent Casting of Porous PLA Scaffolds—In Biomedical and Tissue Engineering Applications. J. Tissue Sci. Eng. 2014, 6, 1. [Google Scholar] [CrossRef]
- Xu, S.; Sang, L.; Zhang, P.; Wang, X.; Li, X. Biological evaluation of human hair keratin scaffolds for skin wound repair and regeneration. Mater. Sci. Eng. C 2013, 33, 648–655. [Google Scholar] [CrossRef]
- Fan, J.; Yu, M.-Y.; Lei, T.-D.; Wang, Y.-H.; Cao, F.-Y.; Qin, X.; Liu, Y. In Vivo Biocompatibility and Improved Compression Strength of Reinforced Keratin/Hydroxyapatite Scaffold. Tissue Eng. Regen. Med. 2018, 15, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Chen, X.; Zhai, D.; Gao, F.; Guo, T.; Li, W.; Hao, S.; Ji, J.; Wang, B. Development of keratin nanoparticles for controlled gastric mucoadhesion and drug release. J. Nanobiotechnology 2018, 16, 24. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, C.; Sotgiu, G.; Sagnella, A.; Varchi, G.; Guerrini, A.; Giuri, D.; Polo, E.; Orlandi, V.T.; Marras, E.; Gariboldi, M.; et al. Wool Keratin 3D Scaffolds with Light-Triggered Antimicrobial Activity. Biomacromolecules 2016, 17, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Yahya, R.; Hassan, A.; Yar, M.; Azzahari, A.D.; Selvanathan, V.; Sonsudin, F.; Abouloula, C.N. pH Sensitive Hydrogels in Drug Delivery: Brief History, Properties, Swelling, and Release Mechanism, Material Selection and Applications. Polymers 2017, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R. Keratins in wound healing. In Wound Healing Biomaterials; Woodhead Publishing: Sawston, UK, 2016; pp. 353–365. [Google Scholar] [CrossRef]
- Havryliak, V.; Mykhaliuk, V. The comparative analysis of the methods for keratin extraction from sheep wool and human hair. Bìol. Tvarin. 2020, 22, 9–12. [Google Scholar] [CrossRef]
- Wong, S.Y.; Lee, C.C.; Ashrafzadeh, A.; Junit, S.M.; Abrahim, N.; Hashim, O.H. A High-Yield Two-Hour Protocol for Extraction of Human Hair Shaft Proteins. PLoS ONE 2016, 11, e0164993. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Nanda, S.; Sood, N.; Reddy, B.V.K.; Markandeywar, T.S. Preparation and Characterization of Poly(vinyl alcohol)-chondroitin Sulphate Hydrogel as Scaffolds for Articular Cartilage Regeneration. Indian J. Mater. Sci. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Balaji, S.; Kumar, R.; Sripriya, R.; Kakkar, P.; Ramesh, D.V.; Reddy, P.N.K.; Sehgal, P. Preparation and comparative characterization of keratin–chitosan and keratin–gelatin composite scaffolds for tissue engineering applications. Mater. Sci. Eng. C 2012, 32, 975–982. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Muthu, M.J.M.; Kavitha, K.; Ruckmani, K.; Shanmuganathan, S. Skimmed milk powder and pectin decorated solid lipid nanoparticle containing soluble curcumin used for the treatment of colorectal cancer. J. Food Process Eng. 2019, 43, 1–15. [Google Scholar] [CrossRef]
- Mohamed, J.M.; Alqahtani, A.; Ahmad, F.; Krishnaraju, V.; Kalpana, K. Pectin co-functionalized dual layered solid lipid nanoparticle made by soluble curcumin for the targeted potential treatment of colorectal cancer. Carbohydr. Polym. 2020, 252, 117180. [Google Scholar] [CrossRef] [PubMed]
- Moideen, M.M.J.; Alqahtani, A.; Venkatesan, K.; Ahmad, F.; Krisharaju, K.; Gayasuddin, M.; Shaik, R.A.; Ibraheem, K.M.M.; Salama, M.E.M.; Abed, S.Y. Application of the Box–Behnken design for the production of soluble curcumin: Skimmed milk powder inclusion complex for improving the treatment of colorectal cancer. Food Sci. Nutr. 2020, 8, 6643–6659. [Google Scholar] [CrossRef]
- Singaravelu, S.; Ramanathan, G.; Muthukumar, T.; Raja, M.D.; Nagiah, N.; Thyagarajan, S.; Aravinthan, A.; P., G.; Natarajan, T.S.; Selva, G.V.N.G.; et al. Durable keratin-based bilayered electrospun mats for wound closure. J. Mater. Chem. B 2016, 4, 3982–3997. [Google Scholar] [CrossRef] [PubMed]
- Escobar, A.; Muzzio, N.E.; Andreozzi, P.; Libertone, S.; Tasca, E.; Azzaroni, O.; Grzelczak, M.; Moya, S.E. Antibacterial Layer-by-Layer Films of Poly(acrylic acid)–Gentamicin Complexes with a Combined Burst and Sustainable Release of Gentamicin. Adv. Mater. Interfaces 2019, 6, 22–1901373. [Google Scholar] [CrossRef]
- Wang, S.; Taraballi, F.; Tan, L.P.; Ng, K.W. Human keratin hydrogels support fibroblast attachment and proliferation in vitro. Cell Tissue Res. 2012, 347, 795–802. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold Ratio | Swelling Index (%) | Water Absorption (%) | Porosity (%) | |
|---|---|---|---|---|
| Size | Weight | |||
| 1:3 | 1.52 ± 0.15 | 26 ± 2.31 | 72.93 ± 9.26 | 68.93 ± 1.33 |
| 1:4 | 2.04 ± 0.09 | 31 ± 2.88 | 83.64 ± 14.29 | 66.05 ± 1.97 |
| Ratio | mg/50 mL (w/v) | |||
|---|---|---|---|---|
| Keratin | PVA (2%) | Alginate Dialdehyde | GS | |
| 1:2 | 4 | 8 | 5 | 4 |
| 1:3 | 4 | 12 | 5 | 4 |
| 1:4 | 4 | 16 | 5 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, J.M.M.; Alqahtani, A.; Al Fatease, A.; Alqahtani, T.; Khan, B.A.; Ashmitha, B.; Vijaya, R. Human Hair Keratin Composite Scaffold: Characterisation and Biocompatibility Study on NIH 3T3 Fibroblast Cells. Pharmaceuticals 2021, 14, 781. https://doi.org/10.3390/ph14080781
Mohamed JMM, Alqahtani A, Al Fatease A, Alqahtani T, Khan BA, Ashmitha B, Vijaya R. Human Hair Keratin Composite Scaffold: Characterisation and Biocompatibility Study on NIH 3T3 Fibroblast Cells. Pharmaceuticals. 2021; 14(8):781. https://doi.org/10.3390/ph14080781
Chicago/Turabian StyleMohamed, Jamal Moideen Muthu, Ali Alqahtani, Adel Al Fatease, Taha Alqahtani, Barkat Ali Khan, B. Ashmitha, and R. Vijaya. 2021. "Human Hair Keratin Composite Scaffold: Characterisation and Biocompatibility Study on NIH 3T3 Fibroblast Cells" Pharmaceuticals 14, no. 8: 781. https://doi.org/10.3390/ph14080781
APA StyleMohamed, J. M. M., Alqahtani, A., Al Fatease, A., Alqahtani, T., Khan, B. A., Ashmitha, B., & Vijaya, R. (2021). Human Hair Keratin Composite Scaffold: Characterisation and Biocompatibility Study on NIH 3T3 Fibroblast Cells. Pharmaceuticals, 14(8), 781. https://doi.org/10.3390/ph14080781