
Phytol and Heptacosane Are Possible Tools to Overcome Multidrug Resistance in an In Vitro Model of Acute Myeloid Leukemia



 , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. In Vitro Cytotoxicity Effects of Phytol and Heptacosane
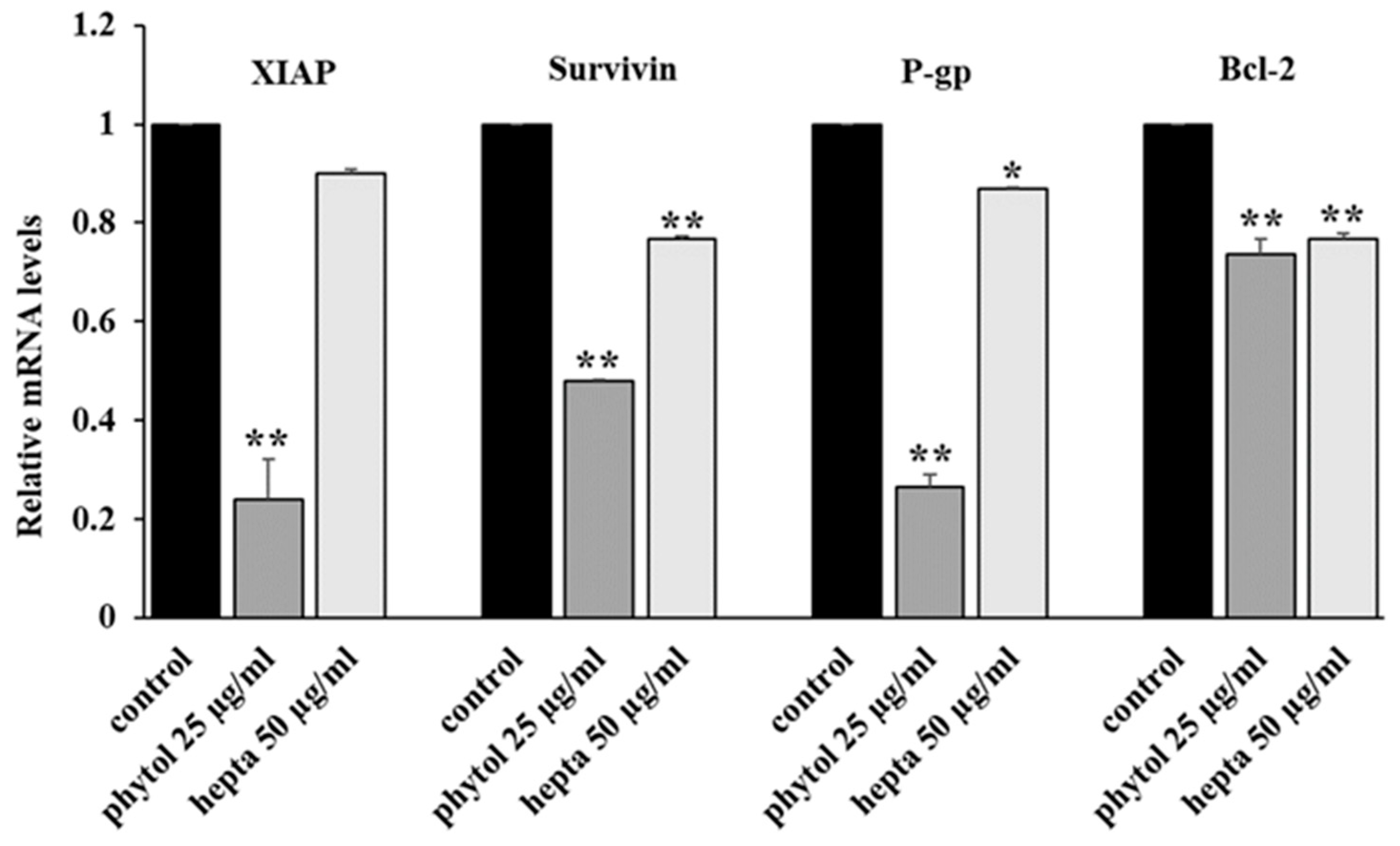
2.2. Effects on the Activation of the NF-κB Signaling Pathway
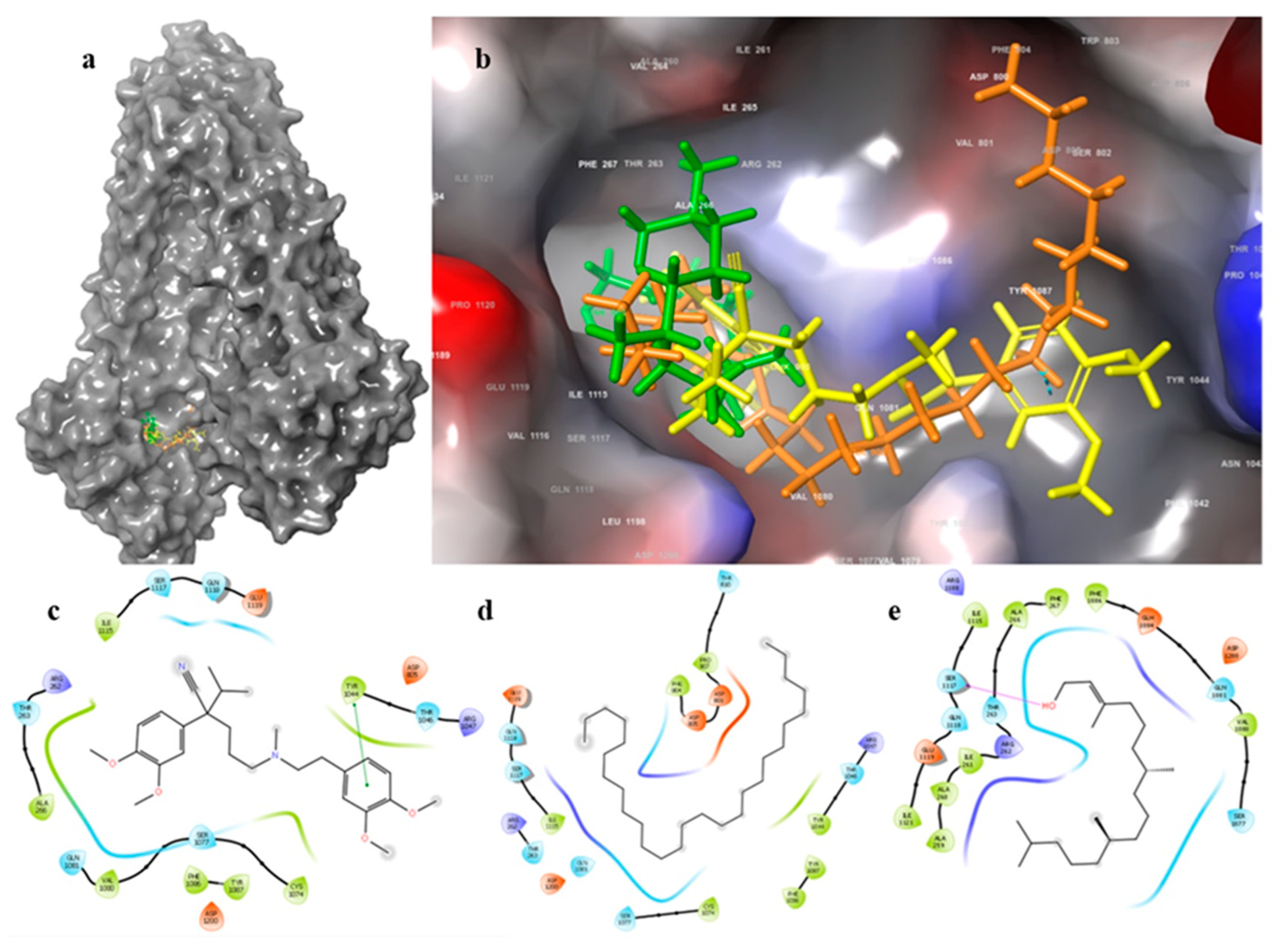
2.3. Binding Site Detection
2.4. Docking and Binding Free Energy Calculation
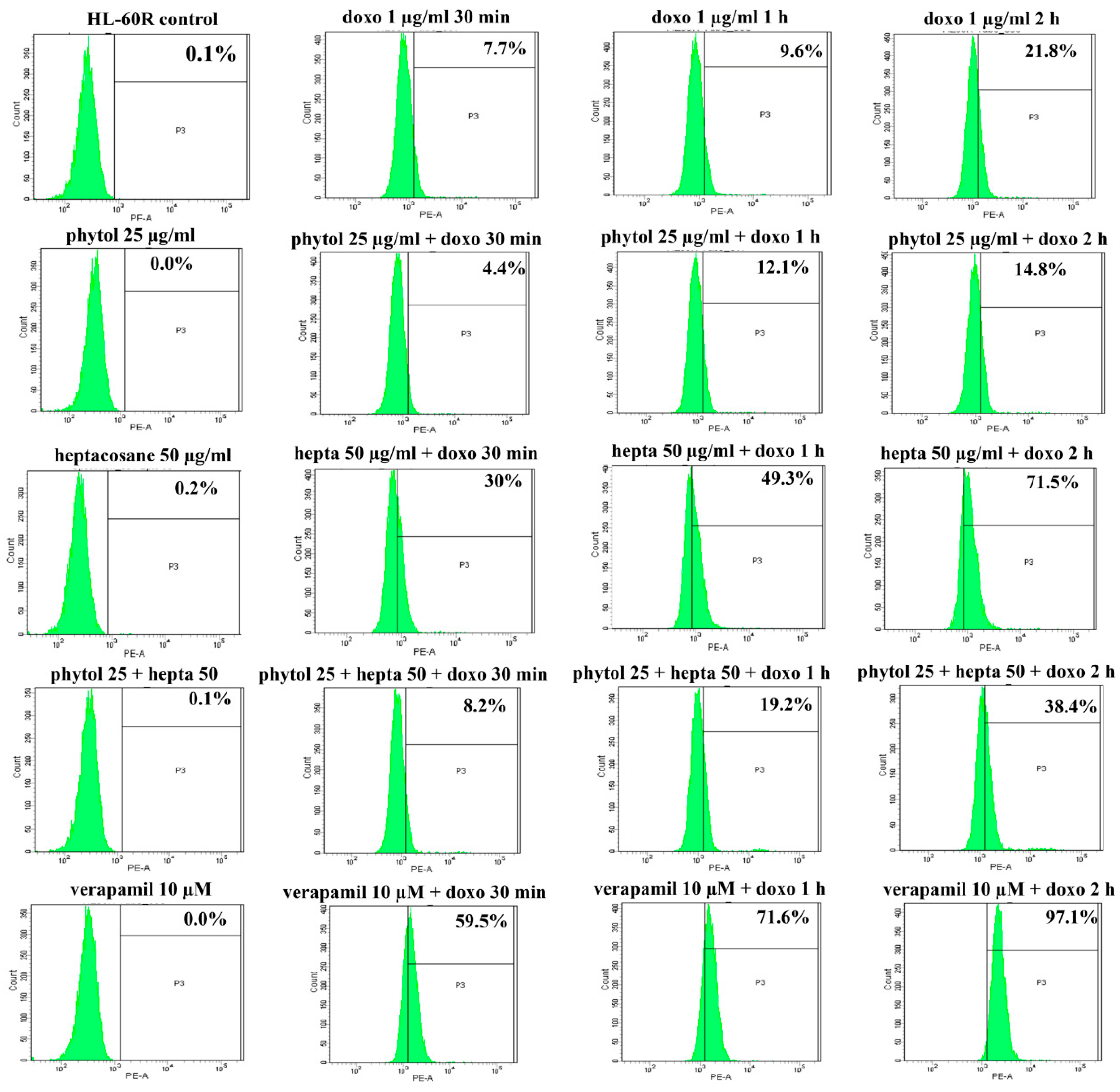
2.5. Effects of Phytol and Heptacosane on Intracellular Accumulation of Doxorubicin in the HL-60R Cell Line
2.6. Cytotoxic Effects of Heptacosane in Combination with Doxorubicin
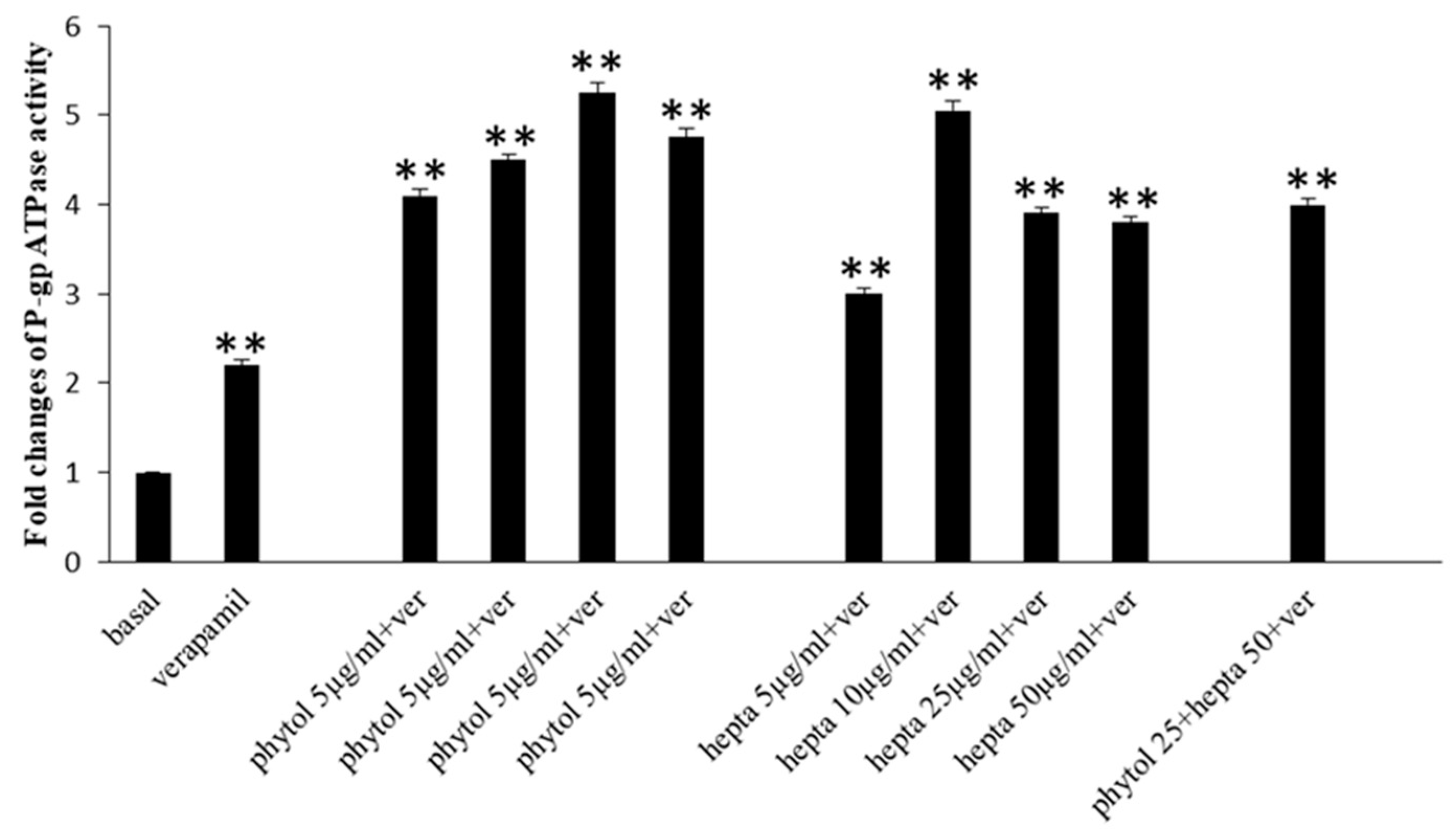
2.7. Effects of Phytol and Heptacosane on P-gp Activity
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Growth Assays
4.3. NF-κB Activation
4.4. Extraction of Cellular RNA and Reverse Transcription-Quantitative PCR (RT-qPCR)
4.5. Western Blotting Analysis
4.6. Active Site Prediction, Ligand Preparation, Docking, and Free Energy Calculation
4.7. Determination of Doxorubicin Accumulation
4.8. P-gp ATPase Activity Determination
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goebel, J.; Chmielewski, J.; Hrycyna, C.A. The roles of the human ATP-binding cassette transporters P-glycoprotein and ABCG2 in multidrug resistance in cancer and at endogenous sites: Future opportunities for structure-based drug design of inhibitors. Cancer Drug Resist. 2021, 4, 784–804. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V.A. Surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Riordan, J.R.; Deuchars, K.; Kartner, N.; Alon, N.; Trent, J.; Ling, V. Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 1985, 316, 817–819. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Seelig, A.; Landwojtowicz, E. Structure-activity relationship of P-glycoprotein substrates and modifiers. Eur. J. Pharm. Sci. 2000, 2, 31–40. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Mollazadeha, S.; Sahebkarb, A.; Hadizadeha, F.; Behravanb, J.; Arabzadeh, S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; De Feo, V.; Zia-Ul-Haq, M. Natural Products as Alternative Choices for P-Glycoprotein (P-gp) Inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef]
- Kumar, A.; Jaita, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef]
- Wang, C.C.; Wang, J.Y.; Lee, T.E.; Chenge, Y.Y.; Morris-Natschkee, S.L.; Leeeg, K.H.; Hungah, C.C. Tenulin and isotenulin inhibit P-glycoprotein function and overcome multidrug resistance in cancer cells. Phytomedicine 2019, 53, 252–262. [Google Scholar]
- Choi, S.U.; Park, S.H.; Kim, K.H.; Choi, E.J.; Kim, S.; Park, W.K.; Zhang, Y.H.; Kim, H.S.; Jung, N.P.; Lee, C.O. The bisbenzylisoquinoline alkaloids, tetrandine and fangchinoline, enhance the cytotoxicity of multidrug resistance-related drugs via modulation of P-glycoprotein. Anti-Cancer Drugs 1998, 9, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Yeo, G.S.; Lim, Y.S.; Kang, C.D.; Kim, C.M.; Chung, B.S. Suppression of multidrug resistance via inhibition of heat shock factor by quercetin in MDR cells. Exp. Mol. Med. 1998, 30, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Zhang, W.; Gupta, P.; Lei, Z.-N.; Wang, J.-Q.; Cai, C.-Y.; Vera, A.A.D.; Zhang, L.; Chen, Z.-S.; Yang, D.-H. Tetrandrine Interaction with ABCB1 Reverses Multidrug Resistance in Cancer Cells Through Competition with Anti-Cancer Drugs Followed by Downregulation of ABCB1 Expression. Molecules 2019, 24, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notarbartolo, M.; Poma, P.; Perri, D.; Dusonchet, L.; Cervello, M.; D’Alessandro, N. Antitumor effects of curcumin, alone or in combination with cisplatin or doxorubicin, on human hepatic cancer cells. Analysis of their possible relationship to changes in NF-κB activation levels and in IAP gene expression. Cancer Lett. 2005, 224, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Barthomeuf, C.; Demeule, M.; Grassi, J.; Saidkhodjaev, A.; Beliveau, R. Conferone from Ferula schtschurowskiana enhances vinblastine cytotoxicity in MDCK-MDR1 cells by competitively inhibiting P-glycoprotein transport. Planta Med. 2006, 72, 634–639. [Google Scholar] [CrossRef]
- Peterson, B.G.; Tan, K.W.; Osa-Andrews, B.; Iram, S.H. High-content screening of clinically tested anticancer drugs identifies novel inhibitors of human MRP1 (ABCC1). Pharmacol Res. 2017, 119, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Simoni, D.; Rizzi, M.; Rondanin, R.; Baruchello, R.; Marchetti, P.; Invidiata, F.P.; Labbozzetta, M.; Poma, P.; Carina, V.; Notarbartolo, M.; et al. Antitumor effects of curcumin and structurally beta-diketone modified analogs on multidrug resistant cancer cells. Bioorg. Med. Chem. Lett. 2008, 18, 845–849. [Google Scholar] [CrossRef]
- Li, Y.; Revalde, J.L.; Reid, G.; Paxton, J.W. Modulatory effects of curcumin on multi-drug resistance-associated protein 5 in pancreatic cancer cells. Cancer Chemother. Pharm. 2011, 68, 603–610. [Google Scholar] [CrossRef]
- Wesołowska, O.; Wiśniewski, J.; Sroda, K.; Krawczenko, A.; Bielawska-Pohl, A.; Paprocka, M.; Duś, D.; Michalak, K. 8-Prenylnaringenin is an inhibitor of multidrug resistance-associated transporters, P-glycoprotein and MRP1. Eur. J. Pharmacol. 2010, 644, 32–40. [Google Scholar] [CrossRef]
- Bi, X.; Yuan, Z.; Qu, B.; Zhou, H.; Liu, Z.; Xie, Y. Piperine enhances the bioavailability of silybin via inhibition of efflux transporters BCRP and MRP2. Phytomedicine 2019, 54, 98–108. [Google Scholar] [CrossRef]
- Fontana, G.; Bruno, M.; Notarbartolo, M.; Labbozzetta, M.; Poma PSpinella, A.; Rosselli, S. Cytotoxicity of oleanolic and ursolic acid derivatives toward hepatocellular carcinoma and evaluation of NF-κB involvement. Bioorg. Chem. 2019, 90, 103054. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Li, L.; Kong, R.; Pan, S.; Ji, L.; Liu, H.; Chen, H.; Sun, B. Hyperoside induces apoptosis and inhibits growth in pancreatic cancer via Bcl-2 family and NF-kappaB signaling pathway both in vitro and in vivo. Tumour Biol. 2016, 37, 7345–7355. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, G.; Gunaseelan, S.; Prasad, N.R. Ferulic acid reverses P-glycoprotein-mediated multidrug resistance via inhibition of PI3K/Akt/NF-κB signaling pathway. J. Nutr. Biochem. 2019, 63, 62–71. [Google Scholar] [CrossRef]
- Shaffer, B.C.; Gillet, J.P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Update 2012, 15, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. 2020, 2, 3233. [Google Scholar] [CrossRef] [PubMed]
- Winer, E.S.; Stone, R.; Novel, M. Therapy in Acute myeloid leukemia (AML): Moving toward targeted approaches. Ther. Adv. Hematol. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Kadia, T.M.; DiNardo, C.D.; Welch, M.A.; Ravandi, F. Acute myeloid leukemia: Treatment and research outlook for 2021 and the MD Anderson approach. Cancer 2021, 127, 1186–1207. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Flore, A.M.; Büsselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef]
- Kumar, S.; Fairmichael, C.; Longley, D.B.; Turkington, R.C. The multiple roles of the IAP super-family in cancer. Pharmacol. Ther. 2020, 214, 107610. [Google Scholar] [CrossRef]
- Poma, P.; Labbozzetta, M.; Ramarosandratana, A.V.; Rosselli, S.; Tutone, M.; Sajeva, M.; Notarbartolo, M. In Vitro Modulation of P-Glycoprotein Activity by Euphorbia intisy Essential Oil on Acute Myeloid Leukemia Cell Line HL-60R. Pharmaceuticals 2021, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Heptacosane (accessed on 22 December 2021).
- Islam, M.T.; Ali, E.S.; Uddin, S.J.; Shaw, S.; Islam, M.A.; Ahmed, M.I.; Chandra Shill, M.; Karmakar, U.K.; Yarla, N.S.; Khan, I.N.; et al. Phytol: A review of biomedical activities. Food Chem. Toxicol. 2018, 121, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Info. Model. 2009, 49, 377. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Saeed, M.E.; Valoti, M.; Frosini, M.; Sgaragli, G.; Efferth, T. Interactions of human P-glycoprotein transport substrates and inhibitors at the drug binding domain: Functional and molecular docking analyses. Biochem. Pharmacol. 2016, 104, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Globisch, C.; Pajeva, I.K.; Wiese, M. Identification of putative binding sites of P-glycoprotein based on its homology model. Chem. Med. Chem. 2008, 3, 280–295. [Google Scholar] [CrossRef]
- Maki, N.; Hafkemeyer, P.; Dey, S. Allosteric modulation of human P-glycoprotein. Inhibition of transport by preventing substrate translocation and dissociation. J. Biol. Chem. 2003, 278, 18132–18139. [Google Scholar] [CrossRef] [Green Version]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981, 41, 1967–1972. [Google Scholar]
- Pang, X.; Wang, L.; Kang, D.; Zhao, Y.; Wu, S.; Liu, A.L.; Du, G.H. Effects of P-glycoprotein on the transport of DL0410, a potential multifunctional anti-Alzheimer agent. Molecules 2017, 22, 1246. [Google Scholar] [CrossRef] [Green Version]
- Zeino, M.; Saeed, M.E.; Kadioglu, O.; Efferth, T. The ability of molecular docking to unravel the controversy and challenges related to P-glycoprotein—A well-known, yet poorly understood drug transporter. Investig. New Drugs 2014, 32, 618–625. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Li-Blatter, X.; Beck, A.; Seelig, A. P-Glycoprotein-ATPase modulation: The molecular mechanisms. Biophys. J. 2012, 102, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.I.; Chen, M.H.; Huang, C.Y.F.; Chang, M.H. Clinical perspective of FDA approved drugs with P-glycoprotein inhibition activities for potential cancer therapeutics. Front. Oncol. 2020, 10, 2336. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Kanimozhi, G.; Pradhapsingh, B.; Khan, H.A.; Alhomida, A.S.; Ekhzaimy, A.; Brindha, G.R.; Rajendra Prasad, N. Phytochemicals reverse P-glycoprotein mediated multidrug resistance via signal transduction pathways. Biomed. Pharmacother. 2021, 139, 2021. [Google Scholar] [CrossRef] [PubMed]
- Poma, P.; Labbozzetta, M.; Notarbartolo, M.; Bruno, M.; Maggio, A.; Rosselli, S.; Sajeva, M.; Zito, P. Chemical composition, in vitro antitumor and pro-oxidant activities of Glandora rosmarinifolia (Boraginaceae) essential oil. PLoS ONE 2018, 13, e0196947. [Google Scholar] [CrossRef] [PubMed]
- Zito, P.; Labbozzetta, M.; Notarbartolo, M.; Sajeva, M.; Poma, P. Essential oil of Cyphostemma juttae (Vitaceae): Chemical composition and antitumor mechanism in triple negative breast cancer cells. PLoS ONE 2019, 14, e0214594. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef]
- Poma, P.; Labbozzetta, M.; Zito, P.; Alduina, R.; Ramarosandratana, A.V.; Bruno, M.; Rosselli, S.; Sajeva, M.; Notarbartolo, M. Essential Oil Composition of Alluaudia procera and in Vitro Biological Activity on Two Drug-Resistant Models. Molecules 2019, 24, 2871. [Google Scholar] [CrossRef] [Green Version]
- Marques, S.M.; Šupolíková, L.; Molčanová, L.; Šmejkal, K.; Bednar, D.; Slaninová, I. Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance. Biomedicines 2021, 9, 357. [Google Scholar] [CrossRef]
- Li, J.; Duan, B.; Guo, Y.; Zhou, R.; Sun, J.; Bie, B.; Yang, S.; Huang, C.; Yang, J.; Li, Z. Baicalein sensitizes hepatocellular carcinoma cells to 5-FU and Epirubicin by activating apoptosis and ameliorating P-glycoprotein activity. Biomed. Pharmacother. 2018, 98, 806–812. [Google Scholar] [CrossRef]
- Di Sotto, A.; Irannejad, H.; Eufemi, M.; Mancinelli, R.; Abete, L.; Mammola, C.L.; Altieri, F.; Mazzanti, G.; Di Giacomo, S. Potentiation of Low-Dose Doxorubicin Cytotoxicity by Affecting P-Glycoprotein through Caryophyllane Sesquiterpenes in HepG2 Cells: An in Vitro and in Silico Study. Int. J. Mol. Sci. 2020, 21, 633. [Google Scholar] [CrossRef] [Green Version]
- Lopez, D.; Martinez-Luis, S. Marine Natural Products with P-Glycoprotein Inhibitor Properties. Mar. Drugs 2014, 12, 525–546. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Lu, Q.; Hu, X. Down-regulation of P-glycoprotein expression in MDR breast cancer cell MCF-7/ADR by honokiol. Cancer Lett. 2006, 243, 274–280. [Google Scholar] [CrossRef]
- Tamm, I.; Richter, S.; Scholz, F.; Schmelz, K.; Oltersdorf, D.; Karawajew, L.; Schoch, C.; Haferlach, T.; Ludwig, W.D.; Wuchter, C. XIAP expression correlates with monocytic differentiation in adult de novo AML: Impact on prognosis. Hematol. J. 2004, 5, 489–495. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Abe, S.; Kurata, M.; Hasegawa, M.; Yamamoto, K.; Inoue, M.; Takemura, T.; Suzuki, K.; Kitagawa, M. IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes. Am. J. Hematol. 2006, 81, 824–831. [Google Scholar] [CrossRef]
- Chen, K.G.; Sale, S.; Tan, T.; Ermoian, R.P.; Sikic, B.I. CCAAT/enhancer-binding protein beta (nuclear factor for interleukin 6) transactivates the human MDR1 gene by interaction with an inverted CCAAT box in human cancer cells. Mol. Pharmacol. 2004, 65, 906–916. [Google Scholar] [CrossRef]
- Maestro v 2018-4 Schrödinger; LLC: New York, NY, USA, 2018.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J.H. Hblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | HL-60 | HL-60R | hTERT RPE-1 | 1-7HB2 |
|---|---|---|---|---|
| IC50 | IC50 | IC50 | IC50 | |
| phytol | >100 µg/mL | >100 µg/mL | >100 µg/mL | >100 µg/mL |
| heptacosane | >100 µg/mL | >100 µg/mL | >100 µg/mL | >100 µg/mL |
| Compounds | Type of Interaction | Key Residue | Binding Free Energy (Kcal/mol) | |
|---|---|---|---|---|
| DBP | doxorubicin | H-Bond H-Bond Pi-stacking | Gln725 Gln990 Phe732 | −52.1 |
| verapamil | H-Bond Hydrophobic | Gln725 Phe72, Tyr307, Phe336, Phe728, Ala729, Phe732, Phe759, Cys956, Tyr953, Phe957, Val982, Phe978, Phe983 | −82.7 | |
| heptacosane | Hydrophobic | Phe72, Phe335, Phe336, Leu339, Ile340, Phe343, Phe732, Leu861, Cys956, Tyr953, Ile868, Phe957, Phe978, Val982 | −114.2 | |
| phytol | 2 H-Bonds Hydrophobic | Gln725 Tyr310, Phe335, Phe336, Ile340, Phe343, Phe728, Ala729, Phe732, Phe759, Phe983 | −75.8 | |
| NBP | verapamil | Pi-stacking | Tyr1044 | −42.3 |
| heptacosane | Hydrophobic | Asp 800, Val801, Ser802, Phe804, Asp805 | −30.2 | |
| phytol | H-bond | Ser1117 | −29.7 |
| Fluorescence % | ||
|---|---|---|
| Treatment | HL-60 | HL-60R |
| Control | 0.05 ± 0.03 | 0.05 ± 0.03 |
| doxo 1 µg/mL 30 min | 98.4 ± 0.42 * | 7.85 ± 0.10 * |
| doxo 1 µg/mL 1 h | 99.9 ± 0.07 * | 9.80 ± 0.14 * |
| doxo 1 µg/mL 2 h | 100 ± 0.0 * | 21.8 ± 0.14 * |
| phytol 25 µg/mL | 0.05 ± 0.03 | 0.05 ± 0.03 |
| heptacosane 50 µg/mL | 0.15 ± 0.03 | 0.25 ± 0.03 |
| phytol 25 µg/mL + heptacosane 50 µg/mL | 0.15 ± 0.03 | 0.05 ± 0.03 |
| verapamil 10 µM | 0.05 ± 0.02 | 0.05 ± 0.03 |
| phytol 25 µg/mL + doxo 1 µg/mL 30 min | 99.7 ± 0.12 * | 4.70 ± 0.20 * |
| phytol 25 µg/mL + doxo 1 µg/mL 1 h | 99.9 ± 0.03 * | 10.8 ± 0.90 * |
| phytol 25 µg/mL + doxo 1 µg/mL 2 h | 100 ± 0.0 * | 17.1 ± 1.80 * |
| heptacosane 50 µg/mL + doxo 1 µg/mL 30 min | 98.9 ± 0.67 * | 31.0 ± 0.70 *,a |
| heptacosane 50 µg/mL + doxo 1 µg/mL L h | 100 ± 0.0 * | 49.6 ± 0.25 *,a |
| heptacosane 50 µg/mL + doxo 1 µg/mL 2 h | 100 ± 0.0 * | 71.7 ± 0.12 *,a |
| phytol 25 µg/mL + hepta 50 µg/mL + doxo 1 µg/mL 30 min | 99.9 ± 0.03 * | 8.35 ± 0.10 * |
| phytol 25 µg/mL + hepta 50 µg/mL + doxo 1 µg/mL 1 h | 100 ± 0.0 * | 19.6 ± 0.30 *,a |
| phytol 25 µg/mL + hepta 50 µg/mL + doxo 1 µg/mL 2 h | 100 ± 0.0 * | 39.2 ± 0.57 *,a |
| verapamil 10 µM + doxo 1 µg/mL 30 min | 99.5 ± 0.35 * | 58.7 ± 0.53 *,a |
| verapamil 10 µM + doxo 1 µg/mL 1 h | 99.0 ± 0.70 * | 70.8 ± 0.57 *,a |
| verapamil 10 µM + doxo 1 µg/mL 2 h | 100 ± 0.0 * | 98.0 ± 0.70 *,a |
| Treatments | Cell Growth Inhibition, % | Expected, % |
|---|---|---|
| heptacosane 20 µg/mL | 9.0 ± 0.7 | |
| heptacosane 50 µg/mL | 17.0 ± 0.7 * | |
| doxo 0.25 µg/mL | 10.5 ± 3.9 | |
| doxo 0.5 µg/mL | 17.5 ± 1.8 * | |
| hepta 20 µg/mL + doxo 0.25 µg/mL | 22.5 ± 5.3 ** | 18.7 ± 3.0 * |
| hepta 20 µg/mL + doxo 0.5 µg/mL | 48.5 ± 0.3 ** | 27.0 ± 1.0 **,b |
| hepta 50 µg/mL + doxo 0.25 µg/mL | 50.5 ± 2.5 ** | 25.5 ± 3.9 ** |
| hepta 50 µg/mL + doxo 0.5 µg/mL | 65.5 ± 0.3 ** | 31.5 ± 2.0 **,a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labbozzetta, M.; Poma, P.; Tutone, M.; McCubrey, J.A.; Sajeva, M.; Notarbartolo, M. Phytol and Heptacosane Are Possible Tools to Overcome Multidrug Resistance in an In Vitro Model of Acute Myeloid Leukemia. Pharmaceuticals 2022, 15, 356. https://doi.org/10.3390/ph15030356
Labbozzetta M, Poma P, Tutone M, McCubrey JA, Sajeva M, Notarbartolo M. Phytol and Heptacosane Are Possible Tools to Overcome Multidrug Resistance in an In Vitro Model of Acute Myeloid Leukemia. Pharmaceuticals. 2022; 15(3):356. https://doi.org/10.3390/ph15030356
Chicago/Turabian StyleLabbozzetta, Manuela, Paola Poma, Marco Tutone, James A. McCubrey, Maurizio Sajeva, and Monica Notarbartolo. 2022. "Phytol and Heptacosane Are Possible Tools to Overcome Multidrug Resistance in an In Vitro Model of Acute Myeloid Leukemia" Pharmaceuticals 15, no. 3: 356. https://doi.org/10.3390/ph15030356
APA StyleLabbozzetta, M., Poma, P., Tutone, M., McCubrey, J. A., Sajeva, M., & Notarbartolo, M. (2022). Phytol and Heptacosane Are Possible Tools to Overcome Multidrug Resistance in an In Vitro Model of Acute Myeloid Leukemia. Pharmaceuticals, 15(3), 356. https://doi.org/10.3390/ph15030356