3.2. Chemistry
3.2.1. 3,6-anhydro-2-deoxy-4,5-O-(1-methylethylidene)-7-fluoro-D-ribo-heptanoic acid methyl ester 6
To a solution of 5 (100 mg, 0.40 mmol) in diglyme (3 mL), 110 µL of DAST (2 eq., 0.81 mmol) were added dropwise at 0 °C under an inert atmosphere. The reaction mixture was stirred for 30 min at 0 °C and 1 h 30 at 110 °C. After cooling at room temperature, the mixture was neutralized by the addition of a saturated solution of NaHCO3 (10 mL), and the solvent was removed under vacuum. The residue was solubilized in water, the aqueous layer was extracted with CH2Cl2 (2 × 20 mL), and the organic layer was dried over MgSO4. The solvent was removed under vacuum, and the crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 60/40) to afford compound 6. Yield: 85% as a colorless oil, Rf = 0.85 (Cycl/EtOAc: 6/4), [α]D = −11.3 (c = 0.10, CHCl3). IR (cm−1): ν = 2988, 2955, 1734, 1437, 1381, 1307, 1267. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.34 (s, 3H, CH3), 1.49 (s, 3H, CH3), 2.72 (dd, 1H, J2a,2b = 16.5 Hz, J2a,3 = 7.0 Hz, H-2a), 2.78 (dd, 1H, J2b,3 = 7.0 Hz, H-2b), 3.71 (s, 3H, OCH3), 4.19 (app dt, 1H, J6,F = 33.0 Hz, J6,7a = J6,7b = 3.0 Hz, H-6), 4.40–4.46 (m, 1H, H-3), 4.46 (ddd, 1H, J7a,F = 47.5 Hz, J7b,7a = 10.5 Hz, J7b,6 = 3.5 Hz, H-7a), 4.58 (ddd, 1H, J7b,F = 47.5 Hz, J7b,7a = 10.5 Hz, J7b,6 = 3.0 Hz, H-7b), 4.80 (dd, 1H, J4,5 = 6.0 Hz, J4,3 = 4.0 Hz, H-4), 4.85 (app brd, 1H, H-5). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 25.1 (CH3), 26.3 (CH3), 36.4 (C-2), 51.9 (OCH3), 78.6 (d, JC3-F = 2 Hz, C-3), 81.6 (C-4), 82.4 (d, JC5-F = 8 Hz, C-5), 82.5 (d, JC6-F = 19 Hz, C-6), 85.1 (d, JC7-F = 172 Hz, C-7), 112.9 (C(CH3)2), 171.6 (C=O). 19F NMR (CDCl3, 376 MHz): δ (ppm) = −229.2. ESI-HRMS [M + Na]+ m/z = 271.0952 (calculated for C11H18FNaO5: 271.0958).
3.2.2. 3,6-anhydro-2-deoxy-4,5-O-(1-methylethylidene)-7-O-(tert-butyldiphenylsilyl)-D-ribo-heptanoic acid methyl ester 7
To a solution of compound 5 (1 g, 4.05 mmol) in dry DMF (10 mL), a solution of 1.58 mL of tert-butyldiphenylsilyl chloride (1.5 eq., 6.07 mmol) and 496 mg of imidazole (1.8 eq., 7.29 mmol) in dry DMF (3mL) was added dropwise at 0 °C under an inert atmosphere. The reaction was stirred overnight at room temperature, and the mixture was then diluted with water (50 mL) and extracted with EtOAc (3 × 80 mL). The organic layer was dried over MgSO4 and filtered, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 90/10) to afford compound 7. Yield: 95% as colorless oil, Rf = 0.47 (Cycl/EtOAc: 8/2), [α]D = +0.4 (c = 0.36, CHCl3). IR (cm−1): ν = 2928, 1742, 1425, 1351, 1210. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.06 (s, 9H, Si-tert-butyl), 1.35 (s, 3H, CH3), 1.49 (s, 3H, CH3), 2.70 (dd, 1H, J2a,2b = 16.5 Hz, J2a,3 = 7.0 Hz, H-2a), 2.76 (dd, 1H, J2b,3 = 7.0 Hz, H-2b), 3.67 (dd, 1H, J7a,7b = 11.0 Hz, J7a,6 = 4.0 Hz, H-7a), 3.71 (s, 3H, COOCH3), 3.77 (dd, 1H, J7b,6 = 4.0 Hz, H-7b), 4.11 (app br t, 1H, H-6), 4.59 (app td, 1H, J3,2a = J3,2b 7.0 Hz, J3,4 = 4.0 Hz, H-3), 4.80 (dd, 1H, J4,5 = 6.0 Hz, H-4), 4.86 (br d, 1H, H-5), 7.36–7.46 (m, 6H, HAr), 7.64–7.68 (m, 4H, HAr). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 19.2 (C-Si), 25.2 (CH3), 26.4 (CH3), 27.0 (Si-tert-butyl), 34.9 (C-2), 51.8 (OCH3), 65.4 (C-7), 78.6 (C-3), 82.0 (C-4), 83.4 (C-5), 84.3 (C-6), 112.5 (C(CH3)2), 128.0 (4CAr), 130.0 (CAr), 130.0 (CAr), 132.9 (CqAr), 133.0 (CqAr), 135.8 (4CAr), 171.8 (C=O). ESI-HRMS [M + K]+ m/z = 523.1877 (calculated for C27H36KO6Si: 523.1913).
3.2.3. 3,6-anhydro-2-deoxy-4,5-O-(1-methylethylidene)-7-fluoro-D-ribo-1-hydroxyl-heptane 8
To a solution of 7 (92 mg, 0.37 mmol, 1.0 eq.) in dry THF (8 mL), 42 mg of LiAlH4 (1.11 mmol, 3.0 eq.) was added at 0 °C under an inert atmosphere, and the mixture was stirred at room temperature for 3 h. The reaction was quenched with the addition of water, and the mixture was filtered off using a Celite® pad. The organic solvent was removed under reduced pressure, and the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 50/50) to afford compound 8. Yield: 82% as yellowish oil, Rf = 0.26 (Cycl/EtOAc: 6/4), [α]D = −3.4 (c = 0.05, CHCl3). IR (cm−1): ν = 3420, 2932, 1456, 1373, 1269, 1234, 1209. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.35 (s, 3H, CH3), 1.51 (s, 3H, CH3), 1.89–2.06 (m, 2H, H-2), 3.76–3.86 (m, 2H, H-1), 4.14–4.21 (m, 1H, H-3), 4.21 (app dt, 1H, J6,F = 27.0 Hz, J6,7 = J6,5 = 2.5 Hz, H-6), 4.44–4.62 (m, 2H, H-7), 4.72 (dd, 1H, J4,5 = 6.0 Hz, J4,3 = 3.5 Hz, H-4), 4.85 (br dt, 1H, H-5). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 25.1 (CH3), 26.4 (CH3), 32.2 (C-2), 60.9 (C-1), 81.6 (d, JC3-F = 2 Hz, C-3), 82.3 (d, JC5-F = 6.0 Hz, C-5), 82.4 (C-4), 82.7 (d, JC6-F = 17.0 Hz, C-6), 85.3 (d, JC7-F = 172.0 Hz, C-7), 112.8 (C(CH3)2). 19F NMR (CDCl3, 376 MHz): δ (ppm) = -228.9. ESI-HRMS [M + Na]+ m/z = 243.1081 (calculated for C10H17FNaO4: 243.1003).
3.2.4. 3,6-anhydro-2-deoxy-4,5-O-(1-methylethylidene)-7-O-(tert-butyldiphenylsilyl)-D-ribo-1-hydroxyl-heptane 9
Prepared from 7 following the procedure described for 8. Yield: 76% as yellowish oil, Rf = 0.18 (Cycl/EtOAc: 8/2), [α]D = +12.68 (c = 0.07, CHCl3). IR (cm−1): ν = 3442, 2929, 2856, 1472, 1427, 1380, 1207, 1111. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.06 (s, 9H, Si-tert-butyl), 1.36 (CH3), 1.51 (CH3), 1.85–1.94 (m, 1H, H-2a), 1.98–2.08 (m, 1H, H-2b), 2.17 (bs, OH), 3.70 (app dd, 1H, J7a,7b = 11.0 Hz, J7a,6 = 4.0 Hz, H-7a), 3.77–3.86 (m, 1H, J7b,6 = 4.0 Hz, H-7b), 3.78–3.87 (m, 2H, H-1), 4.13 (app t, 1H, H-6), 4.29 (m, 1H, H-3), 4.70 (dd, 1H, J4,5 = 6.0 Hz, J4,3 = 4.0 Hz, H-4), 4.86 (dd, 1H, J5,6 = 1.0 Hz, H-5), 7.36–7.47 (m, 6H, HAr), 7.63–7.68 (m, 4H, HAr). 13C NMR (CDCl3, 100.6 MHz): 19.2 (C, Si-C), 25.2 (CH3), 26.5 (CH3), 27.0 (3C, Si-C(CH3)3), 32.3 (C-2), 61.2 (C-1), 65.4 (C-7), 81.8 (C-3), 82.7 (C-4), 83.2 (C-5), 84.5 (C-6), 112.5 (C(CH3)2), 128.0 (4CAr), 130.0 (CAr), 130.1 (CAr), 133.0 (CqAr), 133.1 (CqAr), 135.6 (2CAr), 135.7 (2CAr). ESI-HRMS [M + Na]+ m/z = 479.2236 (calculated for C26H36NaO5Si: 479.2224).
3.2.5. 3,6-anhydro-2-deoxy-4,5-O-(1-methylethylidene)-7-fluoro-D-ribo-(prop-2-yne-1-yloxy)heptane 10
To a suspension of NaH 60% in mineral oil (150 mg, 3.91 mmol, 2.0 eq.) in dry DMF (4 mL), a solution of 431 mg of 8 (0.48 mmol, 1.0 eq.) in dry DMF (4 mL) was added at 0 °C under an inert atmosphere. After 1 h, 654 µL of propargyl bromide with 80% in toluen (5.88 mmol, 3.0 eq.) was added, and the mixture was stirred at room temperature for 16 h. The reaction was quenched with the addition of an aqueous saturated solution of NH4Cl. The solvent was removed under reduced pressure, and the resultant residue was dissolved in water. The aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 50/50) to afford compound 10 Yield: 95% as yellowish oil. Rf = 0.77(Cycl/EtOAc: 6/4), [α]D = −5.86 (c = 0.45, CHCl3). IR (cm−1): ν = 3265, 2986, 2122, 1718, 1375, 1232, 1209. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.34 (s, 3H, CH3), 1.49 (s, 3H, CH3), 1.98 (app q, 2H, J2,3 = J2,1 = 6.5 Hz, H-2), 2.41 (t, 1H, J10,8 = 2.5 Hz, H-10), 3.62–3.72 (m, 2H, H-1), 4.10–4.15 (m, 1H, H-3), 4.15 (d, 2H, H-8), 4.14–4.23 (m, 1H, H-6), 4.42–4.59 (m, 2H, H-7), 4.68 (dd, 1H, J4,5 = 6.0 Hz, J4,3 = 4.0 Hz, H-4), 4.82 (br d, 1H, H-5). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 25.2 (CH3), 26.4 (CH3), 29.7 (C-2), 58.3 (C-8), 67.3 (C-1), 74.3 (C-9), 79.5 (d, JC3-F = 2 Hz, C-3), 80.0 (C-10), 82.0 (C-4), 82.4 (d, JC5-F = 6.0 Hz, C-5), 82.4 (d, JC6-F = 18.0 Hz, C-6), 84.9 (d, JC7-F = 172.0 Hz, C-7), 112.7 (C(CH3)2). 19F NMR (CDCl3, 376 MHz): δ (ppm) = −228.7. ESI-HRMS [M + Na]+ m/z = 281.1137 (calculated for C13H19FNaO4: 281.1160).
3.2.6. 3,6-anhydro-2-deoxy-4,5-O-(1-methylethylidene)-7-O-(tert-butyldiphenylsilyl)-D-ribo-(prop-2-yne-1-yloxy)heptane 11
To a suspension of NaH 60% in mineral oil (27 mg, 0.66 mmol, 1.2 eq.) in dry THF (1 mL), a solution of 250 mg of 9 (0.55 mmol, 1.0 eq.) in dry THF (2 mL) was added at 0 °C under an inert atmosphere. After 10 min, 183 µL of propargyl bromide 80% in toluene (1.64 mmol, 3.0 eq.) was added, and the mixture was stirred at room temperature for 6 h. The reaction was quenched with the addition of an aqueous saturated solution of NH4Cl. The solvent was removed under reduced pressure, and the residue was dissolved in water. The aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 60/40) to afford compound 11. Yield: 70% as colorless oil, Rf = 0.77 (Cycl/EtOAc: 8/2), [α]D = +9.29 (c = 0.10, CHCl3). IR (cm−1): ν = 2926, 2855, 2116, 1670, 1472, 1427, 1361, 1103. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.06 (s, 9H, Si-tert-butyl), 1.36 (CH3), 1.51 (CH3), 1.99 (m, 2H, H-2), 2.38 (t, 1H, J10–8 = 2.5 Hz, H-10), 3.61–3.78 (m, 4H, H-1 and H-7), 4.09 (app t, 1H, H-6), 4.14 (d, 2H, J = 2.5 Hz, H-8), 4.24–4.29 (td, 1H, J4,3 = 4.0 Hz, H-3), 4.67 (dd, 1H, J4,5 = 6.0 Hz, H-4), 4.85 (dd, 1H, J5,6 = 1.0 Hz, H-5), 7.35–7.44 (m, 6H, HAr), 7.66–7.70 (m, 4H, HAr). 13C NMR (CDCl3, 100.6 MHz): 19.3 (C, Si-C), 25.3 (CH3), 26.5 (CH3), 27.0 (3C, Si-C(CH3)3), 29.9 (C-2), 58.2 (C-8), 65.3 (C-7), 67.5 (C-1), 74.3 (C-10), 79.4 (C-3) 80.1 (C-9), 82.4 (C-4), 83.4 (C-5), 84.3 (C-6), 112.3 (C(CH3)2), 127.9 (2CAr), 128.0 (2CAr), 129.9 (CAr), 129.8 (CAr), 133.1 (CqAr), 133.2 (CqAr), 135.0 (2CAr), 135.7 (2CAr). ESI-HRMS [M + Na]+ m/z = 517.2482 (calculated for C29H38NaO5Si: 517.2381).
3.2.7. 3,6-anhydro-2-deoxy-4,5-hydroxy-7-fluoro-D-ribo-(prop-2-yne-1-yloxy)heptane 12
A solution of 10 (500 mg, 1.94 mmol) in a mixture of TFA/H2O 6/4 (v/v) (20 mL) was stirred for 3 h at room temperature. The solvent was removed under vacuum, and the crude residue was purified using a column chromatography on silica gel (eluent: EtOAc/MeOH 100/0 to 90/10) to afford compound 12. Yield: 84% colorless oil, Rf = 0.13 (Cycl/EtOAc: 7/3), [α]D = −2.6 (c = 0.32, CHCl3). IR (cm−1): ν = 3385, 3283 2951, 289, 2116, 1717, 1356. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.93–2.02 (m, 1H, H-2a), 2.08–2.18 (m, 1H, H-2b), 2.47 (t, 1H, J10,8 = 2.5 Hz, H-10), 3.56 (ddd, 1H, J1a,1b = 10.5 Hz, J1a,2b = 9.5 Hz, J1a,2a = 2.5 Hz, H-1a), 3.72 (ddd, 1H, J1b,2b = 3.5 Hz, J1b,2a = 5.0 Hz, H-1b), 3.94 (dddd, 1H, J6,F = 27.0 Hz, J6,5 = 7.0 Hz, J6,7a = 4.0 Hz, J6,7b = 2.5 Hz, H-6), 4.08–4.13 (m, 1H, H-4), 4.14–4.19 (m, 1H, H-3), 4.15 (dd, 1H, J8a,8b = 12.5 Hz, H-8a), 4.20 (dd, 1H, H-8b), 4.23 (dd, 1H, J5,6 = 7.0 Hz, J5,4 = 5.0 Hz H-5), 4.51 (ddd, 1H, J7a,F = 47.5 Hz, J7a,7b = 10.5 Hz, H-7a), 4.63 (ddd, 1H, J7b,F = 48.0 Hz, H-7b). 13C NMR (CDCl3, 100.6 MHz): 29.9 (C-2), 58.6 (C-8), 66.7 (C-1), 72.1 (d, JC3-F = 1.5 Hz, C-3), 72.3 (d, JC5-F = 7.5 Hz, C-5), 75.3 (C-10), 79.0 (C-9), 80.6 (d, JC6-F = 18.0 Hz, C-6), 80.7 (C-4), 83.1 (d, JC7-F = 172.0 Hz, C-7). 19F NMR (CDCl3, 376 MHz): δ (ppm) = −232.1. ESI-HRMS [M + Na]+ m/z = 241.0818 (calculated for C10H15FNaO4: 241.0847).
3.2.8. Compound 13
A solution of 11 (70 mg, 0.14 mmol, 1.0 eq.) in AcOH 80% in water (1.5 mL) was stirred at 80 °C for 4 h. The mixture was cooled at 0 °C, and the reaction wash quenched with the addition of water (5 mL) and solid NaHCO3 until pH = 7. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL), the combined organic layers were dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 60/40) to afford compound 13. Yield: 54% colorless oil, Rf = 0.13 (Cycl/EtOAc: 8/2), [α]D = +33.25 (c = 0.04, CHCl3). IR (cm−1): ν = 3379, 3302, 2927, 2852, 2119, 1666, 1462, 1103. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.06 (s, 9H, Si-tert-butyl), 1.94–2.02 (m, 1H, H-2a), 2.04–2.15 (m, 1H, H-2b), 2.44 (t, 1H, J10–8 = 2.5 Hz, H-10), 3.58 (app td, 1H, J1a,1b = J1a,2a = 9.5 Hz, J1a,2b = 3.5 Hz, H-1a), 3.69–3.76 (m, 1H, H-1b), 3.81 (dd, 1H, J7a,7b = 11.5 Hz, J7a,6 = 4.5 Hz, H-7a), 3.84–3.90 (m, 2H, H-6 and H-7b), 4.09–4.14 (m, 1H, H-3), 4.14–4.18 (m, 3H, H-4 and H-8), 4.43 (dd, 1H, J = 6.5 Hz, J = 5.0 Hz, H-5), 7.35–7.45 (m, 6H, HAr), 7.66–7.73 (m, 4H, HAr). 13C NMR (CDCl3, 100.6 MHz): 19.4 (C, Si-C), 27.0 (3C, Si-C(CH3)3), 30.0 (C-2), 58.5 (C-8), 64.4 (C-7), 66.9 (C-1), 72.7 (C-4), 73.6 (C-5), 75.0 (C-10), 79.3 (C-9), 80.1 (C-3), 82.2 (C-6), 127.8 (2CAr), 127.9 (2CAr), 129.8 (CAr), 129.9 (CAr), 133.5 (CqAr), 133.6 (CqAr), 135.8 (2CAr), 135.8 (2CAr). ESI-HRMS [M + Na]+ m/z = 477.2088 (calculated for C26H34NaO5Si: 477.2068).
3.2.9. 3,6-anhydro-2-deoxy-4,5-O-(3-(triisopropylsilyl)prop-2-yne)-7-fluoro-D-ribo-(prop-2-yne-1-yloxy)heptane 14
To a suspension of NaH 60% in mineral oil (88 mg, 2.20 mmol, 3.0 eq.) in dry DMF (1 mL), a solution of 160 mg of 12 (0.73 mmol, 1.0 eq.) in dry DMF (3 mL) was added at 0 °C under an inert atmosphere. After 45 min, 1.2 g of 3-bromo-1-(triisopropylsilyl)-1-propyne (4.38 mmol, 6.0 eq.) was added, and the mixture was stirred at room temperature for 5 h. The reaction was quenched with the addition of an aqueous saturated solution of NH4Cl. The solvent was removed under vacuum, and the residue was dissolved in water. The aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 50/50) to afford compound 12. Yield: 94% as yellowish oil, Rf = 0.59 (Cycl/EtOAc: 8/2), [α]D = +24.2 (c = 0.10, CHCl3). IR (cm−1): ν = 3298, 2941, 2864, 1717, 1458, 1383, 1227. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.07 (br s, 42H, Si-isopropyl and Si-isopropyl), 1.97 (app q, 2H, J2,1 = J2,3 = 6.5 Hz, H-2), 2.39 (t, 1H, J16,14 = 2.5 Hz, H-16), 3.60–3.69 (m, 2H, H-1), 4.03–4.12 (m, 1H, H-6), 4.10 (dd, 1H, J14a,14b = 15.0 Hz, H-14a), 4.15 (dd, 1H, H-14b), 4.18–4.23 (td, 1H, J3,4 = 4.0 Hz, H-3), 4.28 (app t, 1H, J4,3 = J4,5 = 4.0 Hz, H-4), 4.30–4.34 (m, 1H, H-5), 4.35 (d, 2H, J = 1.0 Hz, H-8 or H-11), 4.45 (ddd, 1H, J7a,F = 47.0 Hz, J7a,7b = 10.0 Hz, J7a,6 = 4.0 Hz, H-7a), 4.45 (d, 2H, J = 1.0 Hz, H-8 or H-11), 4.56 (ddd, 1H, J7b,F = 48.0 Hz, J7b,6 = 2.5 Hz, H-7b). 13C NMR (CDCl3, 100.6 MHz): 11.3 (3C, Si-CH-(CH3)2), 11.3 (3C, Si-CH-(CH3)2), 18.7 (6C, Si-CH-(CH3)2), 18.7 (6C, Si-CH-(CH3)2), 30.1 (C-2), 58.2 (C-14), 58.7 (C-8 or C-11), 59.2 (C-8 or C-11), 67.0 (C-1), 76.0 (C-4), 77.8 (C-3), 77.9 (d, JC5-F = 6.0 Hz, C-5), 78.7 (d, JC6-F = 18.5 Hz, C-6), 80.1 (C-15), 82.8 (d, JC7-F = 173.5 Hz, C-7), 88.6 (C-10 or C-13), 89.0 (C-10 or C-13), 102.5 (C-9 or C-12), 103.2 (C-9 or C-12). 19F NMR (CDCl3, 376 MHz): δ (ppm) = −231.0. ESI-HRMS [M + K]+ m/z = 645.3541 (calculated for C34H59FKO4Si2: 645.3567).
3.2.10. 3,6-anhydro-2-deoxy-4,5-O-(3-(triisopropylsilyl)prop-2-yne)-7-O-(tert-butyldiphenylsilyl)-D-ribo-(prop-2-yne-1-yloxy)heptane 15
To a suspension of NaH 60% in mineral oil (12 mg, 0.29 mmol, 2.2 eq.) in dry THF (1 mL), a solution of 60 mg of 13 (0.13 mmol, 1.0 eq.) in dry THF (1 mL) was added at 0 °C under an inert atmosphere. After 10 min, 218 mg of 3-bromo-1-(triisopropylsilyl)-1-propyne (0.79 mmol, 6.0 eq.) was added, and the mixture was stirred at room temperature for 5 h. The reaction was quenched with the addition of an aqueous saturated solution of NH4Cl. The organic solvent was removed under reduced pressure. The aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: cyclohexane/EtOAc 100/0 to 50/50) to afford compound 15. Yield: 78% as colorless oil, Rf = 0.59 (Cycl/EtOAc: 8/2), [α]D = +20.34 (c = 0.09, CHCl3). IR (cm−1): ν = 2939, 2889, 2862, 2253, 2167, 2115, 1462, 1103. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.05 (s, 21H, Si-isopropyl), 1.06 (s, 9H, Si-tert-butyl), 1.07 (s, 21H, Si-isopropyl), 1.93–2.01 (m, 2H, H-2), 2.34 (t, 1H, J10–8 = 2.5 Hz, H-16), 3.59–3.72 (m, 3H, H-1 and H-7a), 3.78 (dd, 1H, J7b,7a = 11.0 Hz, J7b,6 = 4.0 Hz, H-7a), 4.01–4.06 (m, 1H, H6), 4.11 (bt, 2H, H-14), 4.15–4.21 (m, 1H, H-3), 4.24–4.33 (m, 2H, H-4 and H-5), 4.26 (d, 1H, J = 16.0 Hz, Hz, H-8a or H-11a), 4.33 (d, 1H, J = 16.0 Hz, Hz, H-8b or H-11b), 4.42 (d, 1H, J = 16.5 Hz, Hz, H-8b or H-11b), 4.49 (d, 1H, J = 16.5 Hz, Hz, H-8b or H-11b), 7.34–7.44 (m, 6H, HAr), 7.66–7.71 (m, 4H, HAr). 13C NMR (CDCl3, 100.6 MHz): 11.3 (3C, Si-CH-(CH3)2), 11.3 (3C, Si-CH-(CH3)2), 18.7 (6C, Si-CH-(CH3)2), 18.7 (6C, Si-CH-(CH3)2), 19.4 (C, Si-C), 27.0 (3C, Si-C(CH3)3), 30.2 (C-2), 58.2 (C-14), 58.6 (C-8 or C-11), 59.2 (C-8 or C-11), 64.7 (C-7), 67.2 (C-1), 74.1 (C-16), 77.4 (C-3 and C-4), 79.4 (C-5), 80.2 (C-15), 81.2 (C-6), 87.9 (C-10 or C-13), 88.0 (C-10 or C-13), 103.3 (C-9 or C-12), 103.6 (C-9 or C-12), 127.8 (2CAr), 127.8 (2CAr), 129.8 (CAr), 129.8 (CAr), 133.5 (CqAr), 133.7 (CqAr), 135.8 (2CAr), 135.8 (2CAr). ESI-HRMS [M + H]+ m/z = 843.5243 (calculated for C50H79O5Si3: 843.5235).
3.2.11. 3H-Indolium,2-[5-(1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)-1,3-pentadien-1-yl]-3,3-dimethyl-1-[6-oxo-6-(3-azidopropylamino)hexyl]-iodonium salt 16
To a solution of commercial
N-hydroxysuccinimide cyanine
5 (90 mg, 0.13 mmol, 1.0 eq.) in DMF (2 mL), 35 mg of 3-azidopropyl-1-amine hydrochloride [
51] (0.26 mmol, 2.0 eq.) and 40 µL of DIPEA (0.39 mmol, 3.0 eq.) were added, and the mixture was stirred at room temperature for 6 h. The reaction mixture was then diluted with CH
2Cl
2 (20 mL), and the organic layer was washed with water (3 × 10 mL). The organic layer was dried over MgSO
4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: DCM/MeOH 100/0 to 90/10) to afford compound
16. Yield: 69% as a blue solid,
Rf = 0.61 (DCM/MeOH: 95/5). IR (cm
−1): ν = 2928, 2095, 1649, 1479, 1452, 1369, 1335, 1219.
1H NMR (CDCl
3, 400 MHz): δ (ppm) = 1.52–1.64 (m, 2H,
H-6), 1.69 (br s, 12H, 4
CH3), 1.76–1.92 (m, 6H,
H-2, H-5, H-7), 2.44 (br t, 2H,
J4,5 = 6.5 Hz,
H-4), 3.29–3.36 (m, 2H,
H-3), 3.38 (t, 2H,
J1,2 = 7.0 Hz,
H-1) 3.59 (s, 3H,
N-CH3), 4.12 (br t, 2H,
J8,7 = 6.5 Hz,
H-8), 6.31 (d, 1H,
J = 14.0 Hz,
H-11 or
H-15), 6.71 (d, 1H,
H-11 or
H-15), 6.99–7.08 (m, 1H,
H-13), 7.06 (d, 1H,
J = 7.5 Hz,
HAr), 7.13 (d, 1H,
J = 7.5 Hz,
HAr), 7.18–7.25 (m, 2H,
HAr), 7.32–7.41 (m, 4H,
HAr), 7.82 (app t, 1H,
J = 10.5 Hz,
H-12 or
H-14), 7.86 (app t, 1H,
J = 10.5 Hz,
H-12 or
H-14), 8.59 (br s, 1H,
NH).
13C NMR (CDCl
3, 100.6 MHz): δ (ppm) = 25.2 (
C-2, C-5 or
C-7), 26.4 (
C-6), 27.0 (
C-2, C-5 or
C-7), 28.2 (2
CH3), 28.3 (2
CH3), 29.1 (
C-2, C-5 or
C-7), 31.5 (N-
CH3), 36.0 (
C-4), 36.9 (
C-3), 44.9 (
C-8), 49.0 (
C-9 or
C-17), 49.5 (
C-1), 49.8 (
C-9 or
C-17), 103.8 (
C-11 or
C-15), 105.5 (
C-11 or
C-15), 110.2 (
CAr), 111.2 (
CAr), 122.2 (
CAr), 122.3 (
CAr), 125.6 (
CAr), 127.1 (
C-13), 128.8 (
CAr), 129.0 (
CAr), 140.7 (
CqAr), 141.3 (
CqAr), 142.0 (
CqAr), 143.0 (
CqAr), 152.2 (
C-12 or
C-14), 153.8 (
C-12 or
C-14), 172.3 (
C-10,
C-16 or
C=O), 173.6 (
C-10,
C-16 or
C=O), 174.6 (
C-10,
C-16 or
C=O). ESI-HRMS [M]
+ m/z = 565.3654 (calculated for C
35H
45N
6O: 565.3649).
3.2.12. Compound 17
To a solution of 16 (27 mg, 0.040 mmol, 1.0 eq.) in ACN (160 µL), 36 mg of 14 (0.060 mmol, 1.5 eq.), 40 µL of sodium ascorbate (1M in water, 0.04 mmol, 1 eq.), and 120 µL of copper (II) acetate (0.3 M in water, 0.036 mmol, 0.9 eq.) were added, and the mixture was stirred at room temperature for 16 h. The organic solvent was removed under reduced pressure, the aqueous layer was extracted with CH2Cl2 (3 × 20 mL) and dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: DCM/MeOH 100/0 to 90/10) to afford compound 17. Yield: 71% as a blue solid, Rf = 0.24 (DCM/MeOH: 95/5), Mp: 119 °C. IR (cm−1): ν = 3341, 2924, 2864, 1653, 1481, 1452, 1369, 1335, 1219. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.06 (s, 21H, Si-isopropyl), 1.06 (s, 21H, Si-isopropyl), 1.53–1.64 (m, 2H, H-20), 1.68 (s, 12H, 4 CH3), 1.71–1.98 (m, 6H, H-2, H-19 and H-21), 2.19 (app qt, 2H, J16,15 = J16,17 = 6.5 Hz, H-16), 2.51 (br t, 2H, J18,19 = 7.0 Hz, H-18), 3.30 (br s, 2H, H-17), 3.57 (s, 3H, N-CH3), 3.63 (br t, 2H, J2,1 = 6.5 Hz, H-1), 4.01–4.12 (m, 1H, H-6), 4.15 (br t, 2H, J22,21 = 7.0 Hz, H-22), 4.16–4.22 (m, 1H, H-3), 4.24–4.33 (m, 2H, H-4, H-5), 4.35 (s, 2H, H-8 or H-11), 4.37–4.51 (m, 2H, H-7), 4.43 (s, 2H, H-8 or H-11), 4.52–4.61 (m, 4H, H-14 and H-15), 6.25 (d, 1H, J = 14.0 Hz, H-25 or H-29), 6.70 (d, 1H, H-25 or H-29), 6.84 (app br t, 1H, J27,26 = J27,28 = 13.0 Hz, H-27), 7.07 (d, 1H, J = 7.5 Hz, HAr), 7.14 (d, 1H, J = 7.5 Hz, HAr), 7.18–7.26 (m, 2H, HAr), 7.31–7.42 (m, 4H, HAr), 7.77 (app t, 1H, H-26 or H-28), 7.81 (app t, 1H, H-26 or H-28), 8.20 (br s, 1H, H-triazole), 9.10 (br s, 1H, NH). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 11.3 (3C, Si-CH(CH3)3), 11.3 (3C, C(CH3)3), 18.7 (6C, C(CH3)3), 18.7 (3C, Si-CH(CH3)3), 25.2 (C-19), 26.3 (C-20), 27.0 (C-21), 28.2 (2 CH3), 28.3 (2 CH3), 30.2 (C-2), 30.6 (C-16), 31.5 (N-CH3), 35.9 (C-18), 36.3 (C-17), 48.3 (C-22), 49.0 (C-15), 58.6 (C-8 or C-11), 59.2 (C-8 or C-11), 64.4 (C-14), 67.4 (C-1), 75.9 (C-4), 77.8 (C-3), 78.0 (d, JC5-F = 5 Hz, C-5), 78.6 (d, JC6-F = 18.5 Hz, C-6), 83.0 (d, JC7-F = 173.0 Hz, C-7), 88.6 (C-10 or C-13), 88.8 (C-10 or C-13), 102.5 (C-9 or C-12), 103.2 (C-9 or C-12), 103.6 (C-25 or C-29), 105.4 (C-25 or C-29), 110.3 (CAr), 111.3 (CAr), 122.2 (CAr), 122.2 (CAr), 124.3 (CH-triazole), 125.0 (CAr), 125.7 (CAr), 126.9 (C-27), 128.9 (CAr), 129.1 (CAr), 140.7 (CqAr), 141.3 (CqAr), 142.0 (CqAr), 143.0 (CqAr), 144.6 (Cq-triazole), 152.4 (C-26 or C-28), 153.6 (C-26 or C-28), 172.4 (C-30 or C-24), 174.7(C-30 or C-24). 19F NMR (CDCl3, 376 MHz): δ (ppm) = −230.7. ESI-HRMS [M + H]2+ m/z = 586.3942 (calculated for C69H105FN6O5Si2: 586.3829).
3.2.13. Compound 18
Prepared from 15 and 16 following the procedure described for 17. Yield: 95% as a blue solid, Rf = 0.43 (DCM/MeOH: 95/5). IR (cm−1): ν = 3660, 2941, 2929, 2862, 2169, 2096, 1654, 1450, 1369, 1332, 1089.1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.03 (s, 21H, Si-isopropyl), 1.04 (s, 9H, Si-tert-butyl), 1.05 (s, 21H, Si-isopropyl), 1.53–1.63 (m, 2H, H-20), 1.68 (s, 12H, 4 CH3), 1.79–1.99 (m, 6H, H-2, H-19 and H-21), 2.18 (app qt, 2H, J16,15 = J16,17 = 6.0 Hz, H-16), 2.56 (t, 2H, J18,19 = 7.0 Hz, H-18), 3.27–3.33 (br t, 2H, H-17), 3.55 (s, 3H, N-CH3), 3.57–3.76 (m, 4H, H-1 and H-7), 4.00–4.04 (m, 1H, H-6), 4.10–4.18 (m, 3H, H-22 and H-3), 4.22–4.29 (m, 2H, H-4, H-5), 4.26 (d, 1H, J = 16.0 Hz, Hz, H-8a or H-11a), 4.32 (d, 1H, J = 16.0 Hz, Hz, H-8b or H-11b), 4.43 (s, 2H, H-8 or H-11), 4.53 (bt, 1H, H-15), 4.55 (d, 1H, J = 12.0 Hz, H-14a), 4.59 (d, 1H, H-14b), 6.23 (d, 1H, J = 13.5 Hz, H-25 or H-29), 6.67 (d, 1H, H-25 or H-29), 6.97 (br t, 1H, J27,26 = J27,28 = 12.0 Hz, H-27), 7.05 (d, 1H, J = 7.5 Hz, HAr), 7.13 (d, 1H, J = 7.5 Hz, HAr), 7.17–7.25 (m, 2H, HAr), 7.30–7.42 (m, 10H, HAr), 7.64–7.70 (m, 4H, HAr), 7.78 (app t, 1H, H-26 or H-28), 7.82 (app t, 1H, H-26 or H-28), 8.07 (s, 1H, H-triazole), 9.37 (br s, 1H, NH). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 11.3 (3C, Si-CH-(CH3)2), 11.3 (3C, Si-CH-(CH3)2), 18.7 (6C, Si-CH-(CH3)2), 18.7 (6C, Si-CH-(CH3)2), 19.4 (C, Si-C), 25.3 (C-19), 26.3 (C-20), 26.9 (C-21), 27.0 (3C, Si-C(CH3)3), 28.2 (2 CH3), 28.3 (2 CH3), 30.3 (C-2), 30.4 (C-16), 31.5 (N-CH3), 35.6 (C-18), 36.4 (C-17), 44.8 (C-22), 48.4 (C-15), 49.0 (C-23 or C-31), 49.5 (C-23 or C-31), 58.5 (C-8 or C-11), 59.1 (C-8 or C-11), 64.2 (C-14), 64.8 (C-7), 67.9 (C-1), 76.8 (C-3 and C-4), 79.3 (C-5), 80.2 (C-6), 87.9 (C-10 or C-13), 88.0 (C-10 or C-13), 103.3 (C-9 or C-12), 103.5 (C-9 or C-12), 103.6 (C-25 or C-29), 105.3 (C-25 or C-29), 110.3 (CAr), 111.3 (CAr), 122.2 (CAr), 122.3 (CAr), 124.2 (CH-triazole), 125.0 (CAr), 125.7 (CAr), 126.8 (C-27), 127.8 (2CAr), 127.8 (2CAr), 128.8 (CAr), 129.1 (CAr), 129.8 (2CAr), 133.5 (CqAr), 133.6 (CqAr), 135.7 (2CAr), 135.8 (2CAr), 140.7 (CqAr), 141.3 (CqAr), 142.0 (2CqAr), 142.9 (CqAr), 144.6 (Cq-triazole), 152.4 (C-26 or C-28), 153.6 (C-26 or C-28), 172.5 (C-30, C-24 or C=O), 173.6 (C-30, C-24 or C=O), 174.9 (C-30, C-24 or C=O). ESI-HRMS [M + H]2+ m/z = 704.9436 (calculated for C85H125N6O6Si3: 704.9470).
3.2.14. Compound 19
To a solution of 17 (30 mg, 0.023 mmol, 1.0 eq.) in THF (1 mL), 54 µL of tetrabutylammonium fluoride (1 M in THF, 0.054 mmol, 2.4 eq.) was added at 0 °C under an inert atmosphere, and the mixture was stirred at 0 °C for 3 h. The organic solvent was removed under reduced pressure, and the residue was solubilized in CH2Cl2 (10 mL). The organic layer was washed with 0.01M HCl solution (2 × 5 mL) and with brine until pH = 7. The organic layer was dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified using a flash chromatography on silica gel (eluent: DCM/MeOH 100/0 to 90/10) to afford compound 19. Yield: 68% as a blue solid, Rf = 0.05 (DCM/MeOH: 95/5), Mp: 136 °C. IR (cm−1): ν = 2970, 2912, 1647, 1477, 1448, 1367, 1331, 1217. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.50–1.61 (m, 2H, H-20), 1.68 (s, 12H, 4 CH3), 1.71–2.00 (m, 6H, H-2, H-19 and H-21), 2.15–2.21 (m, 2H, H-16), 2.48 (br t, 2H, J18,19 = 7.5 Hz, H-18), 2.48–2.52 (m, 2H, H-10 and H-13), 3.24–3.31 (m, 2H, H-17), 3.56 (s, 3H, N-CH3), 3.57–3.63 (m, 2H, H-1), 3.96–4.08 (m, 1H, H-6), 4.12 (br t, 2H, J22,21 = 7.0 Hz, H-22), 4.15–4.20 (m, 3H, H-3, H-4 and H-5), 4.27 (d, 2H, J = 2.5 Hz, H-8 or H-11), 4.37 (d, 2H, J = 2.5 Hz, H-8 or H-11), 4.37–4.51 (m, 2H, H-7), 4.51–4.63 (m, 4H, H-14 and H-15), 6.22 (d, 1H, J = 13.5 Hz, H-25 or H-29), 6.60 (d, 1H, H-25 or H-29), 6.91 (app br t, 1H, J27,26 = J27,28 = 12.0 Hz, H-27), 7.07 (d, 1H, J = 8.0 Hz, HAr), 7.14 (d, 1H, J = 8.0 Hz, HAr), 7.19–7.26 (m, 2H, HAr), 7.32–7.41 (m, 4H, HAr), 7.77 (app t, 1H, H-26 or H-28), 7.81 (app t, 1H, H-26 or H-28), 8.22 (s, 1H, H-triazole), 9.00 (br s, 1H, NH). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 25.3 (C-19), 26.4 (C-20), 27.0 (C-21), 28.2 (2 CH3), 28.3 (2 CH3), 29.7 (C-2), 30.4 (C-16), 31.9 (N-CH3), 35.8 (C-18), 36.2 (C-17), 44.9 (C-22), 48.4 (C-15), 58.4 (C-8 or C-11), 58.5 (C-8 or C-11), 64.1 (C-14), 67.2 (C-1), 75.1 (C-10 or C-13), 75.5 (C-10 or C-13), 76.4 (C-4), 77.9 (C-3), 78.0 (d, JC6-F = 18.5 Hz, C-6), 79.2 (d, JC5-F = 5 Hz, C-5), 79.3 (C-9 or C-12), 79.9 (C-9 or C-12), 82.7 (d, JC7-F = 172.0 Hz, C-7), 103.8 (C-25 or C-29), 105.4 (C-25 or C-29), 110.2 (CAr), 111.2 (CAr), 122.1 (CAr), 122.1 (CAr), 124.9 (CH-triazole), 125.0 (CAr), 125.6 (CAr), 126.4 (C-27), 128.7 (CAr), 129.0 (CAr), 140.6 (CqAr), 141.1 (CqAr), 141.8 (CqAr), 142.8 (CqAr), 144.1 (Cq-triazole), 152.3 (C-26 or C-28), 153.5 (C-26 or C-28), 172.3 (C-30 or C-24), 174.7 (C-30 or C-24). 19F NMR (CDCl3, 376 MHz): δ (ppm) = −231.1. ESI-HRMS [M + H]2+ m/z = 430.2592 (calculated for C51H64FN6O5: 430.2495). Analytical HPLC analyses were performed using a waters system. Condition du gradient Rt = 4. 4 min, ACN/H2O 60/40 (v/v) with 0.1% TFA in isocratic conditions, flow rate of 1.5 mL/min, and UV detection (650 nm).
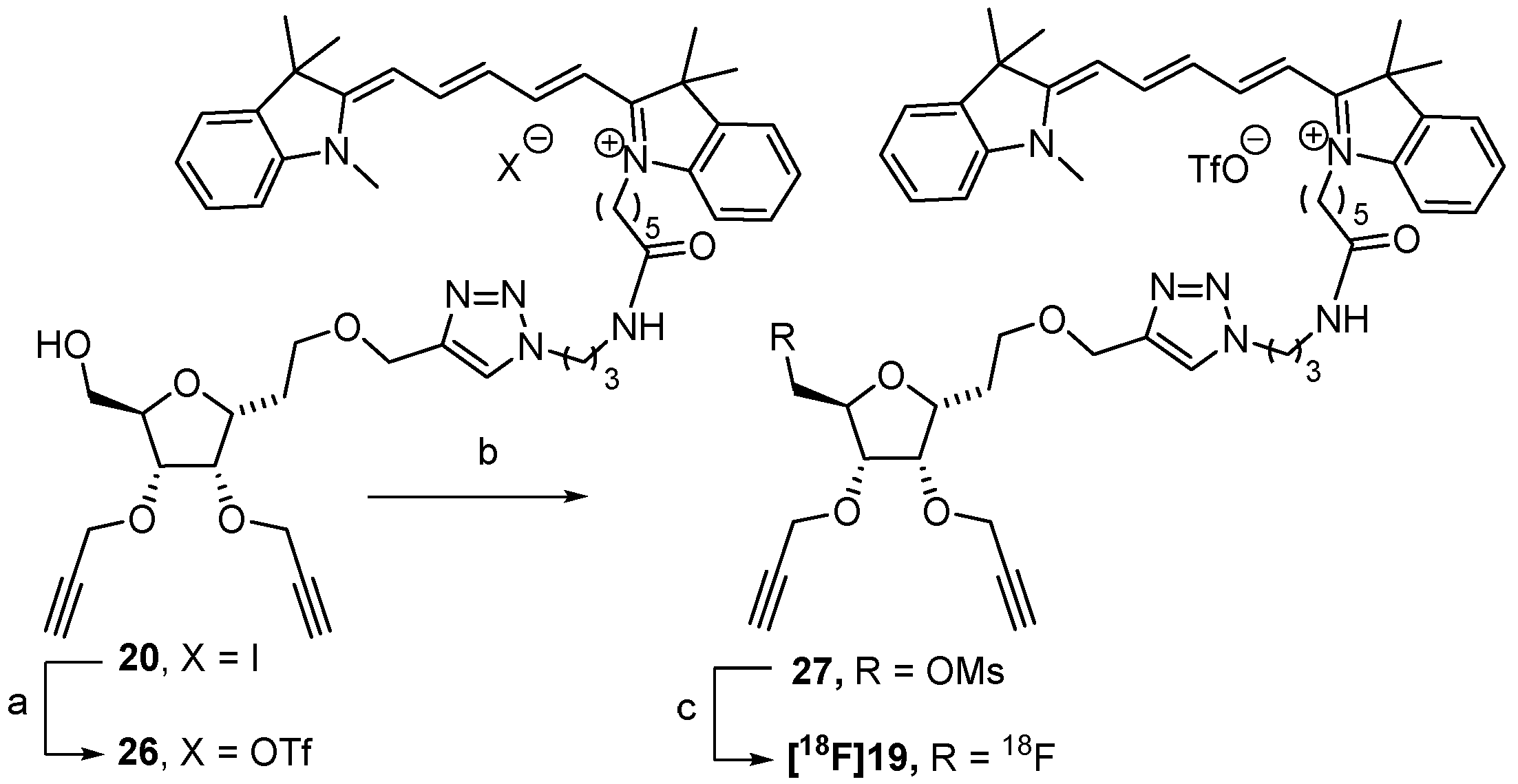
3.2.15. Compound 20
Prepared from 18 following the procedure described for 19. Yield: 69% as a blue solid, Rf = 0.15 (DCM/MeOH: 9/1). IR (cm−1): ν = 3236, 2926, 2162, 1651, 1495, 1495, 1454, 1371, 1091. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.57 (app qt, 2H, J20,19 = J20,21 = 6.5 Hz, H-20), 1.68 (s, 12H, 4 CH3), 1.78–1.94 (m, 6H, H-2, H-19 and H-21), 2.19 (app qt, 2H, J16,15 = J16,17 = 6.5 Hz, H-16), 2.44 (t, 1H, J = 2.0 Hz, H-10 or H-13), 2.47 (t, 1H, J = 2.0 Hz, H-10 or H-13), 2.50 (br t, 2H, J18,19 = 7.0 Hz, H-18), 2.59 (br s, 1H, OH), 3.25–3.32 (m, 2H, H-17), 3.56 (s, 3H, N-CH3), 3.57–3.66 (m, 2H, H-1), 3.66 (dd, 1H, J7a,7b = 12.0 Hz, J7a,6 = 3.5 Hz, H-7a), 3.83 (dd, 1H, J7a,7b = 12.0 Hz, J7b,6 = 2.5 Hz, H-7b), 3.95 (app dt, 1H, J6,5 = 7.5 Hz, H-6), 4.13 (br t, 2H, J22,21 = 7.0 Hz, H-22), 4.16 (br t, 1H, J4,3 = J4,5 = 4.0 Hz, H-4), 4.22–4.28 (m, 2H, H-3, H-5), 4.30 (d, 2H, H-8 or H-11), 4.38 (d, 2H, H-8 or H-11), 4.57 (br t, 2H, J15,16 = 6.5 Hz, H-15), 4.58 (d, 1H, J14a,14b = 12.5 Hz, H-14a), 4.68 (d, 1H, H-14b), 6.21 (d, 1H, J = 13.5 Hz, H-25 or H-29), 6.61 (d, 1H, H-25 or H-29), 6.91 (app br t, 1H, J27,26 = J27,28 = 12.5 Hz, H-27), 7.07 (d, 1H, J = 8.0 Hz, HAr), 7.15 (d, 1H, J = 8.0 Hz, HAr), 7.19–7.28 (m, 2H, HAr), 7.31–7.42 (m, 4H, HAr), 7.75 (app t, 1H, H-26 or H-28), 7.79 (app t, 1H, H-26 or H-28), 8.37 (s, 1H, H-triazole), 9.09 (br s, 1H, NH). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) = 25.1 (C-19), 26.2 (C-20), 26.9 (C-21), 28.1 (2 CH3), 28.1 (2 CH3), 29.9 (C-2), 30.3 (C-16), 31.3 (N-CH3), 35.8 (C-18), 36.0 (C-17), 44.7 (C-22), 48.2 (C-15), 48.9 (C-23 or C-31), 49.4 (C-23 or C-31), 58.4 (C-8 or C-11), 58.5 (C-8 or C-11), 61.9 (C-7), 64.3 (C-14), 66.8 (C-1), 74.6 (C-10 or C-13), 75.1 (C-10 or C-13), 77.2 (C-3), 77.3 (C-4), 79.9 (C-9 or C-12), 80.0 (C-5), 80.1 (C-6), 80.2 (C-9 or C-12), 103.3 (C-25 or C-29), 105.0 (C-25 or C-29), 110.2 (CAr), 111.2 (CAr), 122.1 (CAr), 122.1 (CAr), 124.6 (CH-triazole), 125.0 (CAr), 125.7 (CAr), 126.4 (C-27), 128.7 (CAr), 129.0 (CAr), 140.5 (CqAr), 141.1 (CqAr), 141.8 (CqAr), 142.8 (CqAr), 142.5 (CqAr), 144.5 (Cq-triazole), 152.2 (C-26 or C-28), 153.4 (C-26 or C-28), 172.5 (C-30, C-24 or C=O), 173.5 (C-30, C-24 or C=O), 174.6 (C-30, C-24 or C=O). ESI-HRMS [M+]2+ m/z = 429.2568 (calculated for C51H65N6O6: 429.2516).
3.2.16. Compound 22
To a solution of c(RGDfK) (10 mg, 14.0 µmol, 1.0 eq.) in DMF (1 mL), 11 mg of azido-PEG4-NHS (28.0 µmol, 2.0 eq.) and 5 µL of Et3N (35.0 µmol, 2.5 eq.) were added, and the mixture was stirred at 30 °C for 16 h. The organic solvent was evaporated under vacuum and the solid residue was washed with diethyl ether. The obtained solid was dried under vacuum to afford compound 22. Yield: 72% as white powder. 1H NMR (D2O, 400 MHz): δ (ppm) = 0.84–0.96 (m, 2H), 1.28–1.40 (m, 3H), 1.44–1.60 (m, 3H), 1.62–1.76 (m, 2H), 1.85–1.94 (m, 1H), 2.53–2.58 (m, 3H), 2.71 (dd, 1H, J = 6.8 Hz, J = 15.8 Hz), 2.93 (dd, 1H, J = 11.0 Hz, J = 12.5 Hz), 3.09–3.26 (m, 4H), 3.48–3.53 (m, 2H), 3.70–3.75 (m, 14H), 3.81 (t, 2H, J = 6.0 Hz), 3.81 (app t, 1H, J = 6.0 Hz), 3.87 (dd, 1H, J = 10.5 Hz, J = 4.0 Hz), 4.24 (dd, 1H, J = 15.0 Hz), 4.43 (dd, 1H, J = 8.5 Hz, J = 5.7 Hz), 4.60 (dd, 1H, J = 10.7 Hz, J = 5.7 Hz), 4.73 (app t, 1H, J = 7.1 Hz), 7.27–7.36 (m, 3H), 7.38–7.42 (m, 2H). HRMS [M]+ m/z = 877.4522 (calculated for C38H61N12O12: 877.4526).
3.2.17. Compound 23
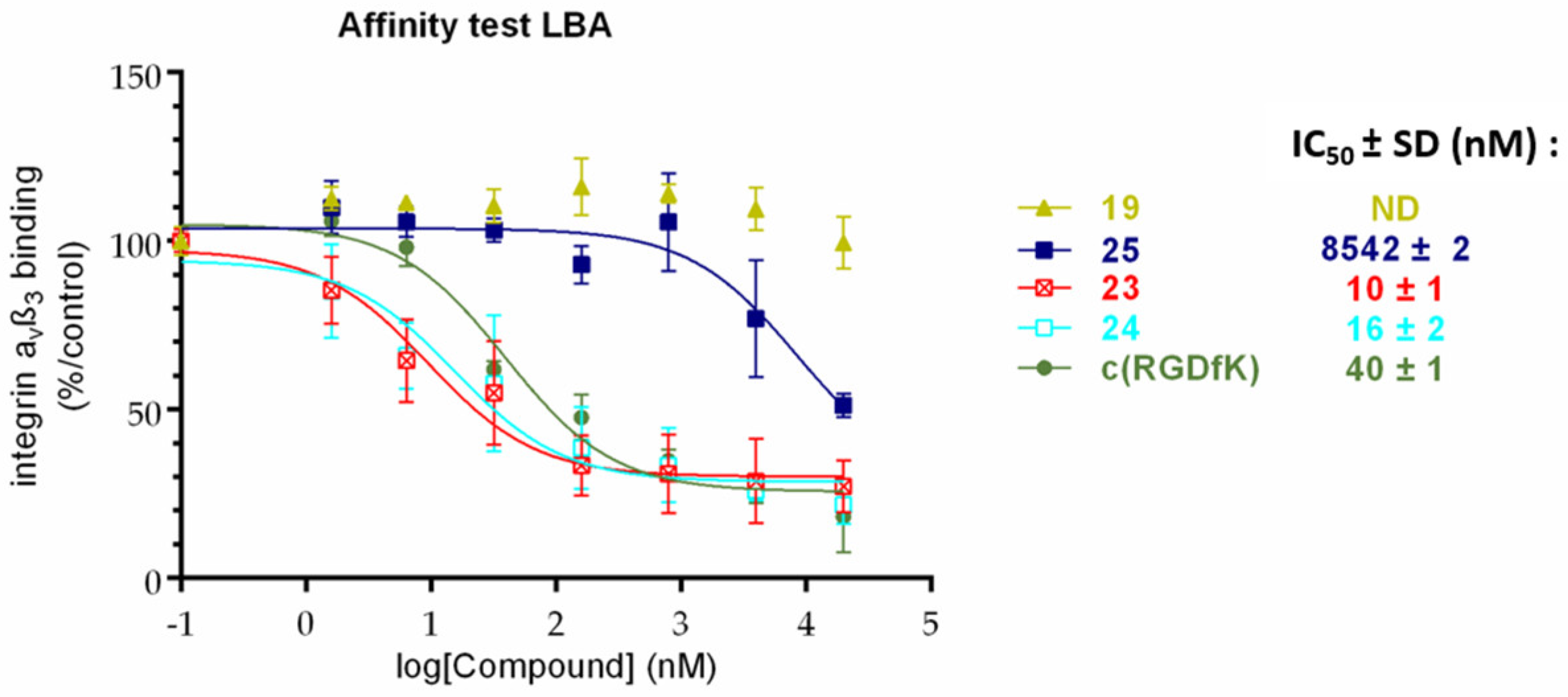
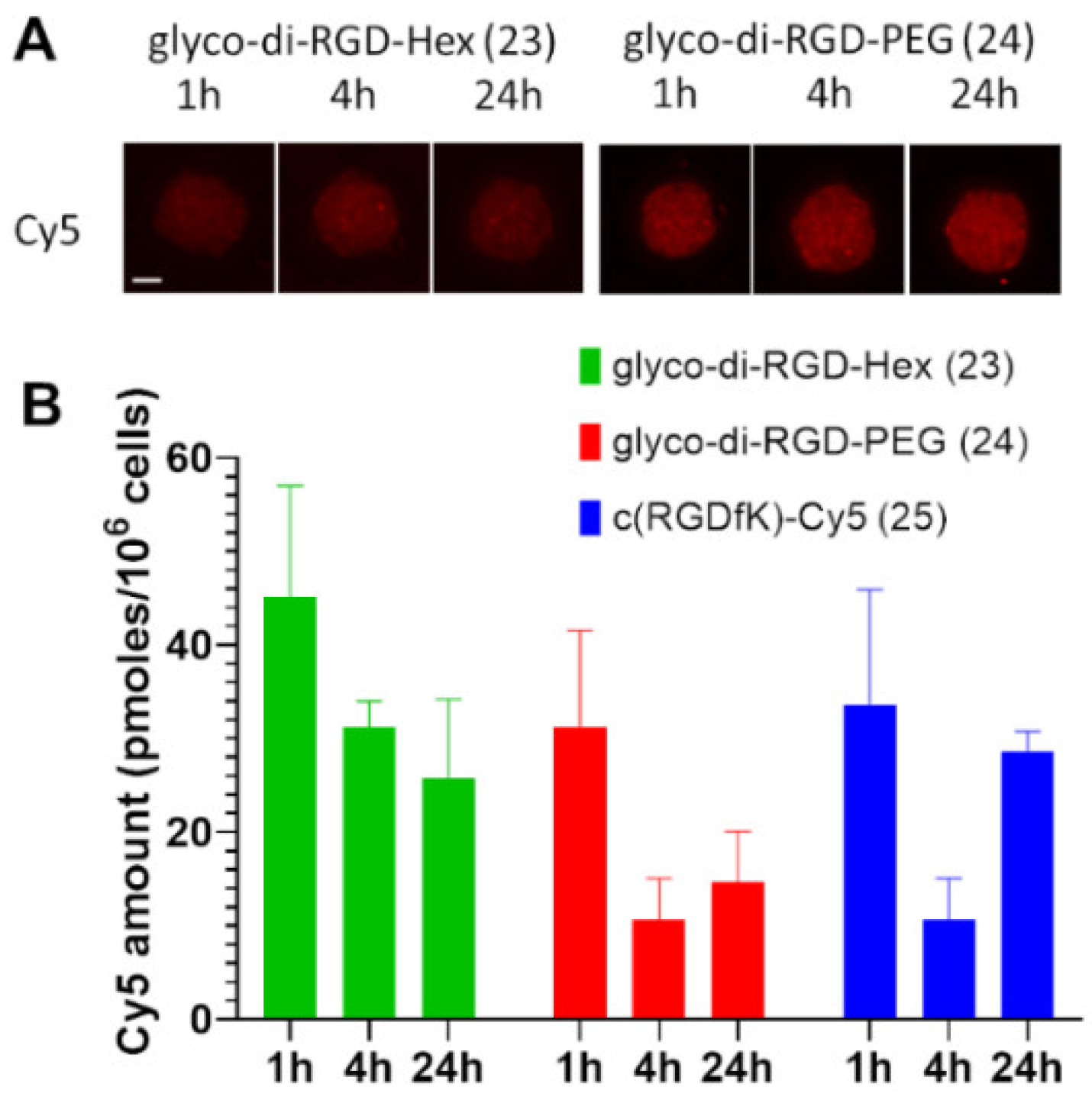
To a solution of 19 (3 mg, 3.04 µmol, 1.0 eq.) in a mixture of water/ACN (3/2.5) (550 µL), 7.8 mg of 21 (9.12 µmol, 3.0 eq.), 9 µL of copper (II) sulphate (1 M in water, 9.12 µmol, 3.0 eq.), and 23 µL of sodium ascorbate (1 M in water, 22.80 µmol, 7.5 eq.) were added, and the mixture was stirred at 40 °C for 24 h. Chelex® 100 resin (100 mg) was then added to the solution, and the suspension was stirred for 10 min. The resin was filtered off and the resulting solution dried under vacuum. The crude product was purified using Sephadex LH20 in water/ACN (7/3) to afford compound 23. Yield: 42% as a blue solid. HRMS [M + H]4+ m/z = 586.8230 (calculated for C117H167FN30O21: 586.8221), [M]3+ m/z = 782.0925 (calculated for C117H166FN30O21: 782.0937).
3.2.18. Compound 24
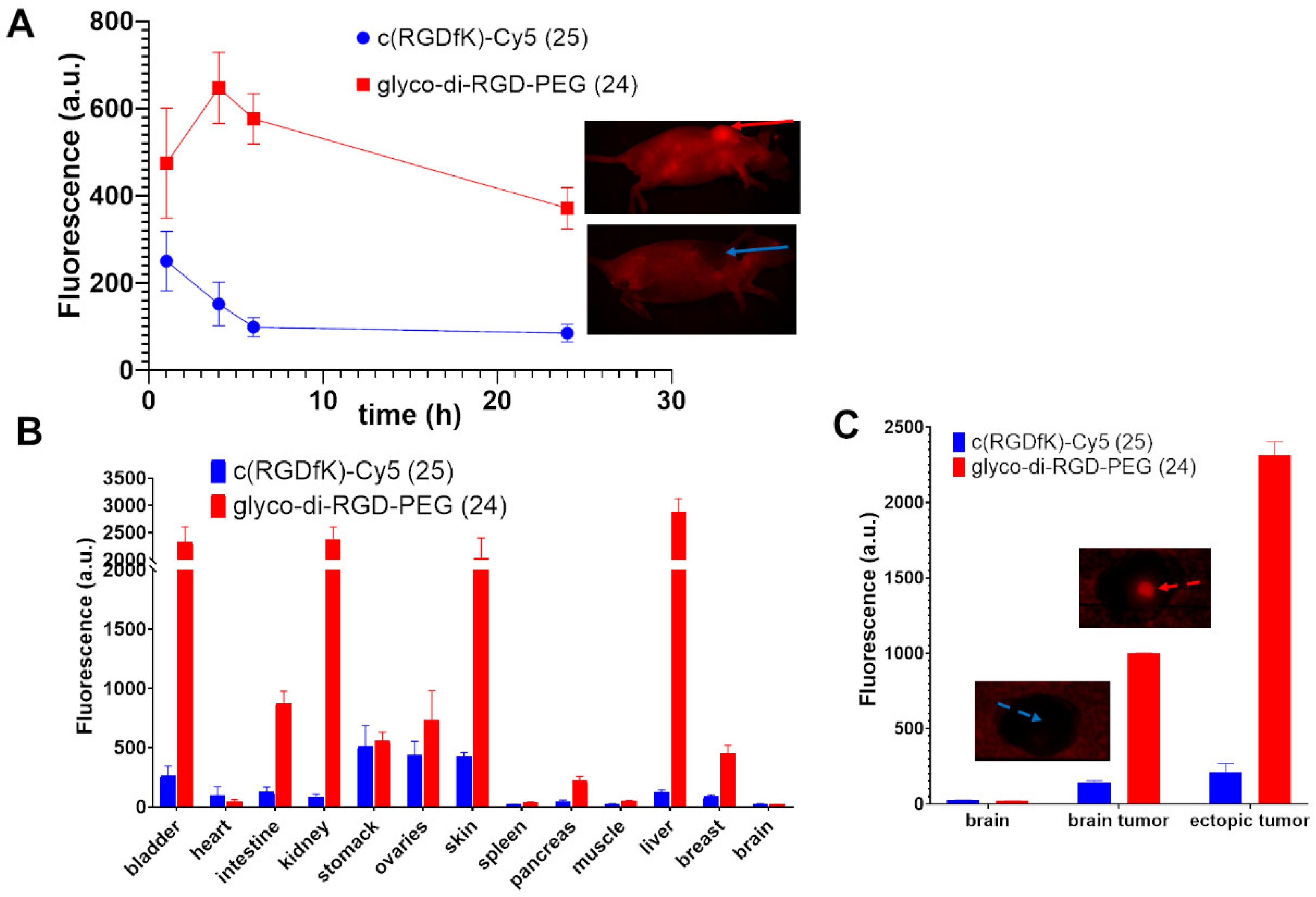
Prepared from 19 and 22 following the procedure described for 23. Yield: 62% as a blue solid. HRMS [M + H]4+ m/z = 653.8509 (calculated for C127H187FN30O29: 653.8511), [M]3+ m/z = 871.4603 (calculated for C127H186FN30O29: 871.4657).
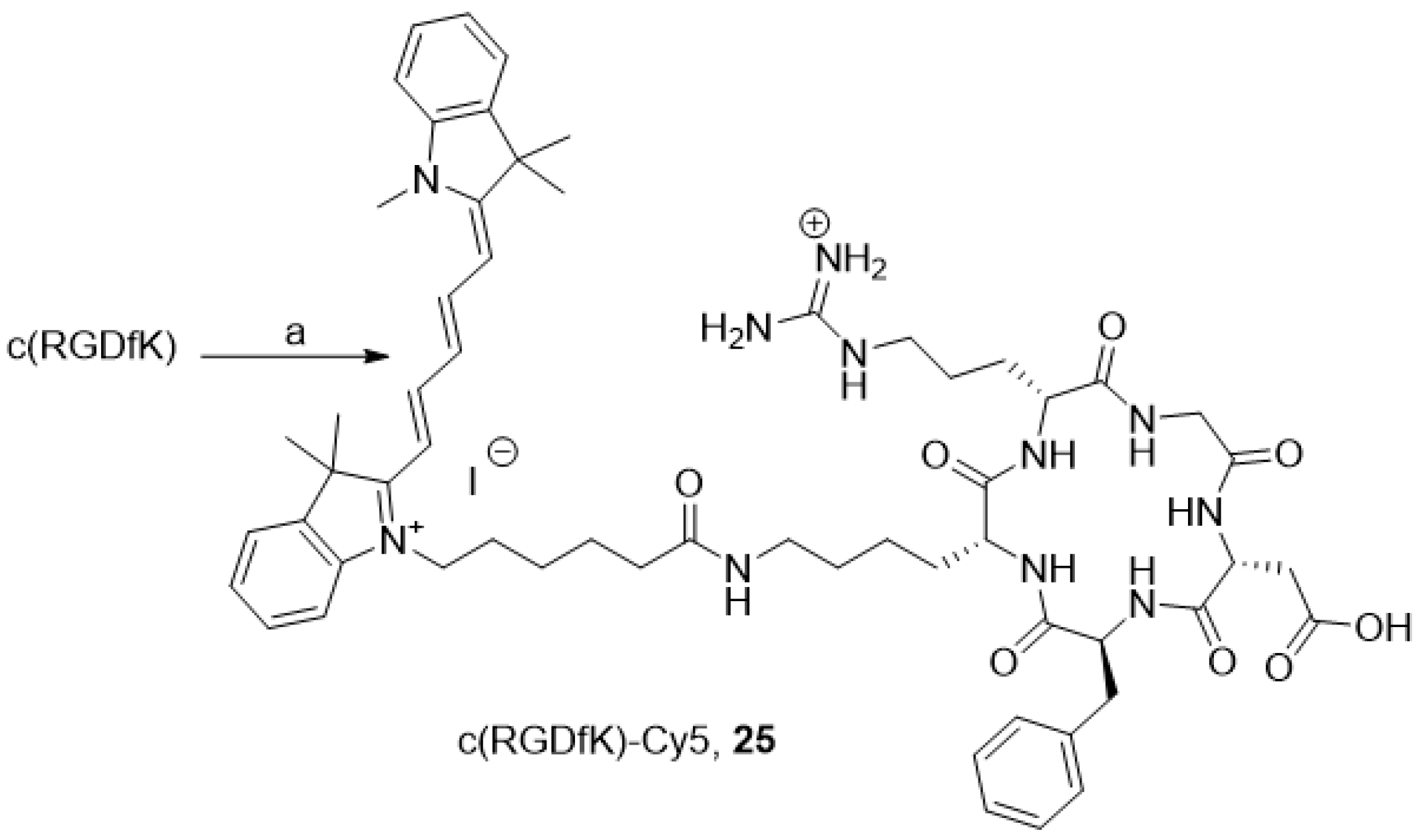
3.2.19. Compound 25
To a solution of c(RGDfK) (9 mg, 12.5 µmol, 1.0 eq.) in DMF (1 mL), 8.8 mg of commercial NHS cyanine 5 (12.5 µmol, 2.0 eq.) and 5 µL of Et3N (50.0 µmol, 4.0 eq.) were added, and the mixture was stirred at 50 °C for 16 h. The organic solvent was evaporated under vacuum, and the crude product was purified using a semi-preparative HPLC with a C18 reversed-phase silica gel: solvent A: 0.1% TFA water; solvent B: ACN; 0 to 2 min: 5% to 20% B, 2 to 5 min, 20% to 30% B, 5 to 20 min, 30% to 100%, 20 to 22 min, 100% to 5% B. Flow rate: 10 mL/min. The resulting solution was freeze-dried to afford compound 25. Yield: 59% as a blue solid, Rf = 0.03 (DCM/MeOH: 8/2), TR = 22.0 min. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 0.79–0.93 (m, 2H), 1.17–1.55 (m, 9H), 1.61 (s, 12H), 1.62–1.72 (m, 2H), 1.78–1.93 (m, 3H), 2.25 (t, 2H, J = 7.0 Hz), 2.64–2.72 (m, 1H), 2.75–2.89 (m, 2H), 2.93–3.06 (m, 3H), 3.09–3.20 (m, 2H), 3.51 (d, 1H, J = 14.5 Hz), 3.59 (s, 3H), 3.74–3.84 (m, 1H), 4.10 (bt, 2H, J = 7.5 Hz), 4.20 (bt, 1H), 4.31–4.71 (m, 3H), 6.13–6.21 (m, 2H), 6.44 (app br t, 1H, J = 12 Hz), 7.16–7.33 (m, 8H), 7.37–7.46 (m, 3H), 7.48–7.55 (m, 2H), 7.89–8.00 (m, 2H). ESI-HRMS [M]2+ m/z = 534.8034 (calculated for C59H79N11O8: 534.8051).
3.2.20. Compound 26
A solution of 20 (9 mg, 9.14 µmol) in ACN (300 µL) was diluted in water (60 mL). The obtained solution was passed through a series of 4 Oasis® MCX cartridges to trap the compound. The cartridges were washed with 100 mL of water (until pH = 7). The product was eluted with a mixture of NaOTf (0.2 M)/ACN (1/9) (100 mL), and the solvent was evaporated under vacuum. The crude product was solubilized in CH2Cl2 (10 mL), washed with water (2 × 5 mL), and dried over MgSO4, and the solvent was removed under vacuum. Compound 26 was obtained quantitatively without further purification. The quantitative yield is a blue solid. 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.47–1.58 (m, 2H, H-20), 1.67 (s, 6H, 4 CH3), 1.68 (s, 6H, 4 CH3), 1.70–1.94 (m, 6H, H-2, H-19 and H-21), 2.06–2.15 (m, 2H, H-16), 2.33 (br t, 2H, J18,19 = 6.5 Hz, H-18), 2.43 (t, 1H, J = 2.0 Hz, H-10 or H-13), 2.48 (t, 1H, J = 2.0 Hz, H-10 or H-13), 3.19–3.26 (m, 2H, H-17), 3.56 (s, 3H, N-CH3), 3.57–3.66 (m, 3H, H-1, H-7a), 3.80 (dd, 1H, J7a,7b = 12.0 Hz, J7b,6 = 2.5 Hz, H-7b), 3.93 (app dt, 1H, J6,5 = 7.5 Hz, H-6), 4.01 (br t, 2H, J22,21 = 7.0 Hz, H-22), 4.15 (br t, 1H, J4,3 = J4,5 = 3.5 Hz, H-4), 4.17–4.24 (m, 2H, H-3, H-5), 4.26 (d, 2H, H-8 or H-11), 4.37 (d, 2H, H-8 or H-11), 4.40 (br t, 2H, J15,16 = 6.0 Hz, H-15), 4.55 (d, 1H, J14a,14b = 12.5 Hz, H-14a), 4.61 (d, 1H, H-14b), 6.24 (d, 1H, J = 13.5 Hz, H-25 or H-29), 6.29 (d, 1H, H-25 or H-29), 6.78 (app br t, 1H, J27,26 = J27,28 = 12.5 Hz, H-27), 7.09 (d, 1H, J = 8.0 Hz, HAr), 7.11 (d, 1H, J = 8.0 Hz, HAr), 7.19–7.27 (m, 2H, HAr), 7.31–7.41 (m, 4H, HAr), 7.44 (br s, 1H, NH), 7.80 (app t, 2H, H-26 and H-28), 7.93 (s, 1H, H-triazole). 19F NMR (CD3CN, 376 MHz: δ −79.27.
3.2.21. Compound 27
To a solution of 26 (1.5 mg, 1.52 µmol, 1.0 eq.) in CH2Cl2 (1 mL), 1.33 μL of DIPEA (7.61 µmol l, 5.0 eq.) and 1.06 mg of methanesulfonic anhydride (6.09 µmol, 4.0 eq.) were added under an inert atmosphere. The mixture was stirred for 16 h at room temperature. The solution was evaporated under vacuum. Yield: 38% as a blue solid. 1H NMR (400 MHz, CD3CN) δ (ppm) = 1.39–1.45 (m, 2H, H-20), 1.64 (app qt, 2H, J19,18 = J19,20 = 6.5 Hz, H-19), 1.68 (s, 12H, 4 CH3), 1.73–1.88 (m, 4H, H-21 and H-2), 2.00 (app qt, 2H, J16,15 = J16,17 = 6.5 Hz, H-16), 2.12–2.17 (m, 2H, H-18), 2.75 (t, 1H, J = 2.0 Hz, H-10 or H-13), 2.79 (t, 1H, J = 2.0 Hz, H-10 or H-13), 3.04 (s, 3H, CH3-Ms), 3.09–3.14 (m, 2H, H-17), 3.54 (s, 5H, H-1, N-CH3), 3.96 (ddd, 1H, J6,5 = 7.5 Hz, J6,7a = 4.5 Hz, J6,7b = 2.5 Hz, H-6), 4.02 (br t, 2H, J22,21 = 7.0 Hz, H-22), 4.06–4.12 (m, 3H, H-3, H-5, H-4), 4.21 (dd, 1H, J7a,7b = 11.5 Hz, J7a,6 = 4.5 Hz, H-7a), 4.24–4.27 (m, 2H, H-8 or H-11), 4.30–4.39 (m, 5H, H-7b, H-15, H-8 or H-11), 4.51 (s, 2H, H-14), 6.19 (d, 1H, J = 13.5 Hz, H-25 or H-29), 6.25 (d, 1H, H-25 or H-29), 6.55 (app br t, 1H, J27,26 = J27,28 = 12.5 Hz, H-27), 6.93 (t, 1H, J = 6.0 Hz, NH), 7.21–7.28 (m, 4H, HAr), 7.33–7.43 (m, 2H, HAr), 7.45–7.50 (m, 2H, HAr), 7.88 (s, 1H, H-triazole), 8.08 (app t, 2H, H-26 and H-28). 13C NMR (CD3CN, 100.6 MHz): δ (ppm) = 26.0 (C-19), 27.0 (C-20), 27.6 (2 CH3), 27.8 (3C, 2 CH3 and C-21), 30.4 (C-2), 31.1 (C-16), 32.0 (N-CH3), 36.5 (C-18), 36.9 (C-17), 37.7 (CH3-Ms), 44.9 (C-22), 48.5 (C-15), 50.1 (C-23 or C-31), 50.2 (C-23 or C-31), 59.0 (C-8 or C-11), 59.4 (C-8 or C-11), 64.8 (C-14), 67.6 (C-1), 71.1 (C-7), 76.0 (C-10 or C-13), 76.6 (C-10 or C-13), 77.8 (C-6), 78.1 (C-4), 78.6 (C-3), 80.6 (C-9 or C-12), 80.9 (C-5), 81.0 (C-9 or C-12), 104.1 (2C, C-25 and C-29), 111.8 (CAr), 112.1 (CAr), 123.1 (CAr), 123.2(CAr), 124.6 (CH-triazole), 125.6 (CAr), 125.9 (CAr), 126.0 (C-27), 129.5 (CAr), 129.5 (CAr), 142.3 (CqAr), 142.4 (CqAr), 143.4 (CqAr), 144.1 (CqAr), 145.6 (CqAr), 144.7 (Cq-triazole), 154.8 (C-26 or C-28), 154.9 (C-26 or C-28), 173.7 (C-30, C-24 or C=O), 174.4 (C-30, C-24 or C=O), 174.9 (C-30, C-24 or C=O). 19F NMR (376 MHz, CD3CN) δ (ppm) = -79.31. ESI-HRMS [M+]+ m/z = 935.4562 and [M+]2+ m/z = 468.2452 (calculated for C52H67N6O8S, respectively: 935.4736 and 468.2404).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









