
In Vitro–In Silico Modeling of Caffeine and Diclofenac Permeation in Static and Fluidic Systems with a 16HBE Lung Cell Barrier



Abstract
:1. Introduction
2. Results
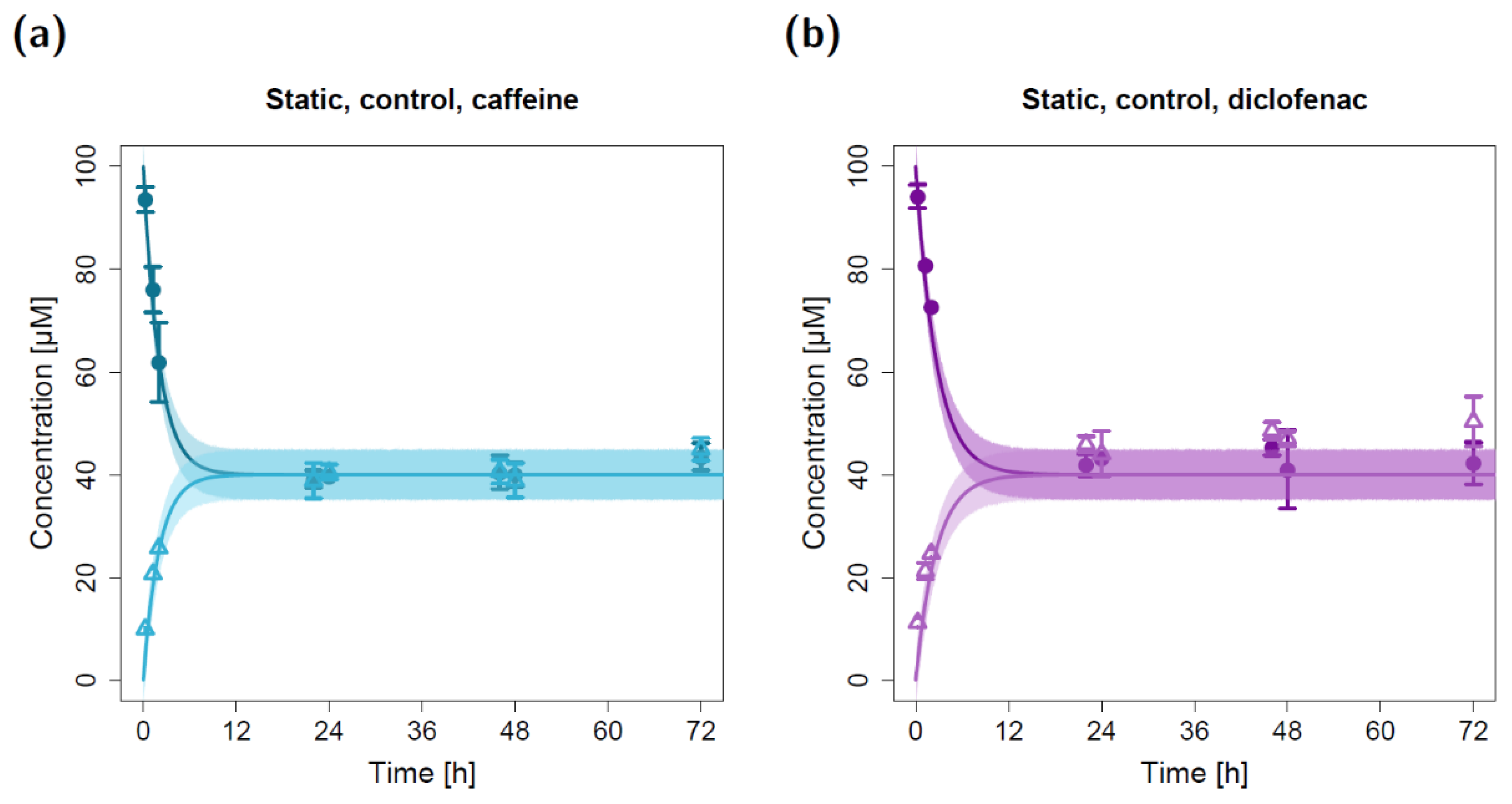
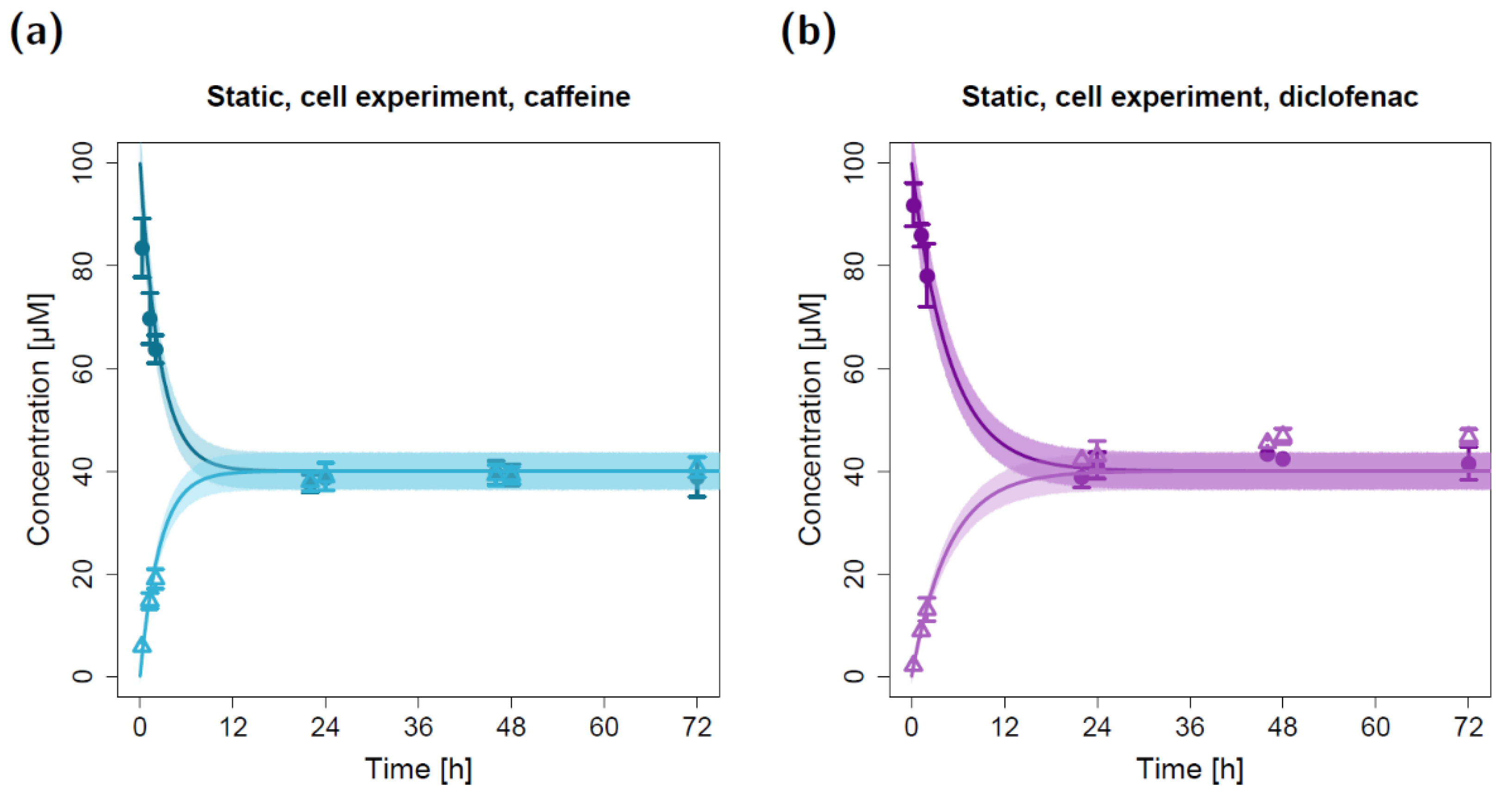
2.1. Static Permeation Studies
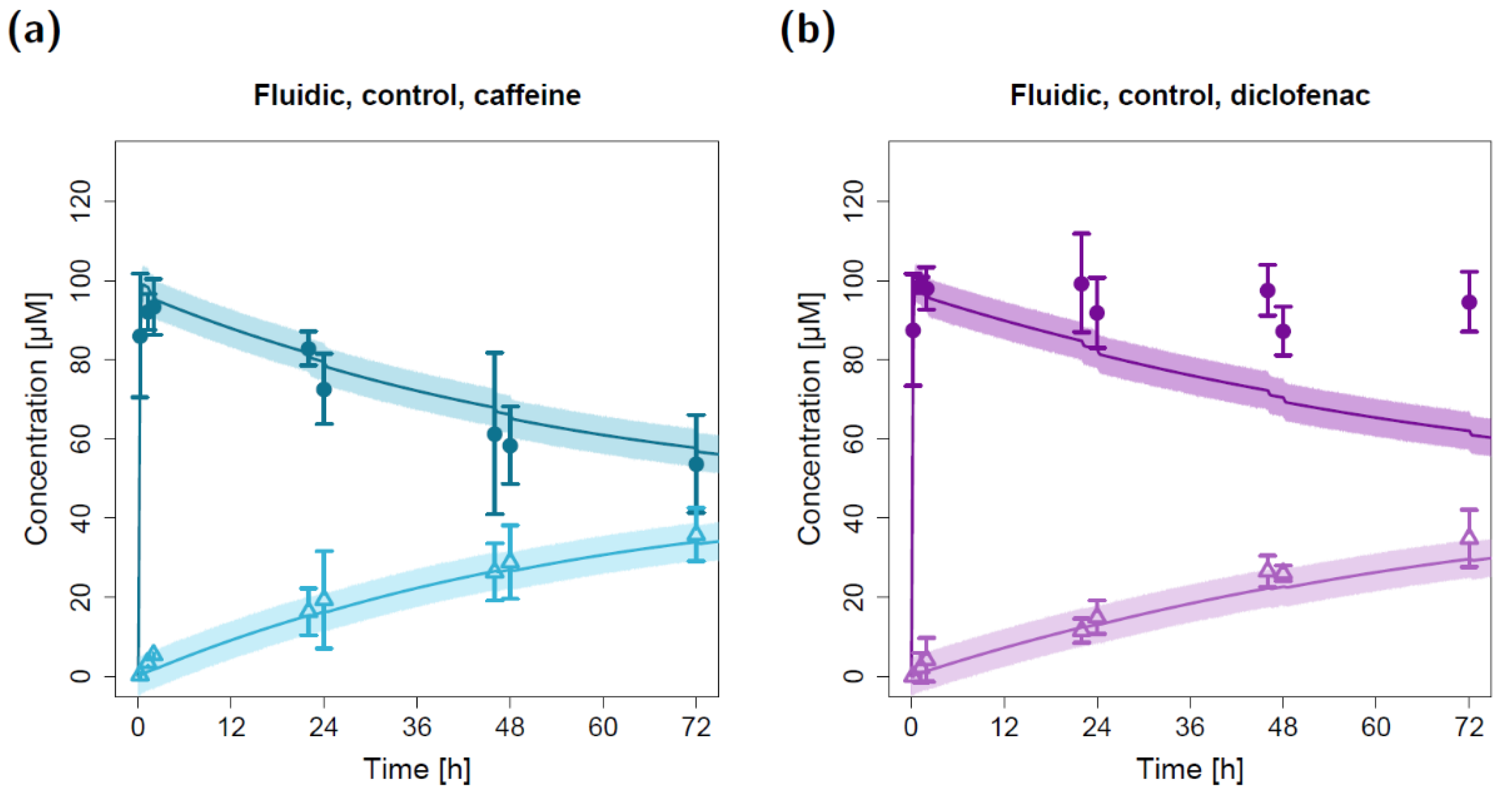
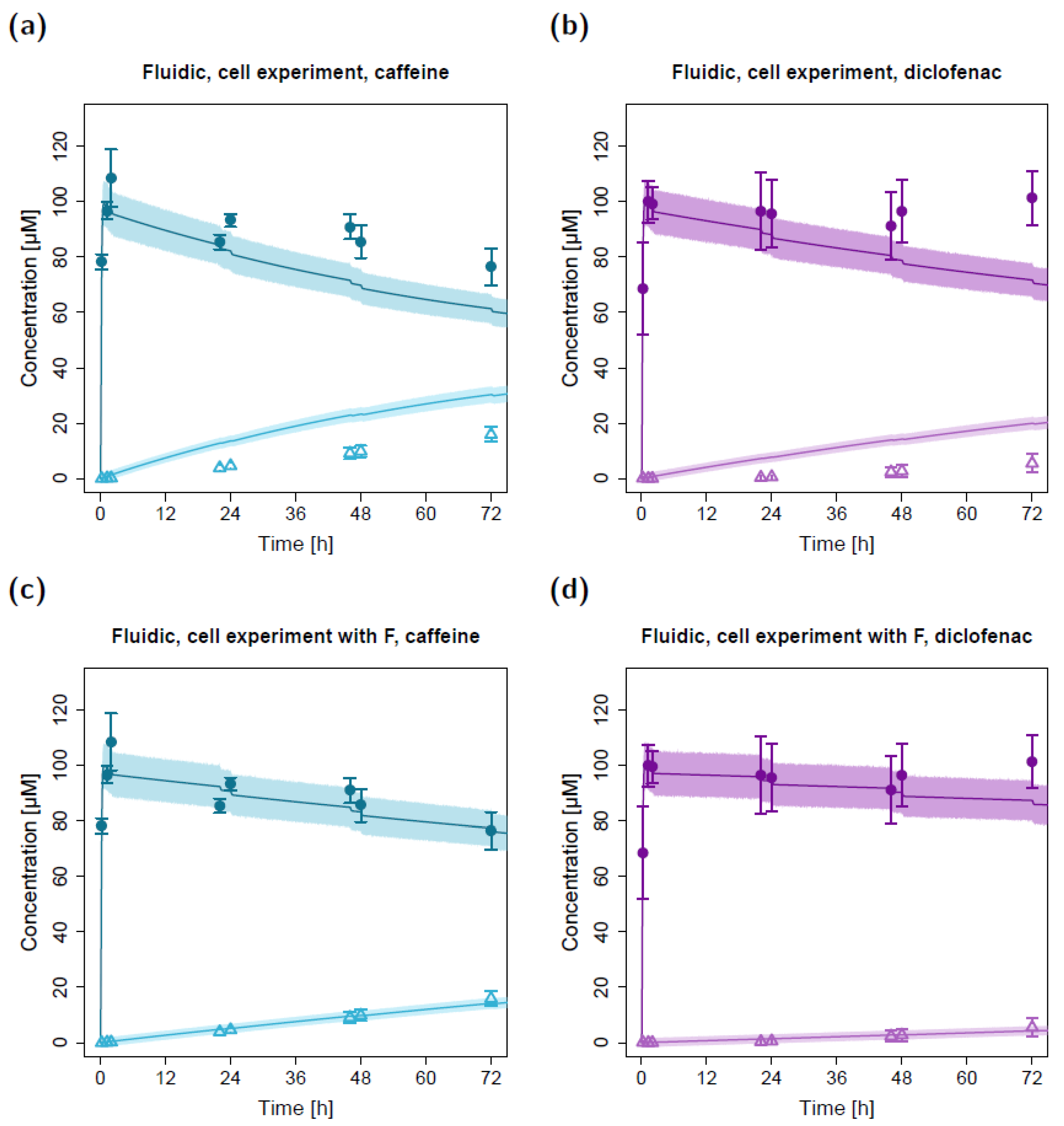
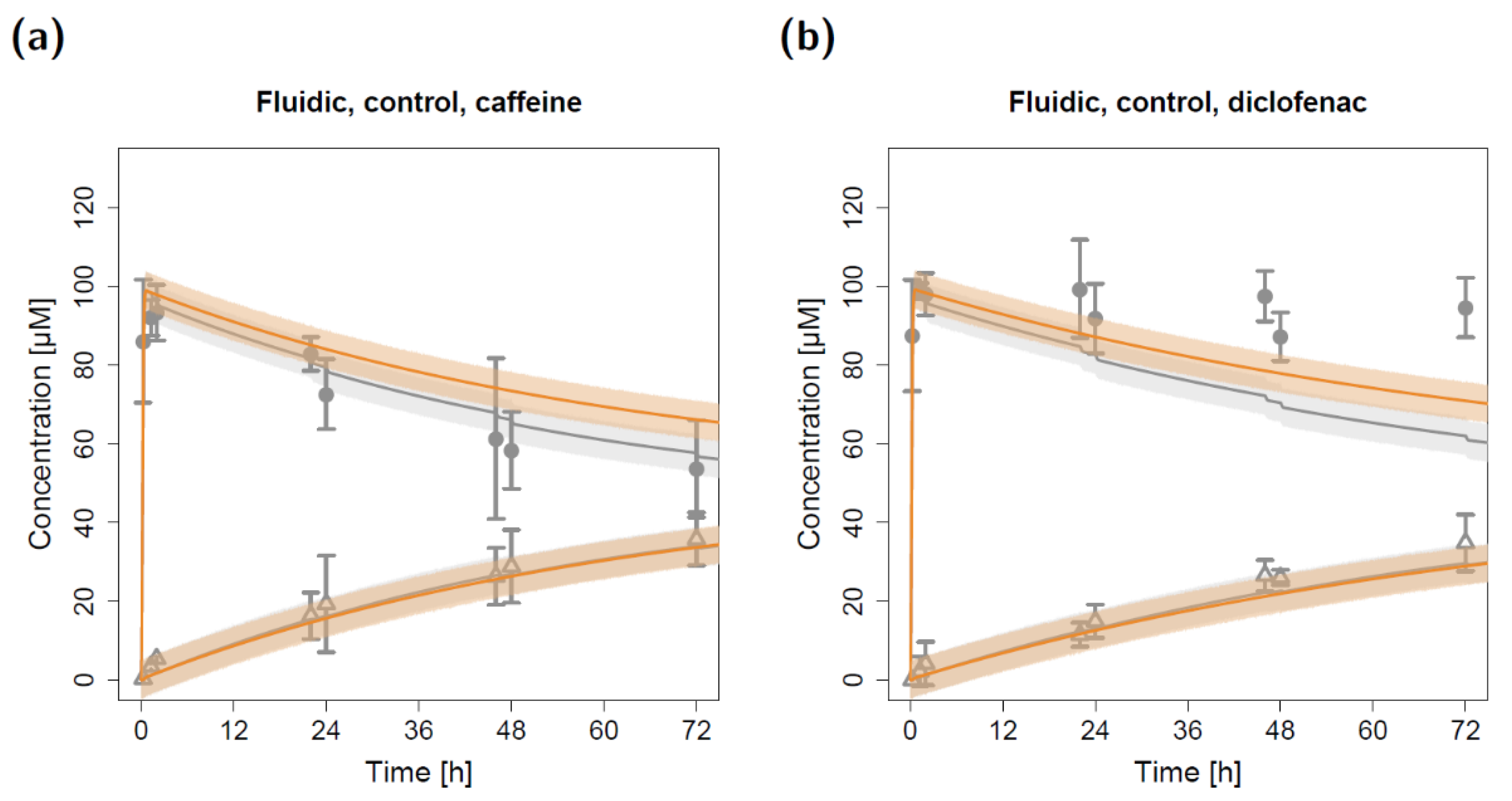
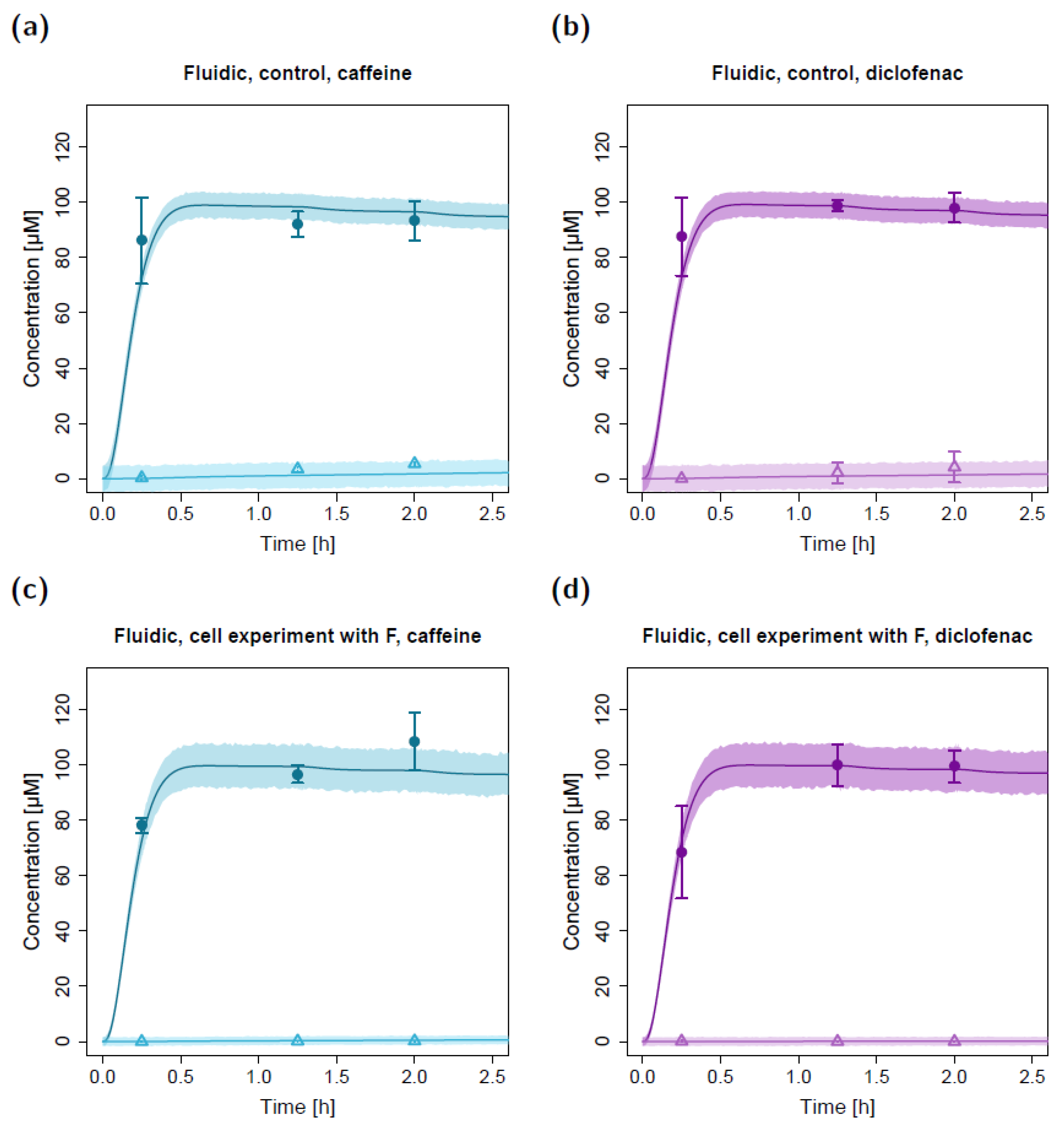
2.2. Fluidic Permeation Studies
3. Discussion
4. Materials and Methods
4.1. Standard Cell Cultivation
4.2. Transepithelial Electric Resistance (TEER) Measurements
4.3. Cell Viability Assay
4.4. Sample Preparation for In Vitro Permeation Studies
4.5. Permeation Studies under Static Conditions
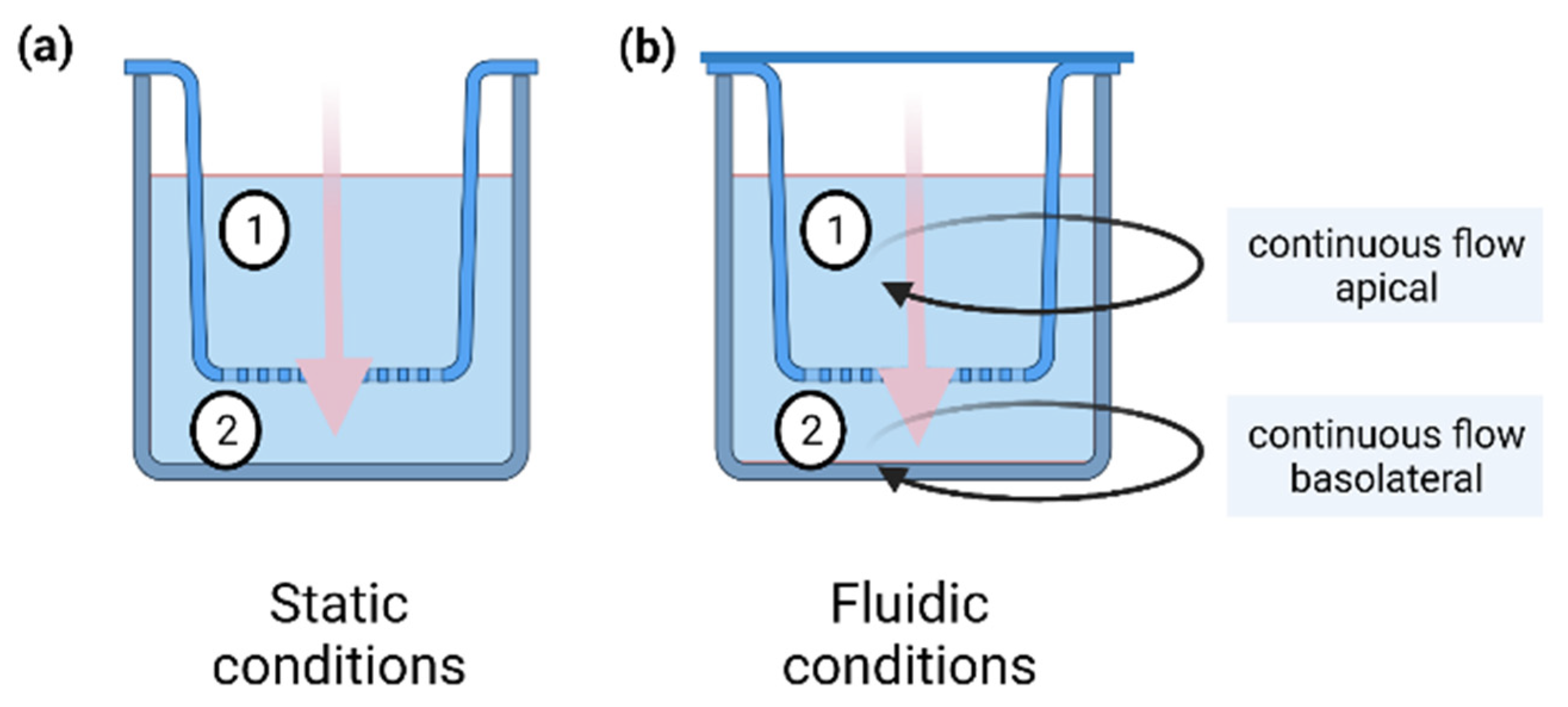
4.6. Permeation Studies under Fluidic Conditions
4.7. High-Performance Liquid Chromatography (HPLC) Analysis
4.8. Software
4.9. In Silico Model for the Static System
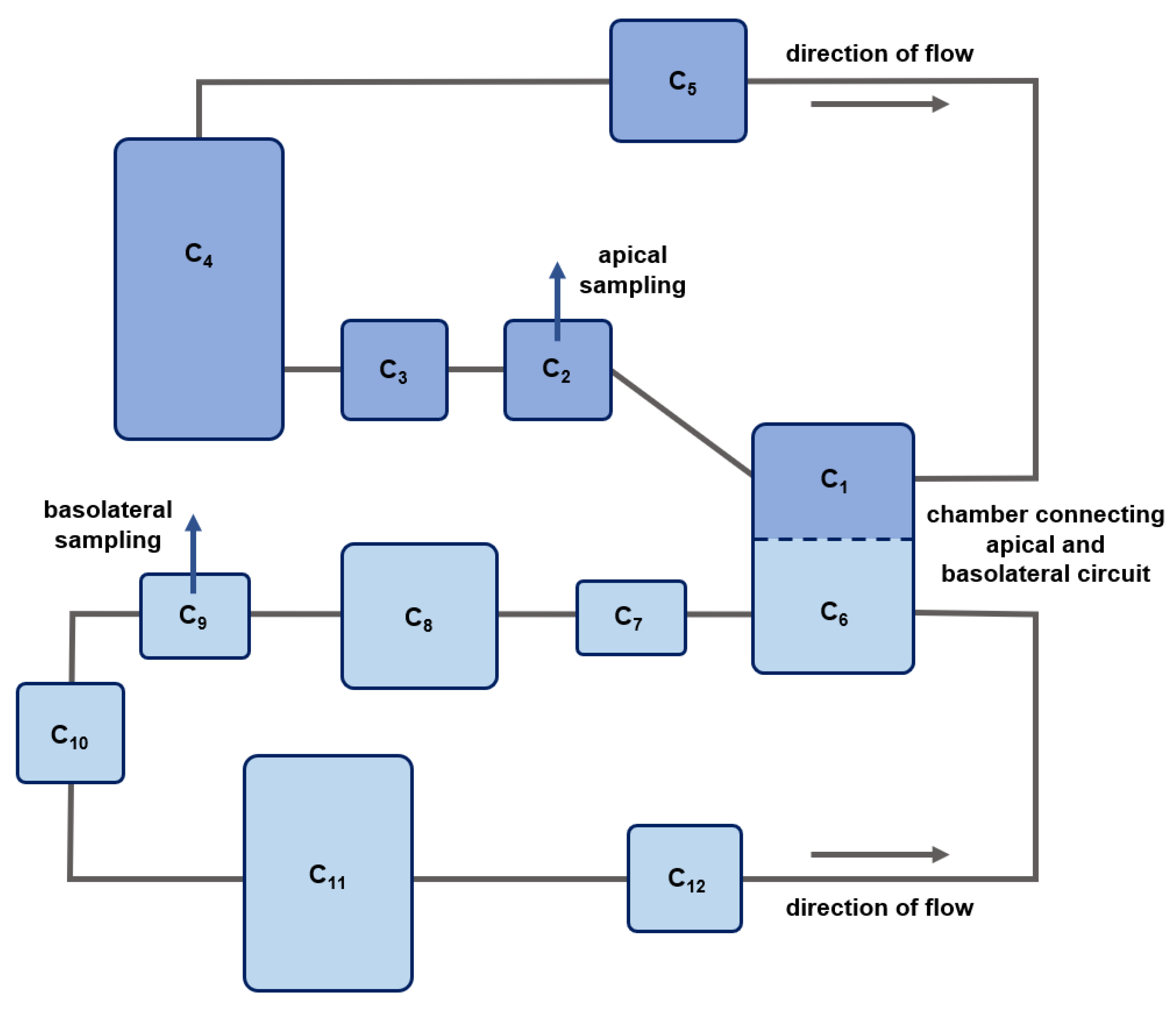
4.10. In Silico Model for the Fluidic System
4.11. Model Parameter Estimation and Simulation
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Directive 2010/63/EU of the European Parliament and of the Coundcil of 22 September 2010 on the Protection of Animals used for Scientific Purposes. Off. J. Eur. Union 2010, L276, 33–79.
- Kirk, R.G.W. Recovering The Principles of Humane Experimental Technique. Sci. Technol. Hum. Values 2018, 43, 622–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movia, D.; Prina-Mello, A. Preclinical Development of Orally Inhaled Drugs (OIDs)—Are Animal Models Predictive or Shall We Move Towards In Vitro Non-Animal Models? Animals 2020, 10, 1259. [Google Scholar] [CrossRef] [PubMed]
- Togami, K.; Chono, S.; Morimoto, K. Transport characteristics of clarithromycin, azithromycin and telithromycin, antibiotics applied for treatment of respiratory infections, in Calu-3 cell monolayers as model lung epithelial cells. Pharmazie 2012, 67, 389–393. [Google Scholar] [CrossRef]
- Mathia, N.R.; Timoszyk, J.; Stetsko, P.I.; Megill, J.R.; Smith, R.L.; Wall, D.A. Permeability characteristics of calu-3 human bronchial epithelial cells: In vitro-in vivo correlation to predict lung absorption in rats. J. Drug Target. 2002, 10, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Avery, M.L.; Yazdanian, M.; Audus, K.L. Characterization of the Calu-3 cell line as a tool to screen pulmonary drug delivery. Int. J. Pharm. 2000, 208, 1–11. [Google Scholar] [CrossRef]
- Tatosian, D.A.; Shuler, M.L. A novel system for evaluation of drug mixtures for potential efficacy in treating multidrug resistant cancers. Biotechnol. Bioeng. 2009, 103, 187–198. [Google Scholar] [CrossRef]
- Balimane, P.V.; Chong, S. Cell culture-based models for intestinal permeability: A critique. Drug Discov. Today 2005, 10, 335–343. [Google Scholar] [CrossRef]
- Heikkinen, A.T.; Korjamo, T.; Lepikkö, V.; Mönkkönen, J. Effects of experimental setup on the apparent concentration dependency of active efflux transport in in vitro cell permeation experiments. Mol. Pharm. 2010, 7, 605–617. [Google Scholar] [CrossRef]
- Heikkinen, A.T.; Mönkkönen, J.; Korjamo, T. Determination of permeation resistance distribution in in vitro cell monolayer permeation experiments. Eur. J. Pharm. Sci. 2010, 40, 132–142. [Google Scholar] [CrossRef]
- Heikkinen, A.T.; Korjamo, T.; Mönkkönen, J. Modelling of Drug Disposition Kinetics in In Vitro Intestinal Absorption Cell Models. Basic Clin. Pharmacol. Toxicol. 2010, 106, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Lozoya-Agullo, I.; Araújo, F.; González-Álvarez, I.; Merino-Sanjuán, M.; González-Álvarez, M.; Bermejo, M.; Sarmento, B. Usefulness of Caco-2/HT29-MTX and Caco-2/HT29-MTX/Raji B Coculture Models To Predict Intestinal and Colonic Permeability Compared to Caco-2 Monoculture. Mol. Pharm. 2017, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, A.T.; Mönkkönen, J.; Korjamo, T. Kinetics of cellular retention during Caco-2 permeation experiments: Role of lysosomal sequestration and impact on permeability estimates. J. Pharmacol. Exp. Ther. 2009, 328, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattei, G.; Giusti, S.; Ahluwalia, A. Design criteria for generating physiologically relevant in vitro models in bioreactors. Processes 2014, 2, 548–569. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, P.; Minieri, M.; Ahluwalia, A. Engineering the Stem Cell Niche and the Differentiative Micro- and Macroenvironment: Technologies and Tools for Applying Biochemical, Physical and Structural Stimuli and Their Effects on Stem Cells. In Stem Cell Engineering; Artmann, G.M., Minger, S., Hescheler, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 41–59. ISBN 978-3-642-11864-7. [Google Scholar]
- Sbrana, T.; Ahluwalia, A. Engineering quasi-vivo® in vitro organ models. Adv. Exp. Med. Biol. 2012, 745, 138–153. [Google Scholar] [CrossRef]
- Chan, C.Y.; Huang, P.-H.; Guo, F.; Ding, X.; Kapur, V.; Mai, J.D.; Yuen, P.K.; Huang, T.J. Accelerating drug discovery via organs-on-chips. Lab Chip 2013, 13, 4697–4710. [Google Scholar] [CrossRef] [Green Version]
- Alborzinia, H.; Can, S.; Holenya, P.; Scholl, C.; Lederer, E.; Kitanovic, I.; Wölfl, S. Real-time monitoring of cisplatin-induced cell death. PLoS ONE 2011, 6, e19714. [Google Scholar] [CrossRef]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Mazzei, D.; Guzzardi, M.A.; Giusti, S.; Ahluwalia, A. A low shear stress modular bioreactor for connected cell culture under high flow rates. Biotechnol. Bioeng. 2010, 106, 127–137. [Google Scholar] [CrossRef]
- Theobald, J.; Abu El Maaty, M.A.; Kusterer, N.; Wetterauer, B.; Wink, M.; Cheng, X.; Wölfl, S. In vitro metabolic activation of vitamin D3 by using a multi-compartment microfluidic liver-kidney organ on chip platform. Sci. Rep. 2019, 9, 4616. [Google Scholar] [CrossRef]
- Theobald, J.; Ghanem, A.; Wallisch, P.; Banaeiyan, A.A.; Andrade-Navarro, M.A.; Taškova, K.; Haltmeier, M.; Kurtz, A.; Becker, H.; Reuter, S.; et al. Liver-Kidney-on-Chip To Study Toxicity of Drug Metabolites. ACS Biomater. Sci. Eng. 2018, 4, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Schimek, K.; Frentzel, S.; Luettich, K.; Bovard, D.; Rütschle, I.; Boden, L.; Rambo, F.; Erfurth, H.; Dehne, E.-M.; Winter, A.; et al. Human multi-organ chip co-culture of bronchial lung culture and liver spheroids for substance exposure studies. Sci. Rep. 2020, 10, 7865. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. A micro cell culture analog (microCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip 2009, 9, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2021, 20, 345–361. [Google Scholar] [CrossRef]
- Marshall, S.; Madabushi, R.; Manolis, E.; Krudys, K.; Staab, A.; Dykstra, K.; Visser, S.A.G. Model-Informed Drug Discovery and Development: Current Industry Good Practice and Regulatory Expectations and Future Perspectives. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 87–96. [Google Scholar] [CrossRef]
- EFPIA MID3 Workgroup; Marshall, S.F.; Burghaus, R.; Cosson, V.; Cheung, S.Y.A.; Chenel, M.; DellaPasqua, O.; Frey, N.; Hamrén, B.; Harnisch, L.; et al. Good Practices in Model-Informed Drug Discovery and Development: Practice, Application, and Documentation. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 93–122. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhu, H.; Madabushi, R.; Liu, Q.; Huang, S.; Zineh, I. Model-Informed Drug Development: Current US Regulatory Practice and Future Considerations. Clin. Pharmacol. Ther. 2019, 105, 899–911. [Google Scholar] [CrossRef]
- Sung, J.H.; Esch, M.B.; Shuler, M.L. Integration of in silico and in vitro platforms for pharmacokinetic–pharmacodynamic modeling. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1063–1081. [Google Scholar] [CrossRef]
- Sung, J.H.; Srinivasan, B.; Esch, M.B.; McLamb, W.T.; Bernabini, C.; Shuler, M.L.; Hickman, J.J. Using physiologically-based pharmacokinetic-guided “body-on-a-chip” systems to predict mammalian response to drug and chemical exposure. Exp. Biol. Med. 2014, 239, 1225–1239. [Google Scholar] [CrossRef] [Green Version]
- Edington, C.D.; Chen, W.L.K.; Geishecker, E.; Kassis, T.; Soenksen, L.R.; Bhushan, B.M.; Freake, D.; Kirschner, J.; Maass, C.; Tsamandouras, N.; et al. Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies. Sci. Rep. 2018, 8, 4530. [Google Scholar] [CrossRef] [Green Version]
- Giusti, S.; Sbrana, T.; La Marca, M.; Di Patria, V.; Martinucci, V.; Tirella, A.; Domenici, C.; Ahluwalia, A. A novel dual-flow bioreactor simulates increased fluorescein permeability in epithelial tissue barriers. Biotechnol. J. 2014, 9, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Chandorkar, P.; Posch, W.; Zaderer, V.; Blatzer, M.; Steger, M.; Ammann, C.G.; Binder, U.; Hermann, M.; Hörtnagl, P.; Lass-Flörl, C.; et al. Fast-track development of an in vitro 3D lung/immune cell model to study Aspergillus infections. Sci. Rep. 2017, 7, 11644. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.C.; Yuan, X.; Stimac, G.; Bannerman, K.; Anderson, J.; Roy, C.; Glykofrydis, F.; Yin, H.; Davies, J.A. Symmetry-breaking in branching epithelia: Cells on micro-patterns under flow challenge the hypothesis of positive feedback by a secreted autocrine inhibitor of motility. J. Anat. 2017, 230, 766–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, P.J.; Ferrick, B.; Rybakovsky, E.; Thomas, S.; Mullin, J.M. Epithelial barrier function properties of the 16HBE14o- human bronchial epithelial cell culture model. Biosci. Rep. 2020, 40, BSR20201532. [Google Scholar] [CrossRef] [PubMed]
- González-Alvarez, I.; Fernández-Teruel, C.; Garrigues, T.M.; Casabo, V.G.; Ruiz-García, A.; Bermejo, M. Kinetic modelling of passive transport and active efflux of a fluoroquinolone across Caco-2 cells using a compartmental approach in NONMEM. Xenobiotica 2005, 35, 1067–1088. [Google Scholar] [CrossRef]
- DrugBank, Entry for Caffeine. Available online: https://go.drugbank.com/drugs/DB00201 (accessed on 7 October 2021).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- DrugBank, Entry for Diclofenac. Available online: https://go.drugbank.com/drugs/DB00586 (accessed on 7 October 2021).
- Prot, J.M.; Aninat, C.; Griscom, L.; Razan, F.; Brochot, C.; Guillouzo, C.G.; Legallais, C.; Corlu, A.; Leclerc, E. Improvement of HepG2/C3a cell functions in a microfluidic biochip. Biotechnol. Bioeng. 2011, 108, 1704–1715. [Google Scholar] [CrossRef]
- Baudoin, R.; Griscom, L.; Prot, J.M.; Legallais, C.; Leclerc, E. Behavior of HepG2/C3A cell cultures in a microfluidic bioreactor. Biochem. Eng. J. 2011, 53, 172–181. [Google Scholar] [CrossRef]
- Snouber, L.C.; Letourneur, F.; Chafey, P.; Broussard, C.; Monge, M.; Legallais, C.; Leclerc, E. Analysis of transcriptomic and proteomic profiles demonstrates improved Madin-Darby canine kidney cell function in a renal microfluidic biochip. Biotechnol. Prog. 2012, 28, 474–484. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, L.; Chow, E.C.Y.; Lin, G.; Zuo, Z.; Pang, K.S. A catenary model to study transport and conjugation of baicalein, a bioactive flavonoid, in the Caco-2 cell monolayer: Demonstration of substrate inhibition. J. Pharmacol. Exp. Ther. 2008, 326, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Kalvass, J.C.; Pollack, G.M. Kinetic considerations for the quantitative assessment of efflux activity and inhibition: Implications for understanding and predicting the effects of efflux inhibition. Pharm. Res. 2007, 24, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirbagheri, M.; Adibnia, V.; Hughes, B.R.; Waldman, S.D.; Banquy, X.; Hwang, D.K. Advanced cell culture platforms: A growing quest for emulating natural tissues. Mater. Horizons 2019, 6, 45–71. [Google Scholar] [CrossRef]
- Bhowmick, R.; Gappa-Fahlenkamp, H. Cells and Culture Systems Used to Model the Small Airway Epithelium. Lung 2016, 194, 419–428. [Google Scholar] [CrossRef]
- van Midwoud, P.M.; Verpoorte, E.; Groothuis, G.M.M. Microfluidic devices for in vitro studies on liver drug metabolism and toxicity. Integr. Biol. 2011, 3, 509–521. [Google Scholar] [CrossRef]
- Pedersen, J.M.; Shim, Y.-S.; Hans, V.; Phillips, M.B.; Macdonald, J.M.; Walker, G.; Andersen, M.E.; Clewell, H.J.; Yoon, M. Fluid Dynamic Modeling to Support the Development of Flow-Based Hepatocyte Culture Systems for Metabolism Studies. Front. Bioeng. Biotechnol. 2016, 4, 72. [Google Scholar] [CrossRef] [Green Version]
- Cozens, A.L.; Yezzi, M.J.; Kunzelmann, K.; Ohrui, T.; Chin, L.; Eng, K.; Finkbeiner, W.E.; Widdicombe, J.H.; Gruenert, D.C. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994, 10, 38–47. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [Green Version]
- Elberskirch, L.; Knoll, T.; Moosmann, A.; Wilhelm, N.; von Briesen, H.; Wagner, S. A novel microfluidic mucus-chip for studying the permeation of compounds over the mucus barrier. J. Drug Deliv. Sci. Technol. 2019, 54, 101248. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Elmokadem, A.; Riggs, M.M.; Baron, K.T. Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling With mrgsolve: A Hands-On Tutorial. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.G. The NLopt Nonlinear-Optimization Package. Available online: https://github.com/stevengj/nlopt (accessed on 14 October 2021).
- Nocedal, J. Updating Quasi-Newton Matrices with Limited Storage. Math. Comput. 1980, 35, 773. [Google Scholar] [CrossRef]
- Liu, D.C.; Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | RSE (%) |
|---|---|---|
| Control experiments (without lung cell barrier) | ||
| P12, caffeine (cm/min) | 3.27 × 10−3 | 0.1 |
| P12, diclofenac (cm/min) | 2.51 × 10−3 | 0 |
| (µM) | 21.4 | 35 |
| 0 (fixed) | - | |
| Cell experiments (with lung cell barrier) | ||
| P12, caffeine (cm/min) | 2.62 × 10−3 | 0.7 |
| P12, diclofenac (cm/min) | 1.38 × 10−3 | 0.9 |
| (µM) | 2.38 | 30 |
| 0.00596 | 23 |
| Time (min) | Solvent A (%) | Solvent B (%) | Solvent C (%) |
|---|---|---|---|
| 0 | 76.5 | 8.5 | 15 |
| 0.5 | 76.5 | 8.5 | 15 |
| 2.5 | 42 | 38 | 20 |
| 4 | 0 | 57.5 | 42.5 |
| 10 | 0 | 57.5 | 42.5 |
| 14 | 76.5 | 8.5 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovar, L.; Wien, L.; Selzer, D.; Kohl, Y.; Bals, R.; Lehr, T. In Vitro–In Silico Modeling of Caffeine and Diclofenac Permeation in Static and Fluidic Systems with a 16HBE Lung Cell Barrier. Pharmaceuticals 2022, 15, 250. https://doi.org/10.3390/ph15020250
Kovar L, Wien L, Selzer D, Kohl Y, Bals R, Lehr T. In Vitro–In Silico Modeling of Caffeine and Diclofenac Permeation in Static and Fluidic Systems with a 16HBE Lung Cell Barrier. Pharmaceuticals. 2022; 15(2):250. https://doi.org/10.3390/ph15020250
Chicago/Turabian StyleKovar, Lukas, Lena Wien, Dominik Selzer, Yvonne Kohl, Robert Bals, and Thorsten Lehr. 2022. "In Vitro–In Silico Modeling of Caffeine and Diclofenac Permeation in Static and Fluidic Systems with a 16HBE Lung Cell Barrier" Pharmaceuticals 15, no. 2: 250. https://doi.org/10.3390/ph15020250
APA StyleKovar, L., Wien, L., Selzer, D., Kohl, Y., Bals, R., & Lehr, T. (2022). In Vitro–In Silico Modeling of Caffeine and Diclofenac Permeation in Static and Fluidic Systems with a 16HBE Lung Cell Barrier. Pharmaceuticals, 15(2), 250. https://doi.org/10.3390/ph15020250