
Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors

Abstract
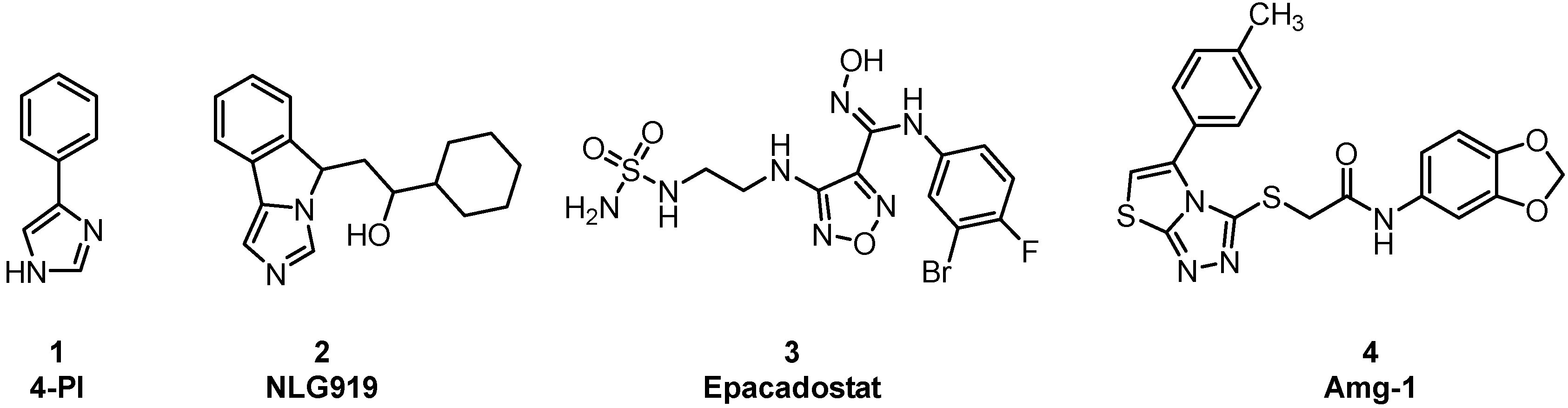
:1. Introduction
2. Results and Discussion
2.1. IDO1 Inhibition Study
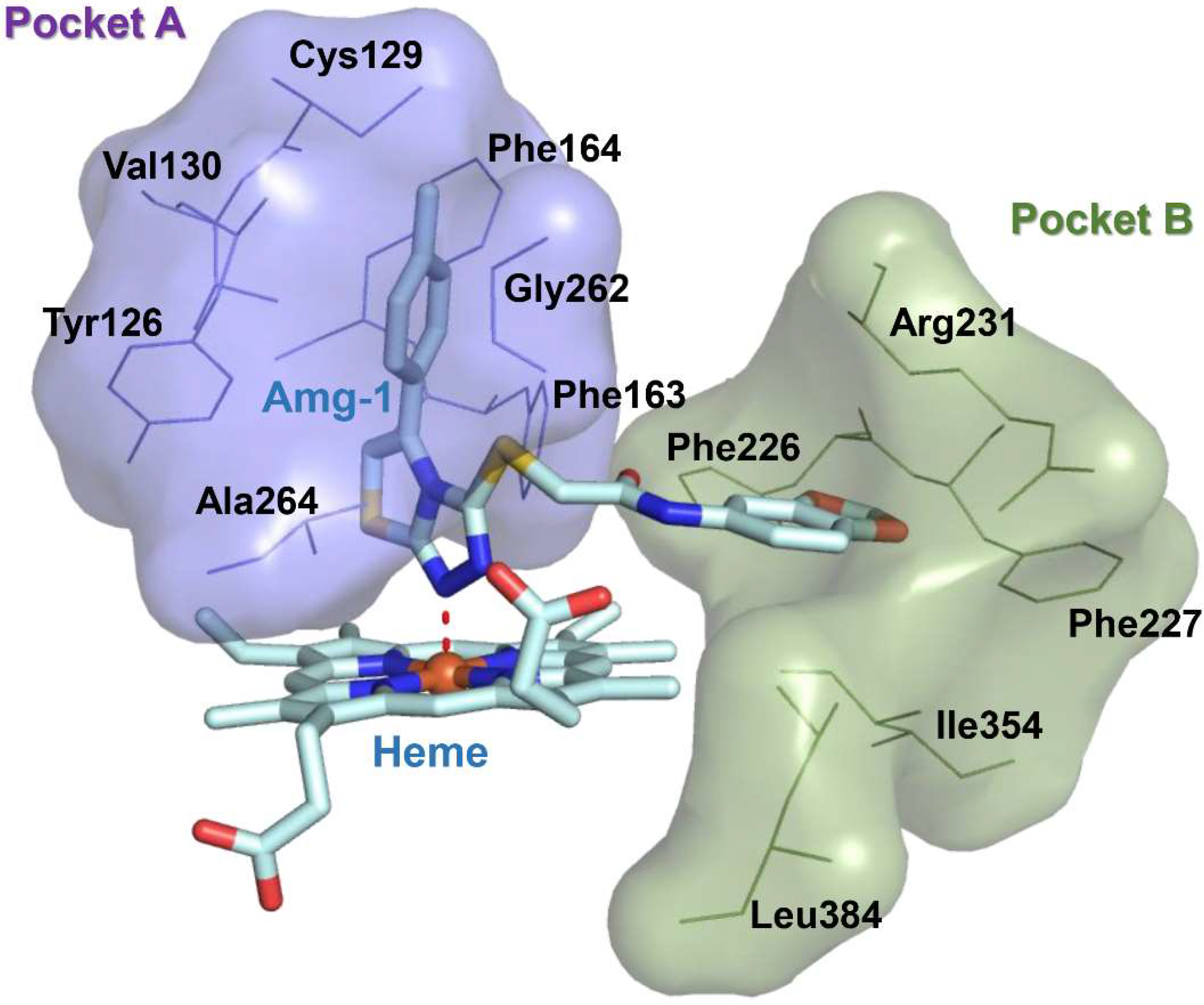
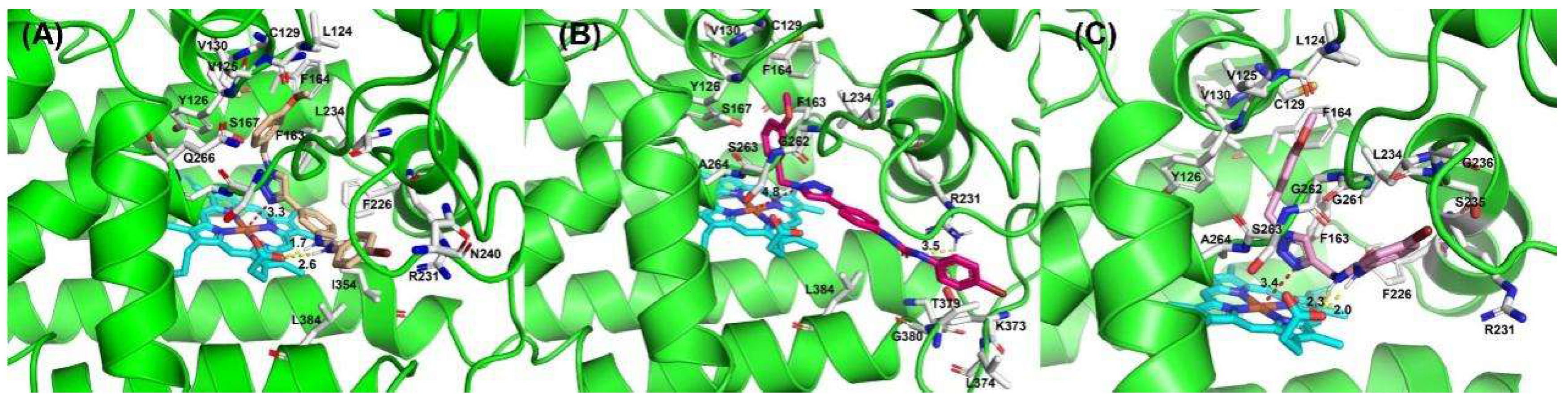
2.2. Molecular Docking Studies of Compounds 3a, 5e and 7f
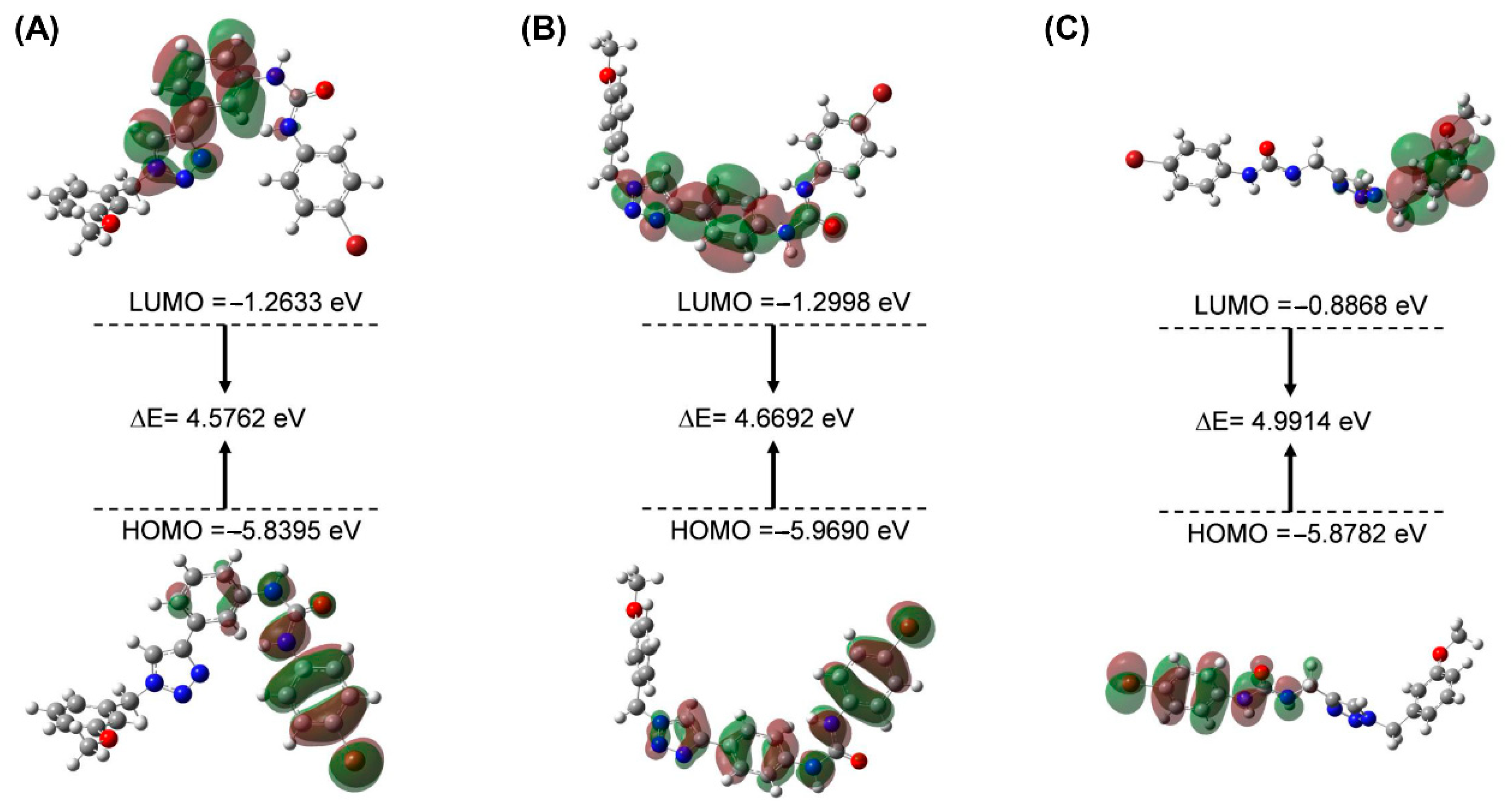
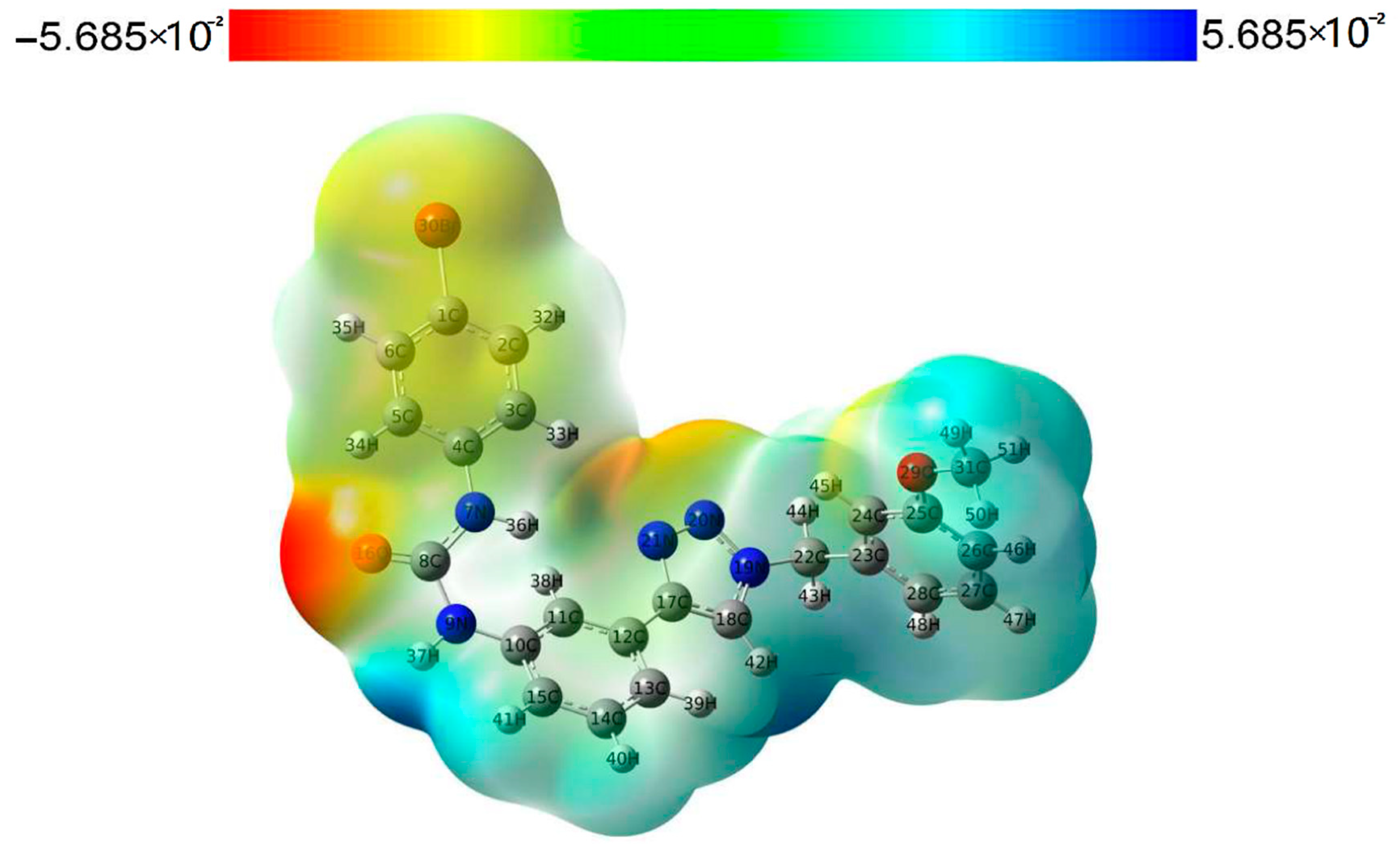
2.3. HOMO-LUMO and MEP Studies
3. Materials and Methods
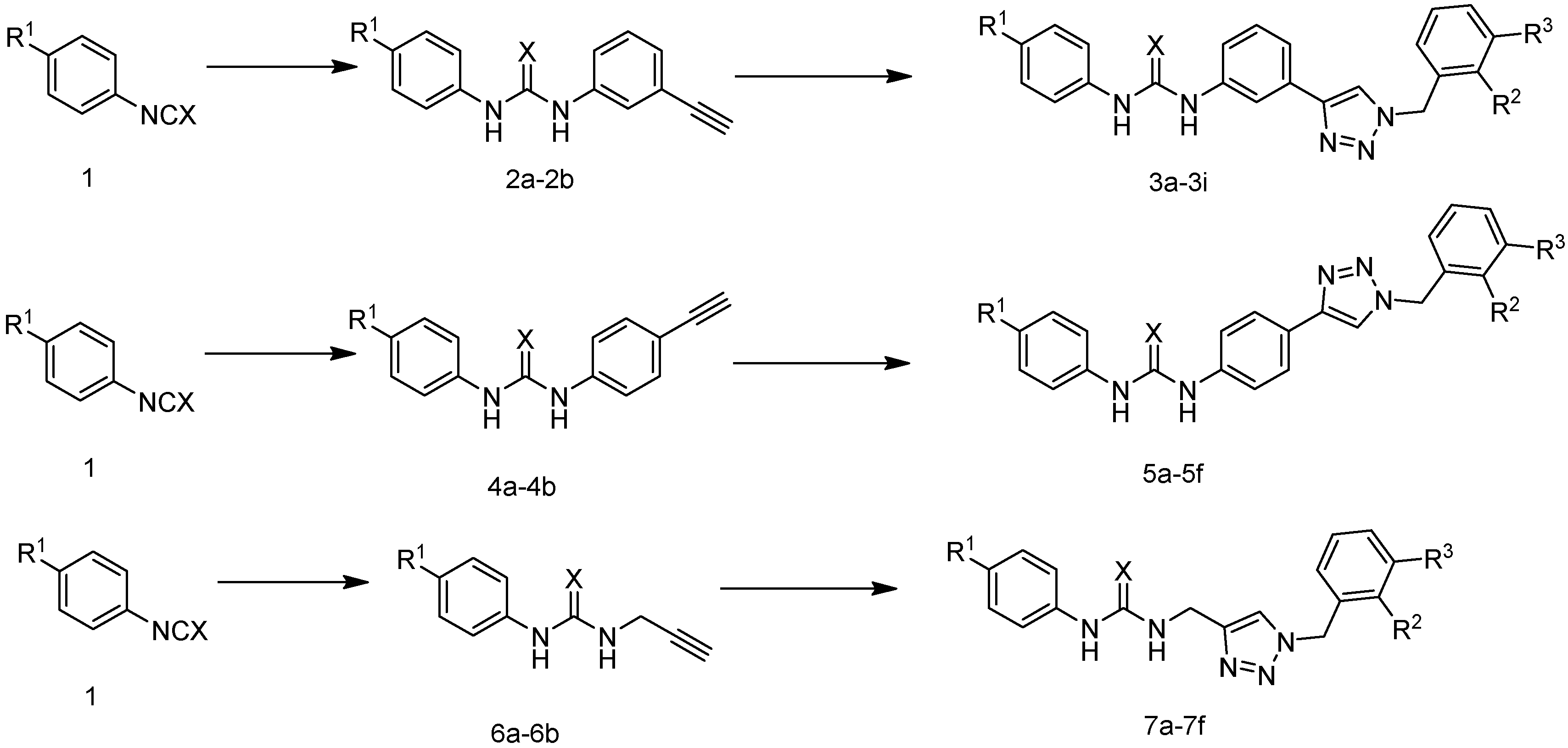
3.1. Materials and Chemistry
3.2. General Procedure for Preparation of Compound 2a (The Method Is Suitable for 2b, 4a, 4b, 6a and 6b)
3.3. General Procedure for Preparation of Compound 3a
3.4. IDO1 Enzymatic Inhibition Assay
3.5. Molecular Docking
3.6. Quantum Mechanical Studies (QM)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Macan, A.M.; Harej, A.; Cazin, I.; Klobucar, M.; Stepanic, V.; Pavelic, K.; Pavelic, S.K.; Schols, D.; Snoeck, R.; Andrei, G.; et al. Antitumor and antiviral activities of 4-substituted 1,2,3-triazolyl-2,3-dibenzyl-L-ascorbic acid derivatives. Eur. J. Med. Chem. 2019, 184, 111739. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.F.; Wang, Y.W.; Zhao, J.; Xu, G.Q.; Yao, X.J.; Li, Y.M. Discovery of Icotinib-1,2,3-Triazole Derivatives as IDO1 Inhibitors. Front. Pharmacol. 2020, 11, 579024. [Google Scholar] [CrossRef] [PubMed]
- Nelp, M.T.; Kates, P.A.; Hunt, J.T.; Newitt, J.A.; Balog, A.; Maley, D.; Zhu, X.; Abell, L.; Allentoff, A.; Borzilleri, R.; et al. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. USA 2018, 115, 3249–3254. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Zhou, Y.; Wang, Q.; Wang, Y.; Tian, C.; Wang, T.; Huang, L.; Nan, J.; Li, L.; Yang, S. Discovery and structure-activity relationship studies of 1-aryl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione derivatives as potent dual inhibitors of indoleamine 2,3-dioxygenase 1 (IDO1) and trytophan 2,3-dioxygenase (TDO). Eur. J. Med. Chem. 2020, 207, 112703. [Google Scholar] [CrossRef] [PubMed]
- Partyka, A.; Chlon-Rzepa, G.; Wasik, A.; Jastrzebska-Wiesek, M.; Bucki, A.; Kolaczkowski, M.; Satala, G.; Bojarski, A.J.; Wesolowska, A. Antidepressant- and anxiolytic-like activity of 7-phenylpiperazinylalkyl-1,3-dimethyl-purine-2,6-dione derivatives with diversified 5-HT(1)A receptor functional profile. Bioorg. Med. Chem. 2015, 23, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, U.F.; Reynaud, A.; Majjigapu, S.R.; Vogel, P.; Pojer, F.; Zoete, V. Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (IDO1). J. Med. Chem. 2019, 62, 8784–8795. [Google Scholar] [CrossRef]
- Serafini, M.; Torre, E.; Aprile, S.; Grosso, E.D.; Gesù, A.; Griglio, A.; Colombo, G.; Travelli, C.; Paiella, S.; Adamo, A.; et al. Discovery of Highly Potent Benzimidazole Derivatives as Indoleamine 2,3-Dioxygenase-1 (IDO1) Inhibitors: From Structure-Based Virtual Screening to in Vivo Pharmacodynamic Activity. J. Med. Chem. 2020, 63, 3047–3065. [Google Scholar] [CrossRef]
- Soliman, H.; Antonia, S.; Sullivan, D.; Vanahanian, N.; Link, C. Overcoming tumor antigen anergy in human malignancies using the novel indeolamine 2,3-dioxygenase (IDO) enzyme inhibitor, 1-methyl-D-tryptophan (1MT). J. Clin. Oncol. 2009, 27, 3004. [Google Scholar] [CrossRef]
- Yue, E.W.; Sparks, R.; Polam, P.; Modi, D.; Douty, B.; Wayland, B.; Glass, B.; Takvorian, A.; Glenn, J.; Zhu, W. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. [Google Scholar] [CrossRef] [Green Version]
- Jr, O.W.; Lew, N.L.; Liu, Y.; Lowrie, E.G.; Lazarus, J.M. The urea reduction ratio and serum albumin concentration as predictors of mortality in patients undergoing hemodialysis. N. Engl. J. Med. 1993, 329, 1001–1006. [Google Scholar]
- Kiser, J.J.; Burton, J.R.; Anderson, P.L.; Everson, G.T. Review and management of drug interactions with boceprevir and telaprevir. Hepatology 2012, 55, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; Carter, C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2007, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L.P. SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fling, S.P.; Gregerson, D.S. Peptide and protein molecular weight determination by electrophoresis using a high-molarity tris buffer system without urea. Anal. Biochem. 1986, 155, 83–88. [Google Scholar] [CrossRef]
- Nauck, M.; Frid, A.; Hermansen, K.; Thomsen, A.B.; During, M.; Shah, N.; Tankova, T.; Mitha, I.; Matthews, D.R. Long-term efficacy and safety comparison of liraglutide, glimepiride and placebo, all in combination with metformin in type 2 diabetes: 2-year results from the LEAD-2 study. Diabetes Obes. Metab. 2013, 15, 204–212. [Google Scholar] [CrossRef]
- Thanthiriwatte, K.S.; De Silva, K.N. Non-linear optical properties of novel fluorenyl derivatives-ab initio quantum chemical calculations. J. Mol. Struct. Theochem. 2002, 617, 169–175. [Google Scholar] [CrossRef]
- Acar Çevik, U.; Celik, I.; Işık, A.; Ahmad, I.; Patel, H.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis, molecular modeling, DFT, ADME and biological evaluation studies of some new 1, 3, 4-oxadiazole linked benzimidazoles as anticancer agents and aromatase inhibitors. J. Biomol. Struct. Dyn. 2022, 6, 1–15. [Google Scholar] [CrossRef]
- Sureshkumar, B.; Mary, Y.S.; Panicker, C.Y.; Suma, S.; Armakovic, S.; Armakovic, S.J.; Alsenoy, C.V.; Narayana, B. Quinolinederivatives as possible lead compounds for anti-malarial drugs: Spectroscopic, DFT and MD study. Arab. J. Chem. 2020, 13, 632–648. [Google Scholar] [CrossRef]
- Sevvanthi, S.; Muthu, S.; Raja, M.; Aayisha, S.; Janani, S. PES, molecular structure, spectroscopic (FT-IR, FT-Raman), electronic (UV-Vis, HOMO-LUMO),quantum chemical and biological (docking) studies on a potent membrane permeable inhibitor:dibenzoxepine derivative. Heliyon 2020, 6, 4724. [Google Scholar] [CrossRef]
- Gaspari, P.; Banerjee, T.; Malachowski, W.P.; Muller, A.J.; Prendergast, G.C.; DuHadaway, J.; Bennett, S.; Donovan, A.M. Structure-Activity Study of Brassinin Derivatives as Indoleamine 2,3-Dioxygenase Inhibitors. J. Med. Chem. 2006, 49, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.X.; Gong, X.Q.; Mao, L.F.; Sun, G.; Yang, J.X. Design, synthesis and biological evaluationof erlotinib-based IDO1 inhibitors. Front. Pharmacol. 2022, 10, 940704. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.F.; Tao, Y.L.; Qu, H.Y.; Wang, C.H.; Yan, F.; Gao, X.J.; Zhang, M.L. Exploration of plant-derived natural polyphenols toward COVID-19 main protease inhibitors: DFT, molecular docking approach, and molecular dynamics simulations. RSC Adv. 2022, 12, 5357–5368. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Gobinath, E. Experimental and theoretical spectroscopic studies, HOMO–LUMO, NBO and NLMO analysis of3,5-dibromo-2,6-dimethoxy pyridine. Spectrochim. Acta A 2012, 97, 215–222. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | X | R1 | R2 | R3 | Yield (%) |
|---|---|---|---|---|---|
| 3a | O | Br | H | OCH3 | 73.9 |
| 3b | O | OCH3 | H | OCH3 | 55.1 |
| 3c | O | F | H | OCH3 | 80.8 |
| 3d | S | H | H | OCH3 | 69.2 |
| 3e | O | Br | H | Br | 85.9 |
| 3f | O | OCH3 | H | Br | 77.4 |
| 3g | O | Br | Br | H | 86.6 |
| 3h | O | F | Br | H | 63.8 |
| 3i | O | OCH3 | Br | H | 49.1 |
| 5a | O | OCH3 | Br | H | 41.7 |
| 5b | O | OCH3 | H | Br | 87.2 |
| 5c | O | Br | Br | H | 39.4 |
| 5d | O | Br | H | Br | 33.4 |
| 5e | O | Br | H | OCH3 | 40.2 |
| 5f | O | Cl | Br | H | 76.4 |
| 7a | O | OCH3 | Br | H | 36.6 |
| 7b | O | OCH3 | H | Br | 43.9 |
| 7c | O | OCH3 | H | OCH3 | 39.8 |
| 7d | O | Br | Br | H | 43.2 |
| 7e | O | Br | H | Br | 86.9 |
| 7f | O | Br | H | OCH3 | 45.4 |
| Compd No. | IC50 (μM) | Compd No. | IC50 (μM) | Compd No. | IC50 (μM) |
|---|---|---|---|---|---|
| IDO1 | IDO1 | IDO1 | |||
| 3a | 0.75 ± 0.27 | 5a | >10 | 7a | >10 |
| 3b | 3.46 ± 0.71 | 5b | 7.43 ± 1.05 | 7b | 3.97 ± 0.92 |
| 3c | 6.21 ± 0.89 | 5c | >10 | 7c | 7.10 ± 1.48 |
| 3d | 4.13 ± 0.42 | 5d | >10 | 7d | >10 |
| 3e | 8.16 ± 1.12 | 5e | >10 | 7e | >10 |
| 3f | 5.93 ± 0.97 | 5f | >10 | 7f | >10 |
| 3g | 2.59 ± 0.13 | ||||
| 3h | 2.58 ± 0.59 | ||||
| 3i | 0.80 ± 0.37 |
| Compd No. | HOMO Energy (eV) | LUMO Energy (eV) | |
|---|---|---|---|
| 3a | −5.8395 | −1.2633 | 4.5762 |
| 5e | −5.9690 | −1.2998 | 4.6692 |
| 7f | −5.8782 | −0.8868 | 4.9914 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, X.; Gong, X.; Mao, L.; Zhao, J.; Yang, J. Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors. Pharmaceuticals 2022, 15, 1316. https://doi.org/10.3390/ph15111316
Hou X, Gong X, Mao L, Zhao J, Yang J. Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors. Pharmaceuticals. 2022; 15(11):1316. https://doi.org/10.3390/ph15111316
Chicago/Turabian StyleHou, Xixi, Xiaoqing Gong, Longfei Mao, Jie Zhao, and Jianxue Yang. 2022. "Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors" Pharmaceuticals 15, no. 11: 1316. https://doi.org/10.3390/ph15111316
APA StyleHou, X., Gong, X., Mao, L., Zhao, J., & Yang, J. (2022). Discovery of Novel 1,2,3-triazole Derivatives as IDO1 Inhibitors. Pharmaceuticals, 15(11), 1316. https://doi.org/10.3390/ph15111316