
Heliciopsides A−E, Unusual Macrocyclic and Phenolic Glycosides from the Leaves of Heliciopsis terminalis and Their Stimulation of Glucose Uptake
 , , and
, , and
Abstract
:
1. Introduction
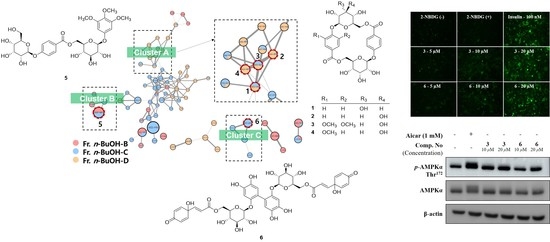
2. Results and Discussion
2.1. Structure Elucidation
2.2. Stimulation of Glucose Uptake
2.3. Compounds 3 and 6 Increased Glucose Uptake by Activating the AMPK Signaling Pathway in Differentiated C2C12 Myoblasts
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. LC-MS/MS Analysis and Molecular Networking
3.4. Extraction and Isolation
3.5. Spectroscopic and Physical Characteristics of Compounds
3.6. Determination of the Absolute Configurations of the Sugars in Compounds 1–3, 5 and 6
3.7. Differentiation of 3T3-L1 Adipocytes
3.8. Cell Viability Assay
3.9. Measurement of Glucose Uptake Using the 2-NBDG Probe
3.10. Differentiation of C2C12 Myoblasts
3.11. Detection of p-AMPKα Thr172 by Western blotting
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, W.Y.; Dall, T.M.; Beronjia, K.; Lin, J.; Semilla, A.P.; Chakrabarti, R.; Hogan, P.F.; Assoc, A.D. Economic Costs of Diabetes in the US in 2017. Diabetes Care 2018, 41, 917–928. [Google Scholar]
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef]
- Funaki, M.; Randhawa, P.; Janmey, P.A. Separation of insulin signaling into distinct GLUT4 translocation and activation steps. Mol. Cell. Biol. 2004, 24, 7567–7577. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef]
- Krook, A.; Wallberg-Henriksson, H.; Zierath, J.R. Sending the signal: Molecular mechanisms regulating glucose uptake. Med. Sci. Sports Exerc. 2004, 36, 1212–1217. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. 2014, 7, 241–253. [Google Scholar]
- O’Neill, H.M. AMPK and exercise: Glucose uptake and insulin sensitivity. Diabetes Metab. J. 2013, 37, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Habegger, K.M.; Hoffman, N.J.; Ridenour, C.M.; Brozinick, J.T.; Elmendorf, J.S. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 2012, 153, 2130–2141. [Google Scholar] [CrossRef]
- Umezawa, S.; Higurashi, T.; Nakajima, A. AMPK: Therapeutic target for diabetes and cancer prevention. Curr. Pharm Des. 2017, 23, 3629–3644. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, H.M.; Park, E.J.; Lee, B.W.; Nghiem, D.T.; Pham, H.T.; Pan, C.H.; Oh, W.K. Triterpenoid saponins from the leaves and stems of Pericampylus glaucus and their insulin mimetic activities. Bioorg. Chem. 2021, 117, 105445. [Google Scholar] [CrossRef]
- Kim, H.W.; Park, E.J.; Cho, H.M.; An, J.P.; Chin, Y.W.; Kim, J.; Sung, S.H.; Oh, W.K. Glucose uptake-stimulating galloyl ester triterpenoids from Castanopsis sieboldii. J. Nat. Prod. 2020, 83, 3093–3101. [Google Scholar] [CrossRef]
- An, J.P.; Park, E.J.; Ryu, B.; Lee, B.W.; Cho, H.M.; Doan, T.P.; Pham, H.T.T.; Oh, W.K. Oleanane triterpenoids from the leaves of Gymnema inodorum and their insulin mimetic activities. J. Nat. Prod. 2020, 83, 1265–1274. [Google Scholar] [CrossRef]
- Lee, B.W.; Ha, T.K.Q.; Pham, H.T.T.; Hoang, Q.H.; Tran, V.O.; Oh, W.K. Hydroxyoleoside-type seco-iridoids from Symplocos cochinchinensis and their insulin mimetic activity. Sci. Rep. 2019, 9, 2270. [Google Scholar] [CrossRef] [Green Version]
- Pham, H.T.T.; Ha, T.K.Q.; Cho, H.M.; Lee, B.W.; An, J.P.; Tran, V.O.; Oh, W.K. Insulin mimetic activity of 3,4-seco and hexanordammarane triterpenoids isolated from Gynostemma longipes. J. Nat. Prod. 2018, 81, 2470–2482. [Google Scholar] [CrossRef]
- Kubitzki, K.; Bayer, C.; Stevens, P.F. Flowering Plants: Eudicots; Berberidopsidales, Buxales, Crossosomatales, Fabales p.p., Geraniales, Gunnerales, Myrtales p.p., Proteales, Saxifragales, Vitales, Zygophyllales, Clusiaceae Alliance, Passifloraceae Alliance, Dilleniaceae, Huaceae, Picramniaceae, Sabiaceae; Springer: Berlin, Germany; New York, NY, USA, 2007. [Google Scholar]
- Giang, P.M.; Thao, D.T.; Nga, N.T.; Trung, B.V.; Anh, D.H.; Viet, P.H. Evaluation of the antioxidant, hepatoprotective, and anti-Inflammatory activities of bisresorcinol isolated from the trunk of Heliciopsis terminalis. Pharm. Chem. J. 2019, 53, 628–634. [Google Scholar] [CrossRef]
- Prommee, N.; Itharat, A.; Panthong, S.; Makchuchit, S.; Ooraikul, B. Ethnopharmacological analysis from Thai traditional medicine called prasachandaeng remedy as a potential antipyretic drug. J. Ethnopharmacol. 2021, 268, 113520. [Google Scholar] [CrossRef]
- Saechan, C.; Nguyen, U.H.; Wang, Z.; Sugimoto, S.; Yamano, Y.; Matsunami, K.; Otsuka, H.; Phan, G.M.; Pham, V.H.; Tipmanee, V.; et al. Potency of bisresorcinol from Heliciopsis terminalis on skin aging: In vitro bioactivities and molecular interactions. PeerJ 2021, 9, e11618. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Choi, H.S.; Ha, T.K.Q.; Seo, J.Y.; Yang, J.L.; Jung, D.W.; Williams, D.R.; Oh, W.K. Anthraquinones from Morinda longissima and their insulin mimetic activities via AMP-activated protein kinase (AMPK) activation. Bioorg. Med. Chem. Lett. 2017, 27, 40–44. [Google Scholar] [CrossRef]
- Qiu, L.; Yuan, H.M.; Liang, J.M.; Cheng, X.L.; Wang, P.; Du, Y.F.; Fu, Q. Clemochinenosides C and D, two new macrocyclic glucosides from Clematis chinensis, J. Asian Nat. Prod. Res. 2018, 20(11), 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.S.; Nakamura, N.; Meselhy, M.R.; Makhboul, M.A.; El-Emary, N.; Hattori, M. Phenolic constituents from Grevillea robusta. Phytochemistry 2000, 53, 149–154. [Google Scholar] [CrossRef]
- Achenbach, H.; Benirschke, G. Joannesialactone and other compounds from Joannesia princeps. Phytochemistry 1997, 45, 149–157. [Google Scholar] [CrossRef]
- Tsao-Jun Su, H.-S.C.; Peng, C.-F.; Lee, S.-J.; Chen, I.-S. Antitubercular resorcinols and cytotoxic alkyl benzoquinones from Ardisia kusukuensis. Taiwan Pharm. J. 2009, 61, 89–105. [Google Scholar]
- Shi, S.P.; Dong, C.X.; Jiang, D.; Tu, P.F. Macrocyclic glycosides from Clematis hexapetala. Helv. Chim. Acta 2006, 89, 3002–3006. [Google Scholar] [CrossRef]
- Wang, X.Y.; Xu, M.; Yang, C.R.; Zhang, Y.J. Phenylpropanoid glycosides from the seeds of Michelia hedyosperma. Food Chem. 2011, 126, 1039–1043. [Google Scholar] [CrossRef]
- Yan, L.H.; Yang, S.L.; Zou, Z.M.; Luo, X.Z.; Xu, L.Z. Two new macrocyclic compounds from the stems of Clematis armandii. Heterocycles 2006, 68, 1917–1924. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Lin, X.L.; Jiang, L.; Huang, J.; Zeng, G.Y.; Deng, X.; Zhou, Y.J. Five macrocyclic glycosides from Schoenoplectus tabernaemontani. Nat. Prod. Res. 2019, 33, 427–434. [Google Scholar] [CrossRef]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Joshi, T.; Singh, A.K.; Haratipour, P.; Sah, A.N.; Pandey, A.K.; Naseri, R.; Juyal, V.; Farzaei, M.H. Targeting AMPK signaling pathway by natural products for treatment of diabetes mellitus and its complications. J. Cell. Physiol. 2019, 234, 17212–17231. [Google Scholar] [CrossRef]
- Uddin, M.N.; Choi, H.S.; Lim, S.-I.; Oh, W.K. AMPK activators from natural products: A patent review. Nat. Prod. Sci. 2013, 19, 1–17. [Google Scholar]
- Yin, J.; Gao, Z.G.; Liu, D.; Liu, Z.J.; Ye, J.P. Berberine improves glucose metabolism through induction of glycolysis. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E148–E156. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.H.; Le, T.V.T.; Kang, H.W.; Chae, J.; Kim, S.K.; Kwon, K.I.; Seo, D.B.; Lee, S.J.; Oh, W.K. AMP-activated protein kinase (AMPK) activators from Myristica fragrans (nutmeg) and their anti-obesity effect. Bioorg. Med. Chem. Lett. 2010, 20, 4128–4131. [Google Scholar] [CrossRef]
- Wang, J.; Ha, T.K.Q.; Shi, Y.P.; Oh, W.K.; Yang, J.L. Hypoglycemic triterpenes from Gynostemma pentaphyllum. Phytochemistry 2018, 155, 171–181. [Google Scholar] [CrossRef]
- Pu, P.; Wang, X.A.; Salim, M.; Zhu, L.H.; Wang, L.; Chen, K.J.; Xiao, J.F.; Deng, W.; Shi, H.W.; Jiang, H.; et al. Baicalein, a natural product, selectively activating AMPKα(2) and ameliorates metabolic disorder in diet-induced mice. Mol. Cell. Endocrinol. 2012, 362, 128–138. [Google Scholar] [CrossRef]
- Deans, B.J.; Kilah, N.L.; Jordan, G.J.; Bissember, A.C.; Smith, J.A. Arbutin derivatives isolated from ancient Proteaceae: Potential phytochemical markers present in Bellendena, Cenarrhenes, and Persoonia Genera. J. Nat. Prod. 2018, 81, 1241–1251. [Google Scholar] [CrossRef]
- Qi, W.Y.; Ou, N.; Wu, X.D.; Xu, H.M. New arbutin derivatives from the leaves of Heliciopsis lobata with cytotoxicity. Chin. J. Nat. Med. 2016, 14, 789–793. [Google Scholar] [CrossRef]
- Yamashita-Higuchi, Y.; Sugimoto, S.; Matsunami, K.; Otsuka, H.; Nakai, T. Grevillosides J−Q, arbutin derivatives from the leaves of Grevillea robusta and their melanogenesis inhibitory activity. Chem. Pharm. Bull. 2014, 62, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Mukhtar, N.; Malik, A.; Riaz, N.; Iqbal, K.; Tareen, R.B.; Khan, S.N.; Nawaz, S.A.; Siddiqui, J. Pakistolides A and B, novel rnzyme inhibitory and antioxidant dimeric 4-(glucosyloxy)benzoates from Berchemia pakistanica. Helv. Chim. Acta 2004, 87, 416. [Google Scholar] [CrossRef]
- Song, C.-Q.; Xu, R.-S. Clemochinenoside A, a macrocyclic compound from Clamatis chinensis. Chin. Chem. Lett. 1992, 3, 119. [Google Scholar]
- Sakurai, N.; Kobayashi, M.; Shigihara, A.; Inoue, T. Berchemolide, a novel dimeric vanillic acid glucoside from Berchemia racemose. Chem. Pharm. Bull. 1992, 40, 851–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, J.; Gong, Y.; Yu, B. Synthesis of oligomeric 4-(glycosyloxy)benzoate macrocyclic glycosides. J. Org. Chem. 2011, 76, 3654–3663. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 5 b | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 122.4 | - | 122.6 | - | 124.6 | - | 153.6 | - |
| 2 | 131.3 | 8.02, d (8.8) | 131.2 | 7.99, d (8.8) | 107.0 | 7.09, d (1.6) | 94.3 | 6.32, s |
| 3 | 115.8 | 7.19, d (8.8) | 115.8 | 7.19, d (8.8) | 153.1 | - | 153.1 | - |
| 4 | 160.6 | - | 160.4 | - | 137.7 | - | 132.6 | - |
| 5 | 115.8 | 7.19, d (8.8) | 115.8 | 7.19, d (8.8) | 152.0 | - | 153.1 | - |
| 6 | 131.3 | 8.02, d (8.8) | 131.2 | 7.99, d (8.8) | 106.7 | 7.52, d (1.6) | 94.3 | 6.32, s |
| 7 | 165.1 | - | 165.0 | - | 164.9 | - | - | |
| d-allo | d-glc | d-glc | d-glc | |||||
| 1′ | 96.8 | 5.39, d (8.0) | 98.4 | 5.22, d (7.2) | 100.9 | 5.36, d (7.2) | 100.2 | 4.98, d (8.0) |
| 2′ | 69.9 | 3.56, dd (8.0, 2.4) | 72.9 | 3.34, m | 73.9 | 3.30, overlap | 73.1 | 3.26, overlap |
| 3′ | 71.5 | 3.99, dd (2.4, 2.4) | 76.7 | 3.38, m | 76.7 | 3.30, overlap | 76.3 | 3.32, overlap |
| 4′ | 68.3 | 3.45, dd (9.6, 2.4) | 70.7 | 3.17, m | 71.1 | 3.12, dd (9.6, 8.8) | 70.0 | 3.26, overlap |
| 5′ | 71.2 | 4.29, ddd (10.4, 9.6, 2.4) | 73.5 | 3.97, m | 73.8 | 3.49, dd (10.4, 2.4) | 73.7 | 3.80, m |
| 6a′ 6b′ | 65.4 | 4.37, dd (11.2, 1.6) 4.11, dd (11.2, 10.4) | 65.0 | 4.40, dd (11.2, 2.4) 4.07, br d (11.2) | 64.9 | 4.40, br d (11.2) 3.89, dd (11.2, 10.4) | 64.2 | 4.58, br d (11.5) 4.23, dd (11.5, 7.0) |
| 1″ | - | - | 122.5 | - | 122.5 | - | 122.9 | - |
| 2″ | - | - | 131.2 | 7.98, d (8.8) | 130.7 | 7.45, d (8.8) | 131.1 | 7.86, d (8.5) |
| 3″ | - | - | 115.8 | 7.18, d (8.8) | 115.9 | 6.98, d (8.8) | 155.9 | 7.09, d (8.5) |
| 4″ | - | - | 160.7 | - | 160.7 | - | 161.4 | - |
| 5″ | - | - | 115.8 | 7.18, d (8.8) | 115.9 | 6.98, d (8.8) | 155.9 | 7.09, d (8.5) |
| 6″ | - | - | 131.2 | 7.98, d (8.8) | 130.7 | 7.45, d (8.8) | 131.1 | 7.86, d (8.5) |
| 7″ | - | - | 165.1 | - | 164.87 | - | 165.3 | - |
| d-allo | d-allo | d-allo | ||||||
| 1‴ | - | - | 96.8 | 5.37, d (8.0) | 97.6 | 5.17, d (8.0) | 98.1 | 5.23, d (7.5) |
| 2‴ | - | - | 69.9 | 3.54, m | 70.0 | 3.53, dd (8.0, 2.4) | 70.2 | 3.46, overlap |
| 3‴ | - | - | 71.5 | 3.97, m | 71.5 | 3.99, m | 71.5 | 3.94, m |
| 4‴ | - | - | 68.2 | 3.43, m | 68.3 | 3.45, m | 66.9 | 3.46, overlap |
| 5‴ | - | - | 71.2 | 4.26, ddd (10.4, 10.4, 1.6) | 71.6 | 4.28, ddd (10.4, 10.4, 1.6) | 74.7 | 3.73, m |
| 6a‴ 6b‴ | - | - | 65.4 | 4.36, dd (11.2, 1.6) 4.09, br d (11.2) | 64.8 | 4.50, dd (11.2, 11.2) 4.40, br d (11.2) | 60.8 | 3.68, m 3.46, overlap |
| 3-OMe | - | - | - | - | 56.0 | 3.61, s | 55.8 | 3.65, s |
| 4-OMe | - | - | - | - | - | - | 60.2 | 3.57, s |
| 5-OMe | - | - | - | - | 56.5 | 3.98, s | 55.8 | 3.65, s |
| No. | δC | δH (J in Hz) | No. | δC | δH (J in Hz) | No. | δC | δH (J in Hz) |
|---|---|---|---|---|---|---|---|---|
| 1 | 148.8 | 1′ | 103.3 | 4.63, d (7.7) | 1″ | 70.4 | - | |
| 2 | 106.7 | 6.66, s | 2′ | 74.8 | 3.27, m | 2″,6″ | 151.2 | 6.89, m |
| 3 | 141.5 | 3′ | 77.7 | 3.33, overlap | 3″,5″ | 128.7 | 6.22, m | |
| 4 | 145.9 | 4′ | 71.4 | 3.33, overlap | 4″ | 187.2 | - | |
| 5 | 119.1 | 6.62, s | 5′ | 75.3 | 3.49, m | 7″ | 148.1 | 6.71, d (16.2) |
| 6 | 121.4 | 6′a 6′b | 64.8 | 4.44, dd (11.2, 2.4) 4.29, dd (11.2, 6.4) | 8″ | 122.9 | 6.30, d (16.2) | |
| 9″ | 167.4 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, B.; Park, E.-J.; Doan, T.-P.; Cho, H.-M.; An, J.-P.; Pham, T.-L.-G.; Pham, H.-T.-T.; Oh, W.-K. Heliciopsides A−E, Unusual Macrocyclic and Phenolic Glycosides from the Leaves of Heliciopsis terminalis and Their Stimulation of Glucose Uptake. Pharmaceuticals 2022, 15, 1315. https://doi.org/10.3390/ph15111315
Ryu B, Park E-J, Doan T-P, Cho H-M, An J-P, Pham T-L-G, Pham H-T-T, Oh W-K. Heliciopsides A−E, Unusual Macrocyclic and Phenolic Glycosides from the Leaves of Heliciopsis terminalis and Their Stimulation of Glucose Uptake. Pharmaceuticals. 2022; 15(11):1315. https://doi.org/10.3390/ph15111315
Chicago/Turabian StyleRyu, Byeol, Eun-Jin Park, Thi-Phuong Doan, Hyo-Moon Cho, Jin-Pyo An, Thi-Linh-Giang Pham, Ha-Thanh-Tung Pham, and Won-Keun Oh. 2022. "Heliciopsides A−E, Unusual Macrocyclic and Phenolic Glycosides from the Leaves of Heliciopsis terminalis and Their Stimulation of Glucose Uptake" Pharmaceuticals 15, no. 11: 1315. https://doi.org/10.3390/ph15111315
APA StyleRyu, B., Park, E. -J., Doan, T. -P., Cho, H. -M., An, J. -P., Pham, T. -L. -G., Pham, H. -T. -T., & Oh, W. -K. (2022). Heliciopsides A−E, Unusual Macrocyclic and Phenolic Glycosides from the Leaves of Heliciopsis terminalis and Their Stimulation of Glucose Uptake. Pharmaceuticals, 15(11), 1315. https://doi.org/10.3390/ph15111315