
Protein-Protein Interaction Inhibitors Targeting the Eph-Ephrin System with a Focus on Amino Acid Conjugates of Bile Acids

 , ,
, ,  ,
,  and
and
Abstract
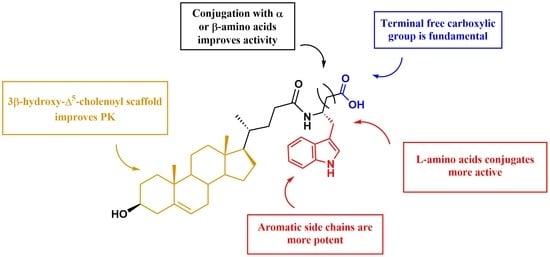
:
1. Introduction
2. Agents Targeting the Eph-Eph System
2.1. Biologics
2.2. Peptides and Peptidomimetics
2.3. Bile Acids Derivatives: An Emerging Class of Small-Molecules Disrupting Eph-ephrin Interaction
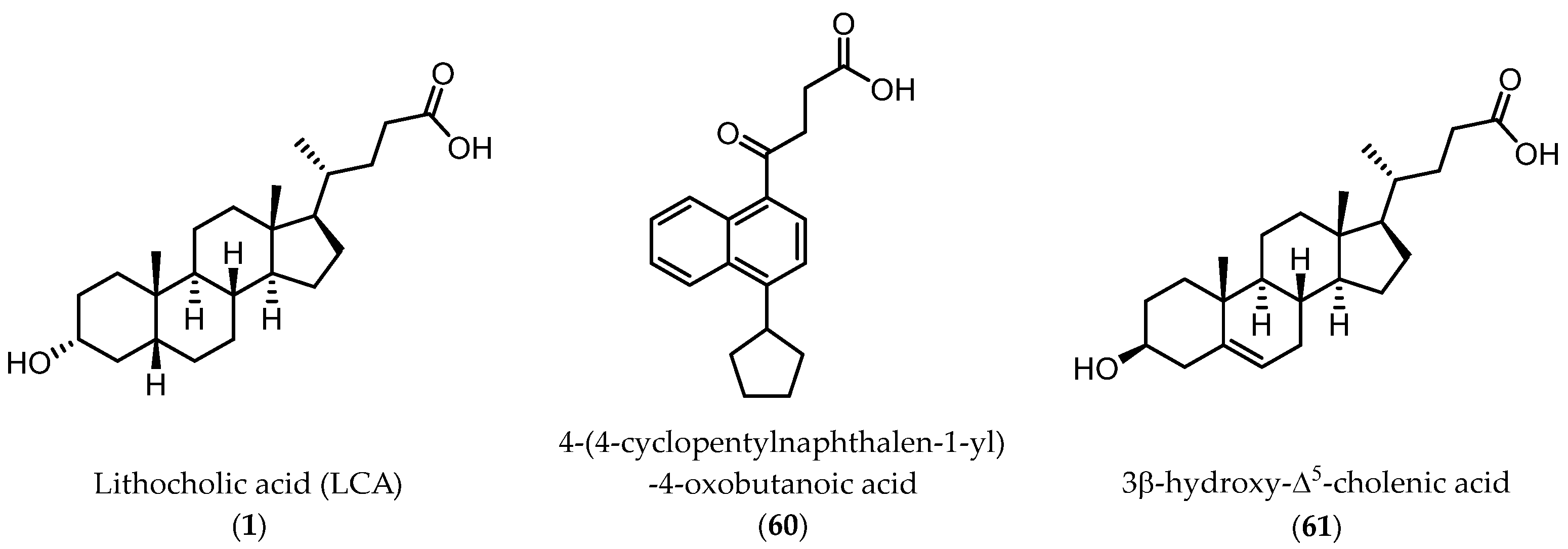
2.3.1. A New Scaffold for EphA2 Receptor Antagonists: Discovery of 3β-Hydroxy-Δ5-Cholenic Acid
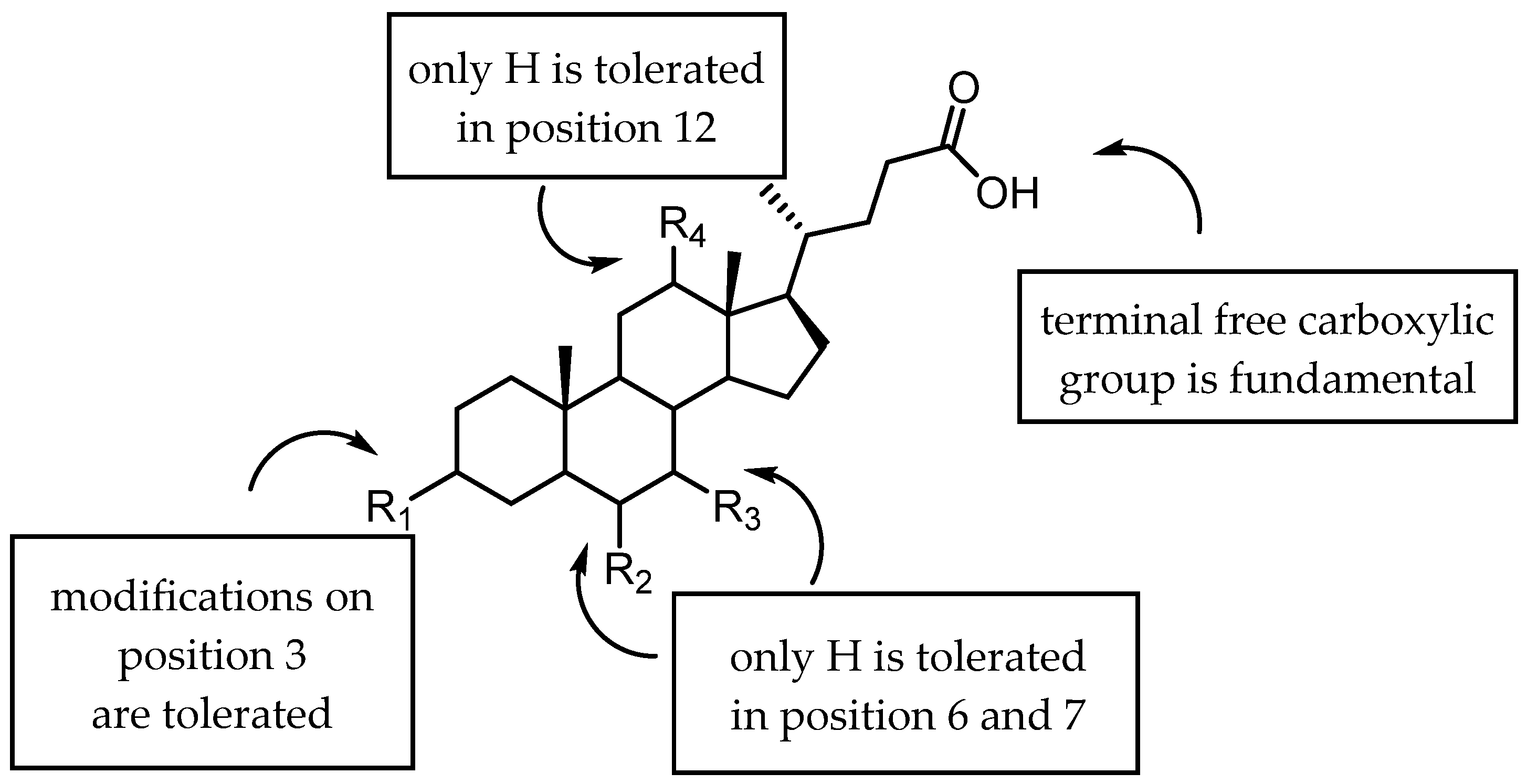
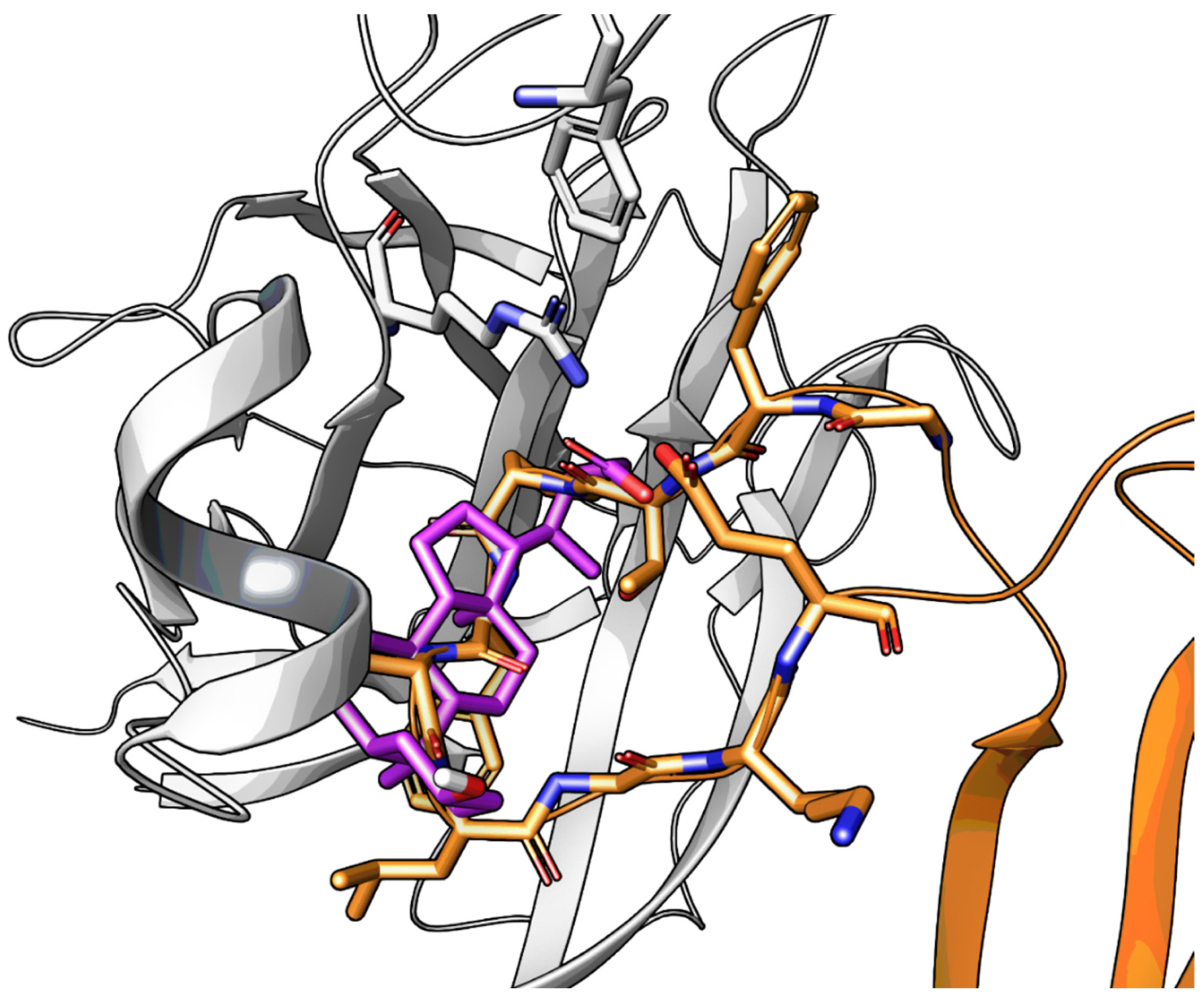
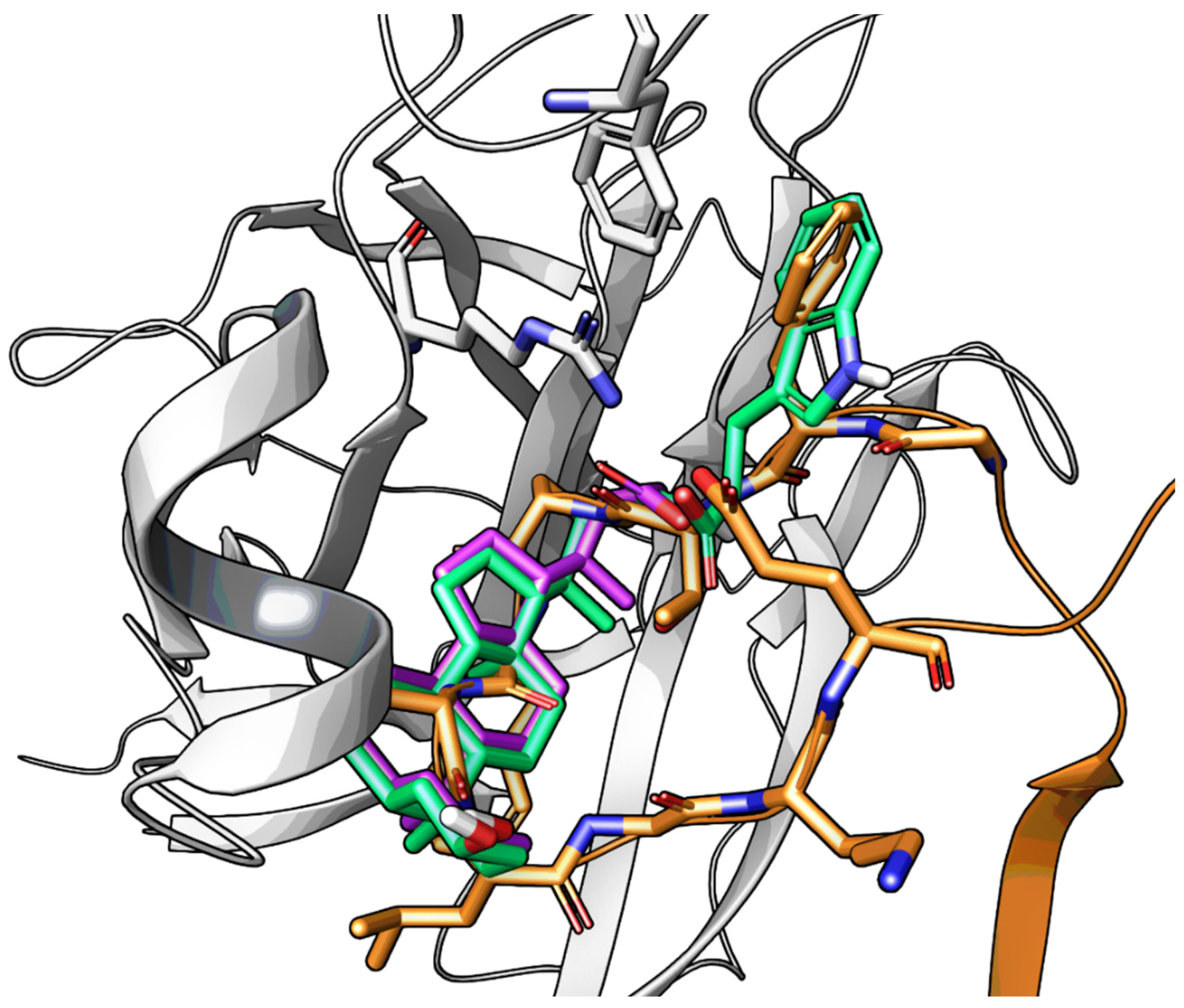
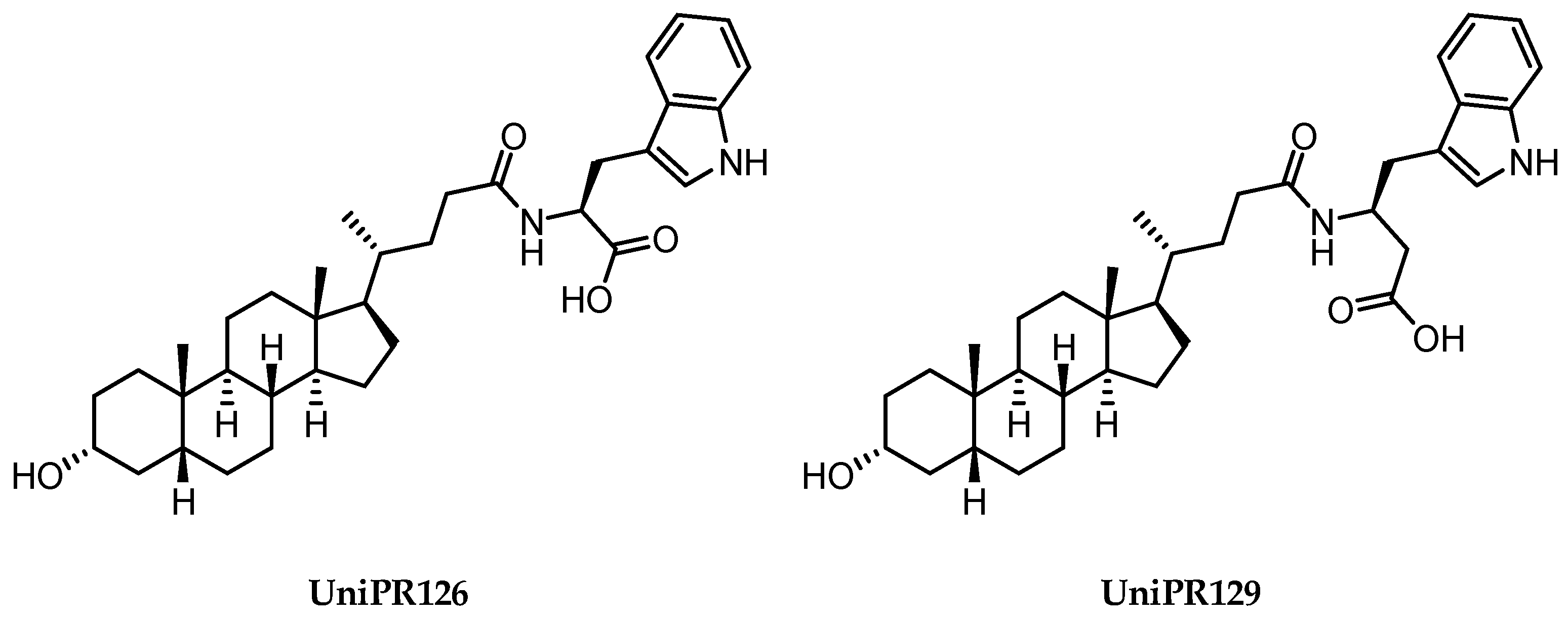
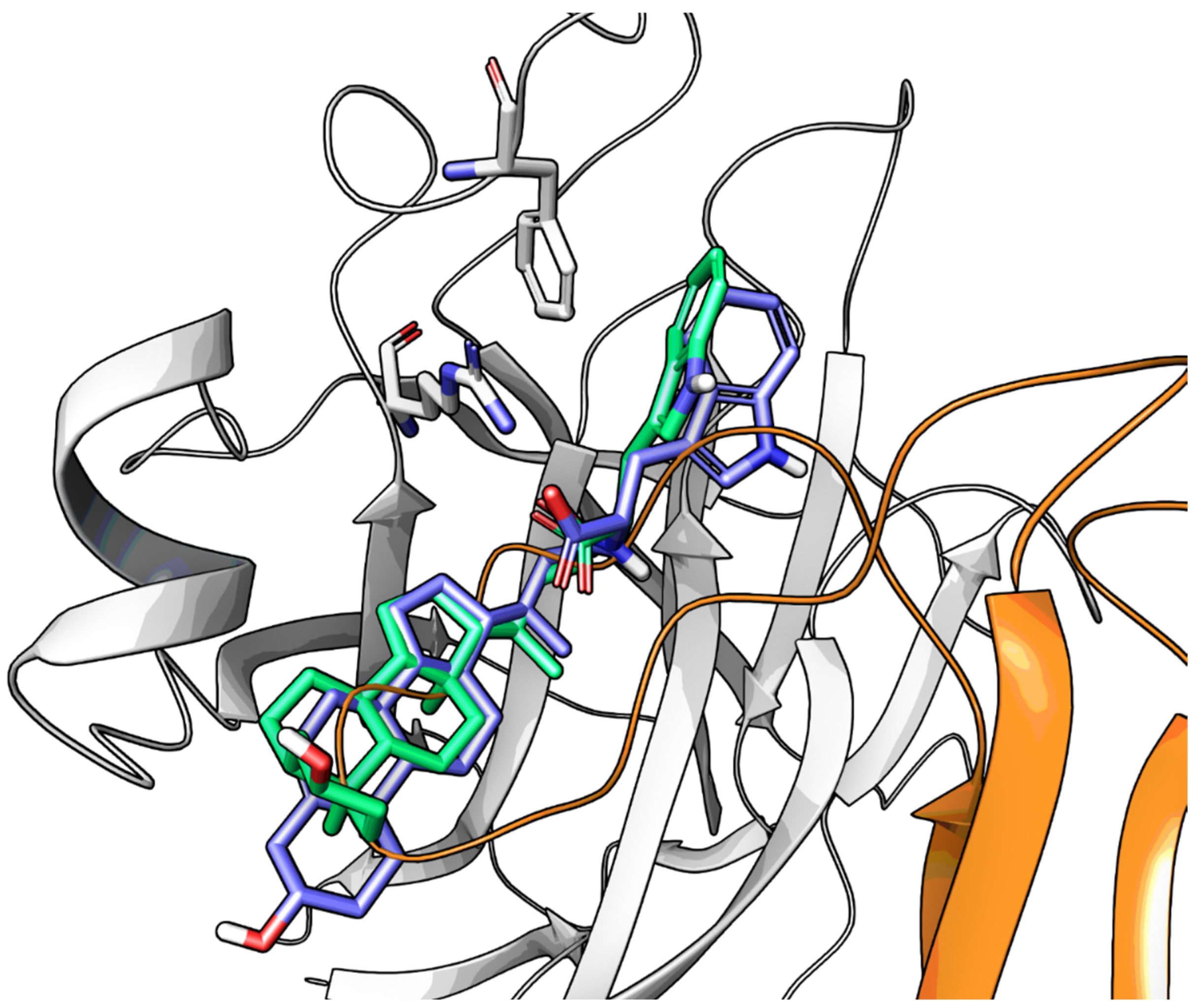
2.3.2. Amino Acid Conjugates of 3β-Hydroxy-Δ5-Cholenic Acid as EphA2 Antagonists
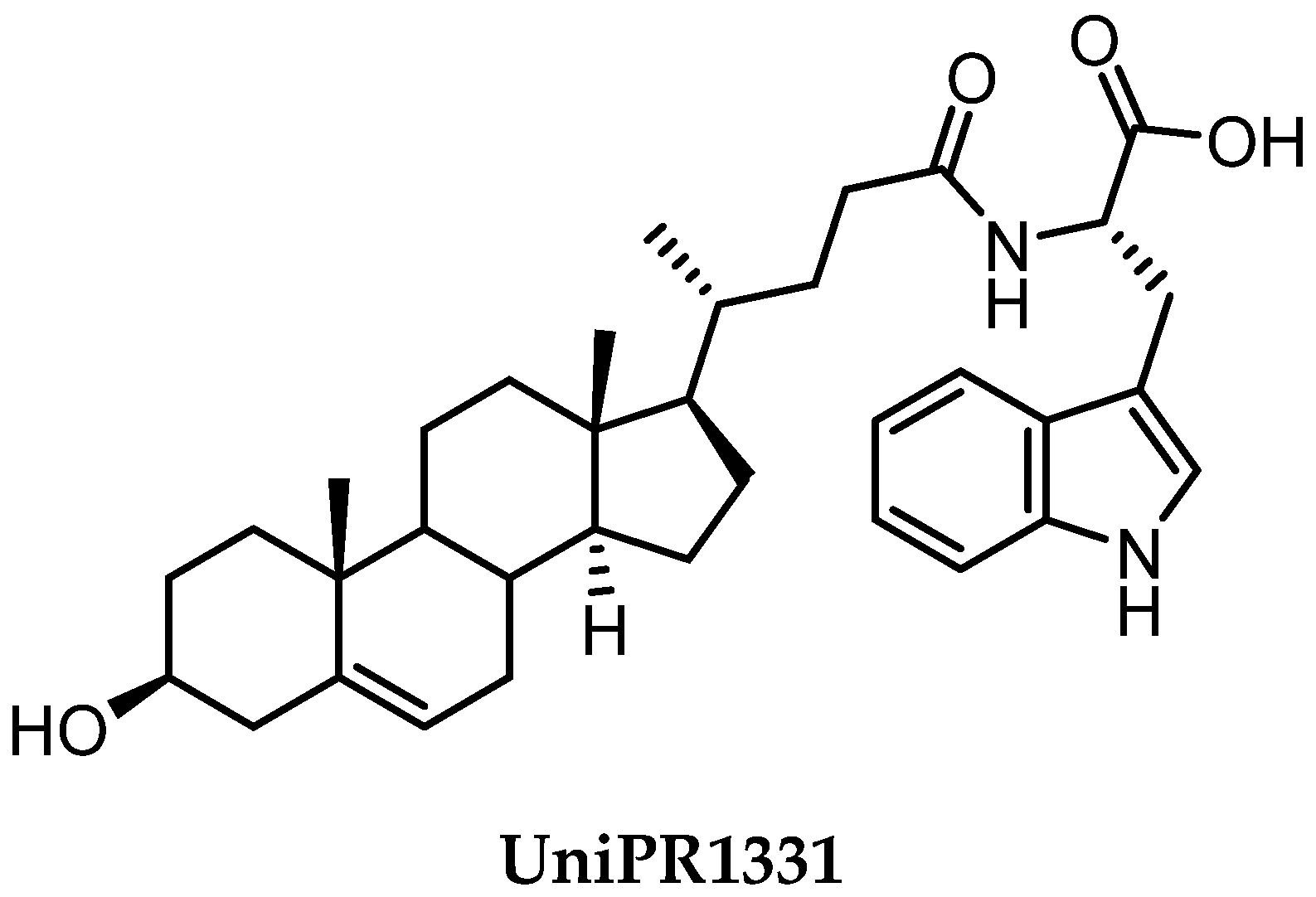
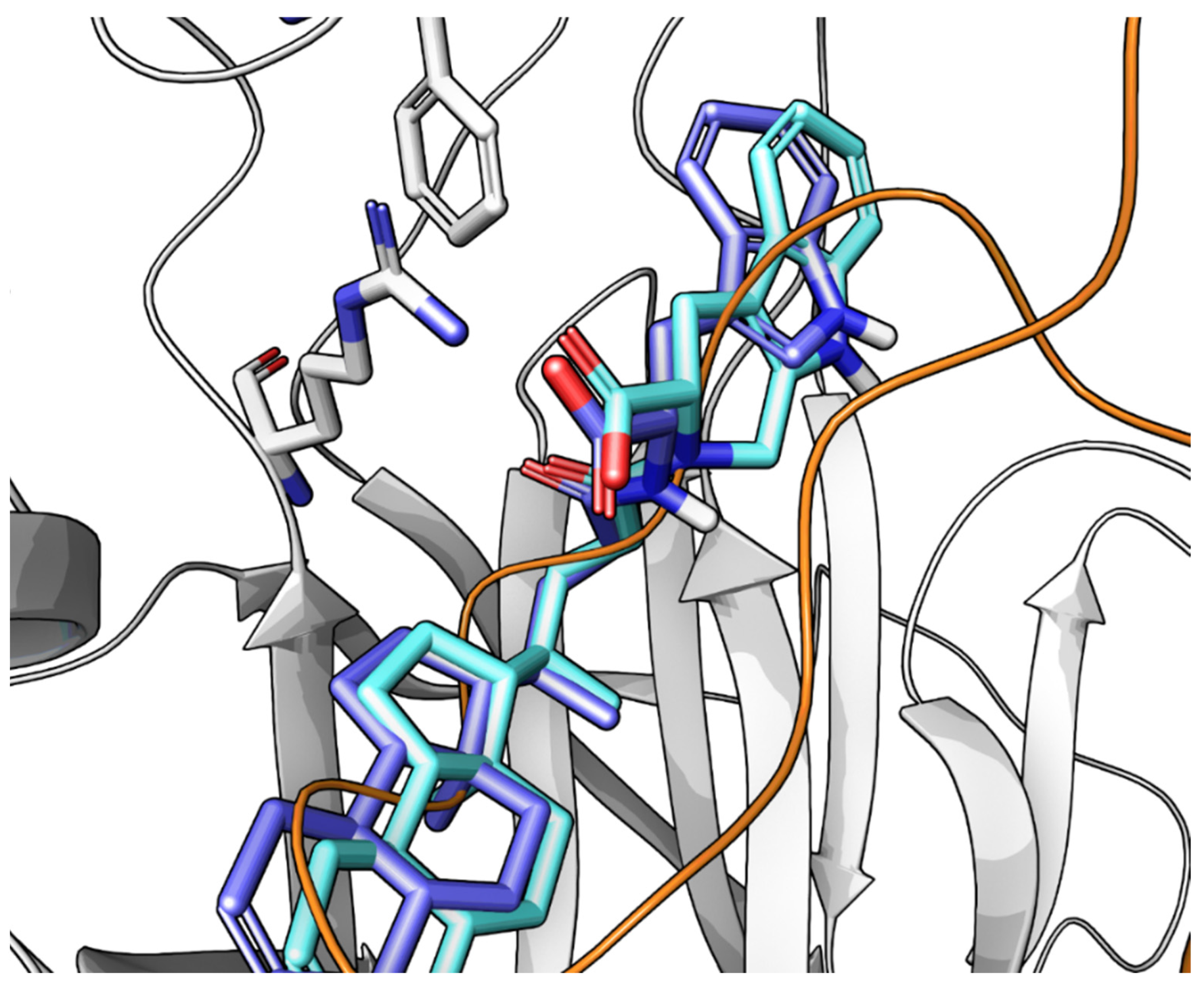
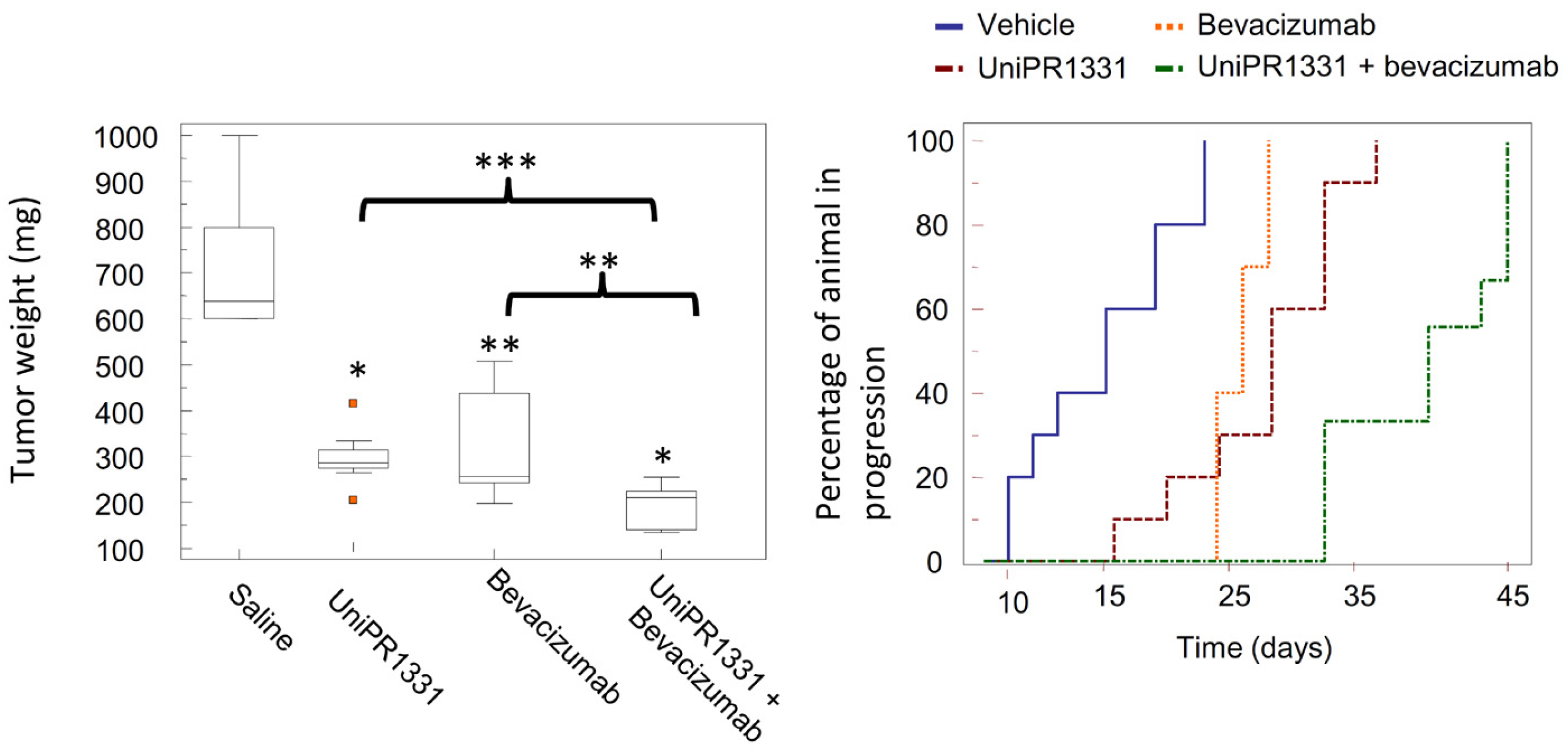
2.3.3. UniPR1331: An Emerging Eph-ephrin Antagonist for In Vivo Study
2.4. Kinase Inhibitors
3. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zisch, A.H.; Pasquale, E.B. The Eph Family: A Multitude of Receptors That Mediate Cell Recognition Signals. Cell Tissue Res. 1997, 290, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Eph Nomenclature Committee. Unified Nomenclature for Eph Family Receptors and Their Ligands, the Ephrins. Cell 1997, 90, 403–404. [Google Scholar] [CrossRef] [Green Version]
- Romero, G.; von Zastrow, M.; Friedman, P.A. Role of PDZ Proteins in Regulating Trafficking, Signaling, and Function of GPCRs: Means, Motif, and Opportunity. Adv. Pharmacol. 2011, 62, 279–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himanen, J.P.; Nikolov, D.B. Eph Receptors and Ephrins. Int. J. Biochem. Cell Biol. 2003, 35, 130–134. [Google Scholar] [CrossRef]
- Himanen, J.P.; Saha, N.; Nikolov, D.B. Cell-Cell Signaling via Eph Receptors and Ephrins. Curr. Opin. Cell Biol. 2007, 19, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Janes, P.W.; Nievergall, E.; Lackmann, M. Concepts and Consequences of Eph Receptor Clustering. Semin. Cell Dev. Biol. 2012, 23, 43–50. [Google Scholar] [CrossRef]
- Ojosnegros, S.; Cutrale, F.; Rodríguez, D.; Otterstrom, J.J.; Chiu, C.L.; Hortigüela, V.; Tarantino, C.; Seriola, A.; Mieruszynski, S.; Martínez, E.; et al. Eph-Ephrin Signaling Modulated by Polymerization and Condensation of Receptors. Proc. Natl. Acad. Sci. USA 2017, 114, 13188–13193. [Google Scholar] [CrossRef] [Green Version]
- Nikolov, D.B.; Xu, K.; Himanen, J.P. Homotypic Receptor-Receptor Interactions Regulating Eph Signaling. Cell Adh. Migr. 2014, 8, 360–365. [Google Scholar] [CrossRef]
- Himanen, J.P.; Nikolov, D.B. Eph Signaling: A Structural View. Trends Neurosci. 2003, 26, 46–51. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Dravis, C.; Yokoyama, N.; Chumley, M.J.; Cowan, C.A.; Silvany, R.E.; Shay, J.; Baker, L.A.; Henkemeyer, M. Bidirectional Signaling Mediated by Ephrin-B2 and EphB2 Controls Urorectal Development. Dev. Biol. 2004, 15, 272–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egea, J.; Diger Klein, R. Bidirectional Eph-Ephrin Signaling during Axon Guidance. Trends Cell Biol. 2007, 17, 230–238. [Google Scholar] [CrossRef]
- Groppa, E.; Brkic, S.; Uccelli, A.; Wirth, G.; Korpisalo-Pirinen, P.; Filippova, M.; Dasen, B.; Sacchi, V.; Muraro, M.G.; Trani, M.; et al. EphrinB2/EphB4 Signaling Regulates Non-sprouting Angiogenesis by VEGF. EMBO Rep. 2018, 19, e45054. [Google Scholar] [CrossRef]
- Mosch, B.; Reissenweber, B.; Neuber, C.; Pietzsch, J. Eph Receptors and Ephrin Ligands: Important Players in Angiogenesis and Tumor Angiogenesis. J. Oncol. 2010, 2010, 135285. [Google Scholar] [CrossRef] [PubMed]
- Kania, A.; Klein, R. Mechanisms of Ephrin-Eph Signalling in Development, Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Genander, M.; Holmberg, J.; Frisén, J. Ephrins Negatively Regulate Cell Proliferation in the Epidermis and Hair Follicle. Stem Cells 2010, 28, 1196–1205. [Google Scholar] [CrossRef]
- Ji, H.; Goode, R.J.A.; Vaillant, F.; Mathivanan, S.; Kapp, E.A.; Mathias, R.A.; Lindeman, G.J.; Visvader, J.E.; Simpson, R.J. Proteomic Profiling of Secretome and Adherent Plasma Membranes from Distinct Mammary Epithelial Cell Subpopulations. Proteomics 2011, 11, 4029–4039. [Google Scholar] [CrossRef]
- Kosinski, C.; Li, V.S.W.; Chan, A.S.Y.; Zhang, J.; Ho, C.; Wai, Y.T.; Tsun, L.C.; Mifflin, R.C.; Powell, D.W.; Siu, T.Y.; et al. Gene Expression Patterns of Human Colon Tops and Basal Crypts and BMP Antagonists as Intestinal Stem Cell Niche Factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.C.; Lin, S.J.; Lin, K.M.; Chou, Y.C.; Lin, C.W.; Yu, S.C.; Chen, C.L.; Shen, T.L.; Chen, C.K.; Lu, J.; et al. Regulation of EBV LMP1-Triggered EphA4 Downregulation in EBV-Associated B Lymphoma and Its Impact on Patients’ Survival. Blood 2016, 128, 1578–1589. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph Receptors and Ephrins in Cancer: Bidirectional Signalling and Beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Barquilla, A.; Pasquale, E.B. Eph Receptors and Ephrins: Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 467–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushansky, A.; Douglass, A.N.; Arang, N.; Vigdorovich, V.; Dambrauskas, N.; Kain, H.S.; Austin, L.S.; Sather, D.N.; Kappe, S.H.I. Malaria Parasites Target the Hepatocyte Receptor EphA2 for Successful Host Infection. Science 2015, 350, 1089–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, E.; Visioli, A.; Giani, F.; Lamorte, G.; Copetti, M.; Pitter, K.L.; Huse, J.T.; Cajola, L.; Zanetti, N.; DiMeco, F.; et al. The EphA2 Receptor Drives Self-Renewal and Tumorigenicity in Stem-like Tumor-Propagating Cells from Human Glioblastomas. Cancer Cell 2012, 22, 765–780. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Li, D.Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 Mediates Ligand-Dependent Inhibition and Ligand-Independent Promotion of Cell Migration and Invasion via a Reciprocal Regulatory Loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; He, S.; Fu, J.; Li, G.; Xu, R.; Lu, H.; Deng, J. Expression of EphrinB2 and EphB4 in Glioma Tissues Correlated to the Progression of Glioma and the Prognosis of Glioblastoma Patients. Clin. Transl. Oncol. 2012, 14, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Ferluga, S.; Debinski, W. Ephs and Ephrins in Malignant Gliomas. Growth Factors 2014, 32, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carles-Kinch, K.; Kilpatrick, K.E.; Stewart, J.C.; Kinch, M.S. Antibody Targeting of the EphA2 Tyrosine Kinase Inhibits Malignant Cell Behavior. Cancer Res. 2002, 62, 2840–2847. [Google Scholar] [PubMed]
- Hammond, S.A.; Lutterbuese, R.; Roff, S.; Lutterbuese, P.; Schlereth, B.; Bruckheimer, E.; Kinch, M.S.; Coats, S.; Baeuerle, P.A.; Kufer, P.; et al. Selective Targeting and Potent Control of Tumor Growth Using an EphA2/CD3-Bispecific Single-Chain Antibody Construct. Cancer Res. 2007, 67, 3927–3935. [Google Scholar] [CrossRef] [Green Version]
- Dall’Acqua, W.F.; Damschroder, M.M.; Zhang, J.; Woods, R.M.; Widjaja, L.; Yu, J.; Wu, H. Antibody Humanization by Framework Shuffling. Methods 2005, 36, 43–60. [Google Scholar] [CrossRef]
- Peng, L.; Oganesyan, V.; Damschroder, M.M.; Wu, H.; Dall’Acqua, W.F. Structural and Functional Characterization of an Agonistic Anti-Human EphA2 Monoclonal Antibody. J. Mol. Biol. 2011, 413, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Stone, R.L.; Lee, S.J.; Nam, E.J.; Roh, J.W.; Nick, A.M.; Han, H.D.; Shahzad, M.M.K.; Kim, H.S.; Mangala, L.S.; et al. EphA2 Targeted Chemotherapy Using an Antibody Drug Conjugate in Endometrial Carcinoma. Clin. Cancer Res. 2010, 16, 2562–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.M.; Kohn, E.C.; LoRusso, P.; Houston, N.D.; Coleman, R.L.; Buzoianu, M.; Robbie, G.; Lechleider, R. Phase 1, Open-Label Study of MEDI-547 in Patients with Relapsed or Refractory Solid Tumors. Investig. New Drugs 2012, 31, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitara, K.; Satoh, T.; Iwasa, S.; Yamaguchi, K.; Muro, K.; Komatsu, Y.; Nishina, T.; Esaki, T.; Hasegawa, J.; Kakurai, Y.; et al. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Afucosylated, Humanized Anti-EPHA2 Antibody DS-8895a: A First-in-Human Phase i Dose Escalation and Dose Expansion Study in Patients with Advanced Solid Tumors. J. Immunother. Cancer 2019, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Greenberg, P.L.; Wei, A.H.; Durrant, S.; Advani, A.S.; Hertzberg, M.S.; Lewis, I.D.; Rivera, G.; Gratzinger, D.; Fan, A.C.; et al. KB004, a First in Class Monoclonal Antibody Targeting the Receptor Tyrosine Kinase EphA3, in Patients with Advanced Hematologic Malignancies: Results from a Phase 1 Study. Leuk. Res. 2016, 50, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Cher, L.; Inglis, P.; Lwin, Z.; Lau, E.; Ackermann, U.; Coombs, N.; Remen, K.; Guo, N.; Ting Lee, S.; et al. Preliminary Findings of a Phase I Safety and Bioimaging Trial of Kb004 (Ifabotuzumab) In Patients with Glioblastoma. Neuro. Oncol. 2019, 21, vi6. [Google Scholar] [CrossRef]
- Hughes, A.; Clarson, J.; Gargett, T.; Yu, W.; Brown, M.P.; Lopez, A.F.; Hughes, T.P.; Yong, A.S.M. Comment on “KB004, a First in Class Monoclonal Antibody Targeting the Receptor Tyrosine Kinase EphA3, in Patients with Advanced Hematologic Malignancies: Results from a Phase 1 Study”. Leuk. Res. 2017, 55, 55–57. [Google Scholar] [CrossRef]
- Combination Therapy With Pembrolizumab and SEphB4-HSA in Previously Treated Urothelial Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02717156 (accessed on 19 January 2022).
- SEphB4-HSA in Treating Patients With Kaposi Sarcoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02799485?term=ephB4&draw=2&rank=2 (accessed on 19 January 2022).
- Koolpe, M.; Dail, M.; Pasquale, E.B. An Ephrin Mimetic Peptide That Selectively Targets the EphA2 Receptor. J. Biol. Chem. 2002, 277, 46974–46979. [Google Scholar] [CrossRef] [Green Version]
- Himanen, J.P.; Rajashankar, K.R.; Lackmann, M.; Cowan, C.A.; Henkemeyer, M.; Nikolov, D.B. Crystal Structure of an Eph Receptor-Ephrin Complex. Nature 2001, 414, 933–938. [Google Scholar] [CrossRef]
- Murai, K.K.; Nguyen, L.N.; Koolpe, M.; McLennan, R.; Krull, C.E.; Pasquale, E.B. Targeting the EphA4 Receptor in the Nervous System with Biologically Active Peptides. Mol. Cell. Neurosci. 2003, 24, 1000–1011. [Google Scholar] [CrossRef]
- Riedl, S.; Pasquale, E. Targeting the Eph System with Peptides and Peptide Conjugates. Curr. Drug Targets 2015, 16, 1031–1047. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Li, Y.; Dai, W.; Guo, Z.; Wang, Z. EphA2 Targeted Doxorubicin Stealth Liposomes as a Therapy System for Choroidal Neovascularization in Rats. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7348–7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Wang, S.; De, S.K.; Barile, E.; Quinn, B.A.; Zharkikh, I.; Purves, A.; Stebbins, J.L.; Oshima, R.G.; Fisher, P.B.; et al. Design and Characterization of Novel EphA2 Agonists for Targeted Delivery of Chemotherapy to Cancer Cells. Chem. Biol. 2015, 22, 876–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, E.; Pellecchia, M. NMR-Based for the Identification and Optimization of Inhibitors of Protein–Protein Interactions. Chem. Rev. 2014, 114, 4749–4763. [Google Scholar] [CrossRef] [PubMed]
- Gambini, L.; Salem, A.F.; Udompholkul, P.; Tan, X.-F.; Baggio, C.; Shah, N.; Aronson, A.; Song, J.; Pellecchia, M. Structure-Based Design of Novel EphA2 Agonistic Agents with Nanomolar Affinity in Vitro and in Cell. ACS Chem. Biol. 2018, 13, 2633–2644. [Google Scholar] [CrossRef]
- Koolpe, M.; Burgess, R.; Dail, M.; Pasquale, E.B. EphB Receptor-Binding Peptides Identified by Phage Display Enable Design of an Antagonist with Ephrin-like Affinity. J. Biol. Chem. 2005, 280, 17301–17311. [Google Scholar] [CrossRef] [Green Version]
- Chrencik, J.E.; Brooun, A.; Recht, M.I.; Nicola, G.; Davis, L.K.; Abagyan, R.; Widmer, H.; Pasquale, E.B.; Kuhn, P. Three-Dimensional Structure of the EphB2 Receptor in Complex with an Antagonistic Peptide Reveals a Novel Mode of Inhibition. J. Biol. Chem. 2007, 282, 36505–36513. [Google Scholar] [CrossRef] [Green Version]
- Chrencik, J.E.; Brooun, A.; Kraus, M.L.; Recht, M.I.; Kolatkar, A.R.; Gye, W.H.; Seifert, J.M.; Widmer, H.; Auer, M.; Kuhn, P. Structural and Biophysical Characterization of the EphB4*ephrinB2 Protein-Protein Interaction and Receptor Specificity. J. Biol. Chem. 2006, 281, 28185–28192. [Google Scholar] [CrossRef] [Green Version]
- Chrencik, J.E.; Brooun, A.; Recht, M.I.; Kraus, M.L.; Koolpe, M.; Kolatkar, A.R.; Bruce, R.H.; Martiny-Baron, G.; Widmer, H.; Pasquale, E.B.; et al. Structure and Thermodynamic Characterization of the EphB4/Ephrin-B2 Antagonist Peptide Complex Reveals the Determinants for Receptor Specificity. Structure 2006, 14, 321–330. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Wang, Z.; Du, Y.; Yuan, H.; Zhang, P.; Zhou, J.; Liu, F.; Li, C.; Hu, F. Specific Tumor Delivery of Paclitaxel Using Glycolipid-like Polymer Micelles Containing Gold Nanospheres. Biomaterials 2013, 34, 4510–4519. [Google Scholar] [CrossRef] [Green Version]
- Noberini, R.; Mitra, S.; Salvucci, O.; Valencia, F.; Duggineni, S.; Prigozhina, N.; Wei, K.; Tosato, G.; Huang, Z.; Pasquale, E.B. PEGylation Potentiates the Effectiveness of an Antagonistic Peptide That Targets the EphB4 Receptor with Nanomolar Affinity. PloS ONE 2011, 6, e28611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tognolini, M.; Lodola, A. Targeting the Eph-Ephrin System with Protein-Protein Interaction (PPI) Inhibitors. Curr. Drug Targets 2015, 16, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Lodola, A.; Incerti, M.; Tognolini, M. On the Use of 2,5-Dimethyl-Pyrrol-1-Yl-Benzoic Acid Derivatives as EPH-Ephrin Antagonists. J. Virol. 2014, 88, 12173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, C.; Mohamed, I.H.; Flammini, L.; Barocelli, E.; Incerti, M.; Lodola, A.; Tognolini, M. Lithocholic Acid Is an Eph-Ephrin Ligand Interfering with Eph-Kinase Activation. PLoS ONE 2011, 6, e18128. [Google Scholar] [CrossRef]
- Tognolini, M.; Incerti, M.; Hassan-Mohamed, I.; Giorgio, C.; Russo, S.; Bruni, R.; Lelli, B.; Bracci, L.; Noberini, R.; Pasquale, E.B.; et al. Structure-Activity Relationships and Mechanism of Action of Eph-Ephrin Antagonists: Interaction of Cholanic Acid with the EphA2 Receptor. ChemMedChem 2012, 7, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Himanen, J.P.; Goldgur, Y.; Miao, H.; Myshkin, E.; Guo, H.; Buck, M.; Nguyen, M.; Rajashankar, K.R.; Wang, B.; Nikolov, D.B. Ligand Recognition by A-Class Eph Receptors: Crystal Structures of the EphA2 Ligand-Binding Domain and the EphA2/Ephrin-A1 Complex. EMBO Rep. 2009, 10, 722–728. [Google Scholar] [CrossRef]
- Incerti, M.; Tognolini, M.; Russo, S.; Pala, D.; Giorgio, C.; Hassan-Mohamed, I.; Noberini, R.; Pasquale, E.B.; Vicini, P.; Piersanti, S.; et al. Amino Acid Conjugates of Lithocholic Acid as Antagonists of the EphA2 Receptor. J. Med. Chem. 2013, 56, 2936–2947. [Google Scholar] [CrossRef]
- Lema Tome, C.M.; Palma, E.; Ferluga, S.; Lowther, W.T.; Hantgan, R.; Wykosky, J.; Debinski, W. Structural and Functional Characterization of Monomeric EphrinA1 Binding Site to EphA2 Receptor. J. Biol. Chem. 2012, 287, 14012–14022. [Google Scholar] [CrossRef] [Green Version]
- Hassan-Mohamed, I.; Giorgio, C.; Incerti, M.; Russo, S.; Pala, D.; Pasquale, E.B.; Zanotti, I.; Vicini, P.; Barocelli, E.; Rivara, S.; et al. UniPR129 Is a Competitive Small Molecule Eph-Ephrin Antagonist Blocking in Vitro Angiogenesis at Low Micromolar Concentrations. Br. J. Pharmacol. 2014, 171, 5195–5208. [Google Scholar] [CrossRef] [Green Version]
- Tognolini, M.; Incerti, M.; Lodola, A. Are We Using the Right Pharmacological Tools to Target EphA4? ACS Chem. Neurosci. 2014, 5, 1146–1147. [Google Scholar] [CrossRef] [Green Version]
- Incerti, M.; Russo, S.; Callegari, D.; Pala, D.; Giorgio, C.; Zanotti, I.; Barocelli, E.; Vicini, P.; Vacondio, F.; Rivara, S.; et al. Metadynamics for Perspective Drug Design: Computationally Driven Synthesis of New Protein-Protein Interaction Inhibitors Targeting the EphA2 Receptor. J. Med. Chem. 2017, 60, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Incerti, M.; Russo, S.; Corrado, M.; Giorgio, C.; Ballabeni, V.; Chiodelli, P.; Rusnati, M.; Scalvini, L.; Callegari, D.; Castelli, R.; et al. Optimization of EphA2 Antagonists Based on a Lithocholic Acid Core Led to the Identification of UniPR505, a New 3α-Carbamoyloxy Derivative with Antiangiogenetic Properties. Eur. J. Med. Chem. 2020, 189, 112083. [Google Scholar] [CrossRef] [PubMed]
- Pala, D.; Castelli, R.; Incerti, M.; Russo, S.; Tognolini, M.; Giorgio, C.; Hassan-Mohamed, I.; Zanotti, I.; Vacondio, F.; Rivara, S.; et al. Combining Ligand- and Structure-Based Approaches for the Discovery of New Inhibitors of the EPHA2-Ephrin-A1 Interaction. J. Chem. Inf. Model. 2014, 54, 2621–2626. [Google Scholar] [CrossRef] [PubMed]
- Castelli, R.; Tognolini, M.; Vacondio, F.; Incerti, M.; Pala, D.; Callegari, D.; Bertoni, S.; Giorgio, C.; Hassan-Mohamed, I.; Zanotti, I.; et al. Δ5-Cholenoyl-Amino Acids as Selective and Orally Available Antagonists of the Epheephrin System. Eur. J. Med. Chem. 2015, 103, 312–324. [Google Scholar] [CrossRef]
- Ferlenghi, F.; Castelli, R.; Scalvini, L.; Giorgio, C.; Corrado, M.; Tognolini, M.; Mor, M.; Lodola, A.; Vacondio, F. Drug-Gut Microbiota Metabolic Interactions: The Case of UniPR1331, Selective Antagonist of the Eph-Ephrin System, in Mice. J. Pharm. Biomed. Anal. 2020, 180, 113067. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Gravina, G.L.; Giorgio, C.; Mancini, A.; Pellegrini, C.; Colapietro, A.; Monache, S.D.; Maturo, M.G.; Sferra, R.; Chiodelli, P.; et al. UniPR1331, a Small Molecule Targeting Eph/Ephrin Interaction, Prolongs Survival in Glioblastoma and Potentiates the Effect of Antiangiogenic Therapy in Mice. Oncotarget 2018, 9, 24347–24363. [Google Scholar] [CrossRef] [Green Version]
- Rusnati, M.; Paiardi, G.; Tobia, C.; Urbinati, C.; Lodola, A.; D’Ursi, P.; Corrado, M.; Castelli, R.; Wade, R.C.; Tognolini, M.; et al. Cholenic Acid Derivative UniPR1331 Impairs Tumor Angiogenesis via Blockade of VEGF/VEGFR2 in Addition to Eph/Ephrin. Cancer Gene Ther. 2021, 1–10. [Google Scholar] [CrossRef]
- Unzue, A.; Lafleur, K.; Zhao, H.; Zhou, T.; Dong, J.; Kolb, P.; Liebl, J.; Zahler, S.; Caflisch, A.; Nevado, C. Three Stories on Eph Kinase Inhibitors: From in Silico Discovery to in Vivo Validation. Eur. J. Med. Chem. 2016, 112, 347–366. [Google Scholar] [CrossRef] [Green Version]
- Neuber, C.; Belter, B.; Mamat, C.; Pietzsch, J. Radiopharmacologist’s and Radiochemist’s View on Targeting the Eph/Ephrin Receptor Tyrosine Kinase System. ACS Omega 2020, 5, 16318–16331. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Cpd. | R1 | R2 | R3 | R4 | RS | Ki (μM) |
| 1 (LCA) |  |  |  |  |  | 49 |
| 2 (CA) |  |  |  |  |  | >100 |
| 3 (DCA) |  |  |  |  |  | >100 |
| 4 (CDCA) |  |  |  |  |  | >100 |
| 5 |  |  |  |  |  | >100 |
| 6 |  |  |  |  |  | >100 |
| 7 |  |  |  |  |  | 25 |
| 8 |  |  |  |  |  | 5.1 |
| 9 |  |  |  |  |  | >100 |
| 10 |  |  |  |  |  | >100 |
| 11 |  |  |  |  |  | >100 |
 | |||||
|---|---|---|---|---|---|
| Cpd. | R | IC50 (μM) | Cpd. | R | IC50 (μM) |
| 1 (LCA) |  | 57 | 22 |  | >100 |
| 12 |  | 49 | 23 |  | >100 |
| 13 |  | < 300 | 24 |  | 28 |
| 14 |  | 20 | 25 |  | 28 |
| 15 |  | 31 | 26 |  | 6.6 |
| 16 |  | 24 | 27 |  | 7.5 |
| 17 |  | 17 | 28 |  | 50 |
| 18 |  | 33 | 29 |  | 100 |
| 19 |  | 60 | 30 |  | 2.0 |
| 20 |  | >100 | 31 |  | 20 |
| 21 |  | >100 | |||
 | ||
|---|---|---|
| Cpd. | R | IC50 (μM) |
| LCA |  | 57 |
| 12 |  | 49 |
| 32 |  | 29 |
| 33 |  | inactive |
| 34 |  | Inactive |
| 35 |  | 72 |
| 36 |  | inactive |
 | ||
|---|---|---|
| Cpd. | R | IC50 (μM) |
| 32 |  | 29 |
| 37 |  | 83 |
| 38 |  | 10 |
| 39 |  | Inactive |
| 40 |  | 14 |
| 41 |  | 18 |
| 42 |  | Inactive |
| 43 |  | 3.9 |
| 44 |  | 1.8 |
| UniPR129 |  | 0.95 |
| 45 |  | 26 |
 | ||
|---|---|---|
| Cpd. | R | IC50 (μM) |
| UniPR129 |  | 0.95 |
| 46 |  | 17 |
| 47 |  | 28 |
| 48 |  | 28 |
| 49 |  | 3.1 |
| 50 |  | 13 |
| 51 |  | 0.8 |
| 52 |  | 5.2 |
 | ||
|---|---|---|
| Cpd. | R | IC50 (μM) |
| UniPR129 |  | 0.91 |
| UniPR502 |  | 0.8 |
| 53 (UniPR505) |  | 0.95 |
| 54 |  | 1.1 |
| 55 |  | 1.5 |
| 56 |  | 15 |
| 57 |  | 7.1 |
| 58 |  | 1.2 |
| 59 |  | 5.5 |
 | |||||
|---|---|---|---|---|---|
| Cpd. | R | IC50 (μM) | Cpd. | R | IC50 (μM) |
| 61 |  | 40 | 69 |  | 4.1 |
| 62 |  | inactive | 70 |  | 8.9 |
| 63 |  | >100 | 71 |  | 22 |
| 64 |  | inactive | 72 |  | 6.0 |
| 65 |  | >100 | 73 |  | 47 |
| 66 |  | 50 | 74 |  | inactive |
| UniPR 1331 |  | 3.5 | 75 |  | 4.2 |
| 67 |  | >100 | 76 |  | 89 |
| 68 |  | 5.0 | 77 |  | 6.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guidetti, L.; Castelli, R.; Scalvini, L.; Ferlenghi, F.; Corrado, M.; Giorgio, C.; Tognolini, M.; Lodola, A. Protein-Protein Interaction Inhibitors Targeting the Eph-Ephrin System with a Focus on Amino Acid Conjugates of Bile Acids. Pharmaceuticals 2022, 15, 137. https://doi.org/10.3390/ph15020137
Guidetti L, Castelli R, Scalvini L, Ferlenghi F, Corrado M, Giorgio C, Tognolini M, Lodola A. Protein-Protein Interaction Inhibitors Targeting the Eph-Ephrin System with a Focus on Amino Acid Conjugates of Bile Acids. Pharmaceuticals. 2022; 15(2):137. https://doi.org/10.3390/ph15020137
Chicago/Turabian StyleGuidetti, Lorenzo, Riccardo Castelli, Laura Scalvini, Francesca Ferlenghi, Miriam Corrado, Carmine Giorgio, Massimiliano Tognolini, and Alessio Lodola. 2022. "Protein-Protein Interaction Inhibitors Targeting the Eph-Ephrin System with a Focus on Amino Acid Conjugates of Bile Acids" Pharmaceuticals 15, no. 2: 137. https://doi.org/10.3390/ph15020137
APA StyleGuidetti, L., Castelli, R., Scalvini, L., Ferlenghi, F., Corrado, M., Giorgio, C., Tognolini, M., & Lodola, A. (2022). Protein-Protein Interaction Inhibitors Targeting the Eph-Ephrin System with a Focus on Amino Acid Conjugates of Bile Acids. Pharmaceuticals, 15(2), 137. https://doi.org/10.3390/ph15020137