1. Introduction
Traditional systemic chemotherapeutics non-specifically kill any proliferating cells, including those that are non-cancerous. Gemcitabine is a common and well-studied chemotherapeutic used as a monotherapy or in combination with other agents to treat many cancers, including pancreatic, bladder, breast, non-small cell lung, and ovarian cancers [
1]. Like many non-targeted chemotherapeutic agents, gemcitabine has dose-limiting side effects related to its off-target activity, including gastrointestinal toxicity, myelosuppression, rash, dyspnea, and fever [
1]. Ligand-targeted therapeutics can minimize these side effects by facilitating the targeted delivery of cytotoxic agents to cancer cells, though highly specific targeting ligands with a robust safety profile are required.
Oligonucleotide aptamers are promising candidates for targeted chemotherapy delivery systems. Generated through a recursive selection process termed the systematic evolution of ligands by exponential enrichment (SELEX), aptamers are short segments of DNA or RNA that form unique 3D structures and bind targets with an antibody-like specificity and affinity [
2]. They are chemically synthesized, readily modifiable using simple chemistry, and inherently non-immunogenic [
3]. The flexibility of aptamer platforms and the relative ease of aptamer selection make them a promising tool for cancer diagnosis and treatment. Due to the fact that oligonucleotide aptamers are smaller than analogous antibodies, they penetrate solid tumors relatively rapidly and effectively [
4]. Numerous aptamer-based therapeutics have been developed, including growth-factor inhibitors [
5], aptamer–drug conjugates [
6], aptamer-labeled nanoparticles [
7,
8,
9], and aptamer-labeled liposomes [
10,
11].
Despite many developments in aptamer therapeutic platforms, only a few are approved for clinical use [
12]. One barrier hindering the translation of aptamer therapeutics from the lab to the clinic is unpredictable in vivo behavior. This unpredictability often stems from the specific techniques used for aptamer drug loading. DNA-intercalating therapeutics suffer from instability and poor tunability due to GC-region scarcity. Nanoparticles functionalized via aptamer labeling may have nephrotoxicity or poor tissue penetrance. Directly conjugated aptamer–drug pairs are often limited by a 1:1 aptamer–drug stoichiometry. Thus, more reliable aptamer–drug loading techniques are needed.
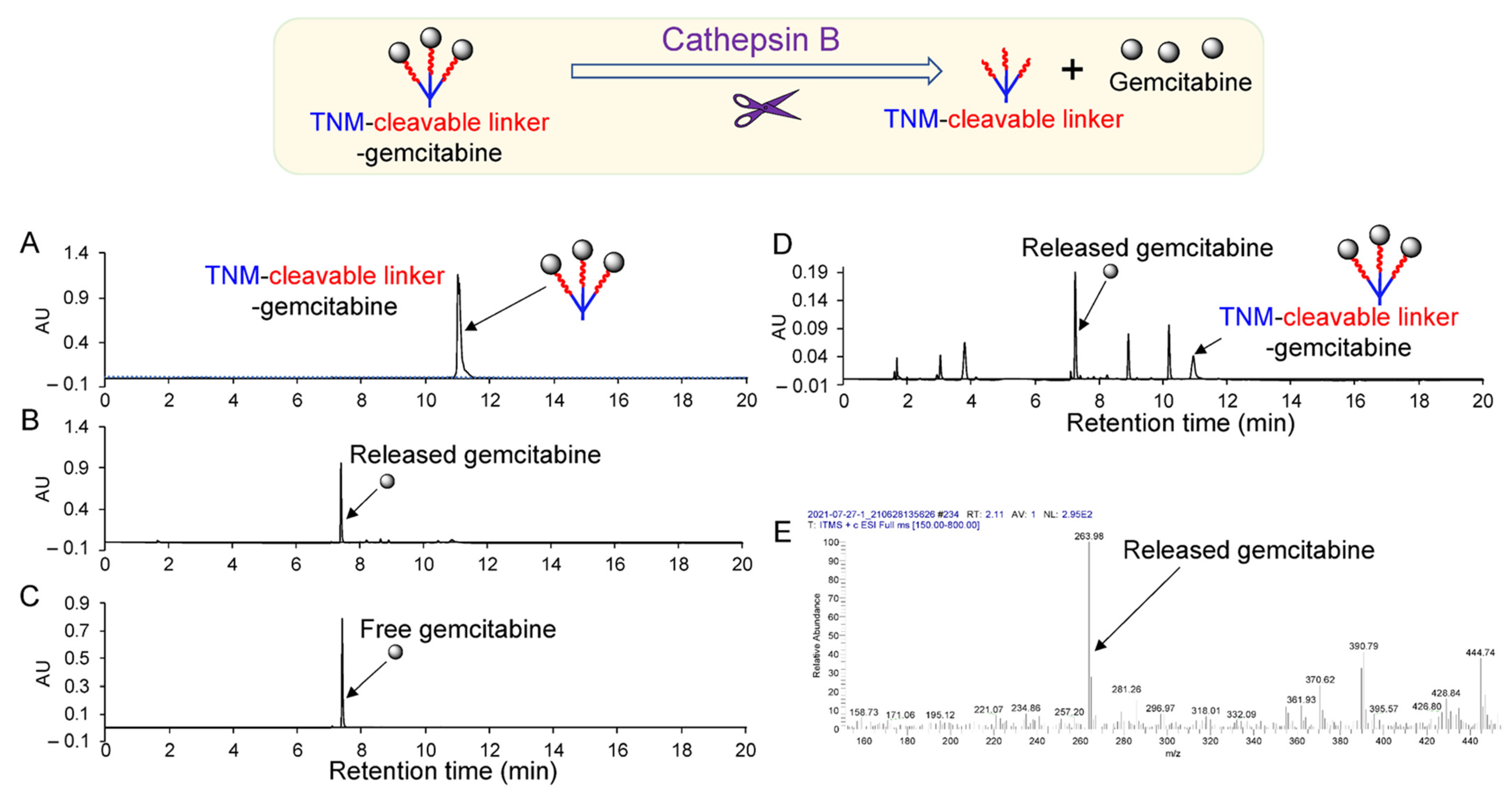
To address these shortcomings, we present a unique aptamer–drug conjugate that incorporates a trifurcated Newkome-type monomer (TNM) to improve drug loading capacity with enzymatically cleavable peptide linkers for endosomal drug release and an ssDNA aptamer to target triple-negative-breast-cancer (TNBC) cells. Aptamer-mediated cell binding allows the intracellular delivery and subsequent release of multiple molecules of gemcitabine (
Figure 1). The liberation of gemcitabine, in turn, provokes tumor-cell death without off-target side effects.
3. Discussion
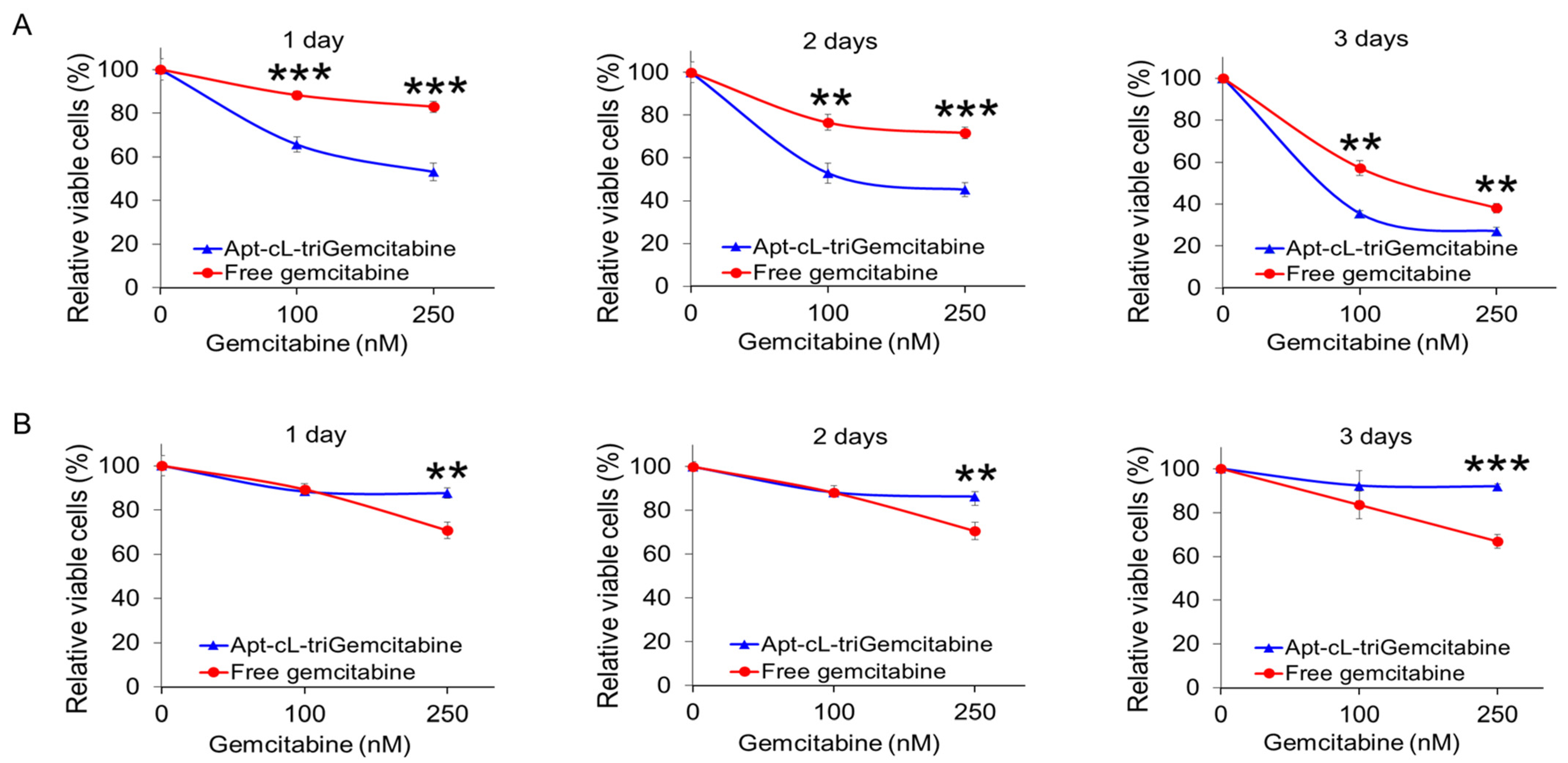
Here, we report a novel aptamer–drug conjugate that potentiates the therapeutic efficacy of gemcitabine in TNBC cells and reduces off-target drug cytotoxicity in non-TNBC cells. Gemcitabine, a deoxycytidine analog that disrupts dividing cells [
13], is a well-studied chemotherapeutic used in the treatment of a variety of cancers. Its mechanism of action allows it to preferentially kill proliferating cell populations, including cancer. Like many non-specific chemotherapeutic agents, however, off-target cytotoxicity can result in deleterious and dose-limiting side effects. Although gemcitabine is regularly used to treat cancers, chemoresistance is common. Chemoresistance can be attributed to the altered expression of nucleoside transporter-1 (hENT1), a key receptor involved in the internalization of free gemcitabine by cancer cells [
14]. Various drug delivery techniques have been employed to overcome gemcitabine resistance, such as incorporating gemcitabine into liposomes or nanoparticles [
15], conjugating gemcitabine to peptide carriers [
16], and incorporating gemcitabine into a G-quadruplex aptamer. Park et al. showed that the gemcitabine-modified aptamer, APTA-12, significantly improved the anti-tumor activity of gemcitabine in nucleolin-overexpressing pancreatic cancer cells [
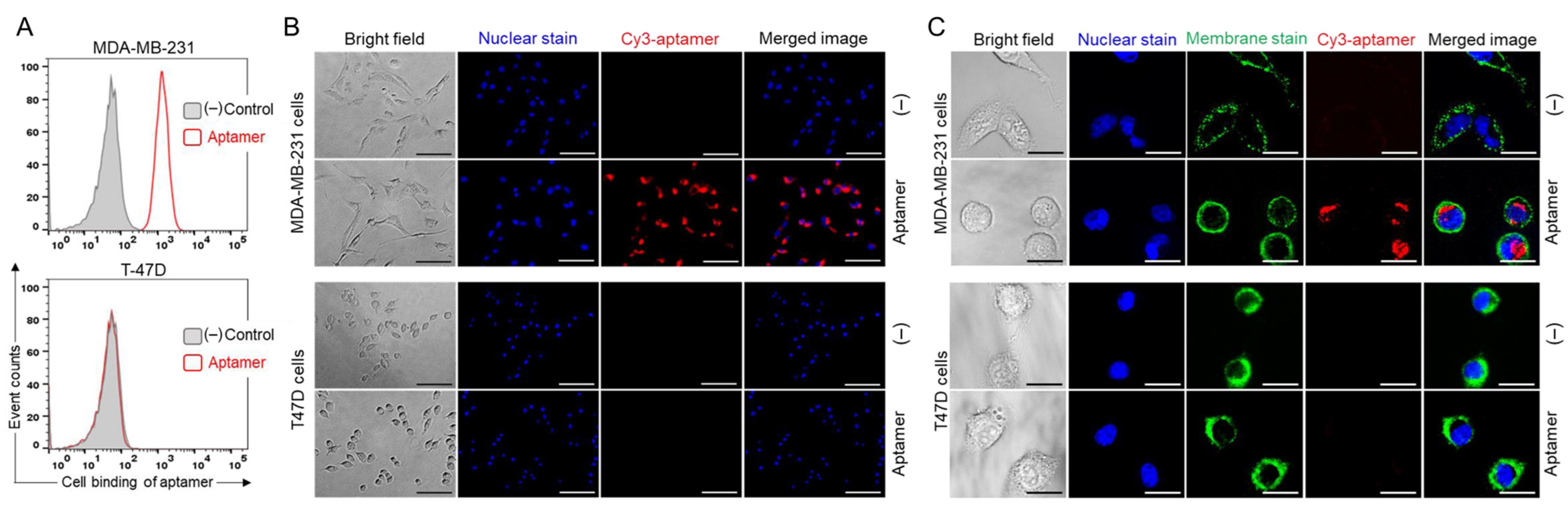
17]. In this study, we demonstrate that the Apt–cL–triGemcitabine conjugate has substantially greater anti-neoplastic activity than free gemcitabine in TNBC cells. We postulate that this improvement stems from efficient aptamer-mediated cell targeting and intracellular delivery of gemcitabine, as evidenced by aptamer internalization by TNBC cells (
Figure 4C). In addition, the conjugation of gemcitabine with an enzymatically cleavable linker facilitated the controlled intracellular drug release by lysosomal cathepsin B protease (
Figure 2 and
Figure 3). Moreover, by incorporating TNM dendrimer structures, three gemcitabine molecules were added to each Apt–cL–triGemcitabine, promoting efficient drug delivery with a high drug payload.
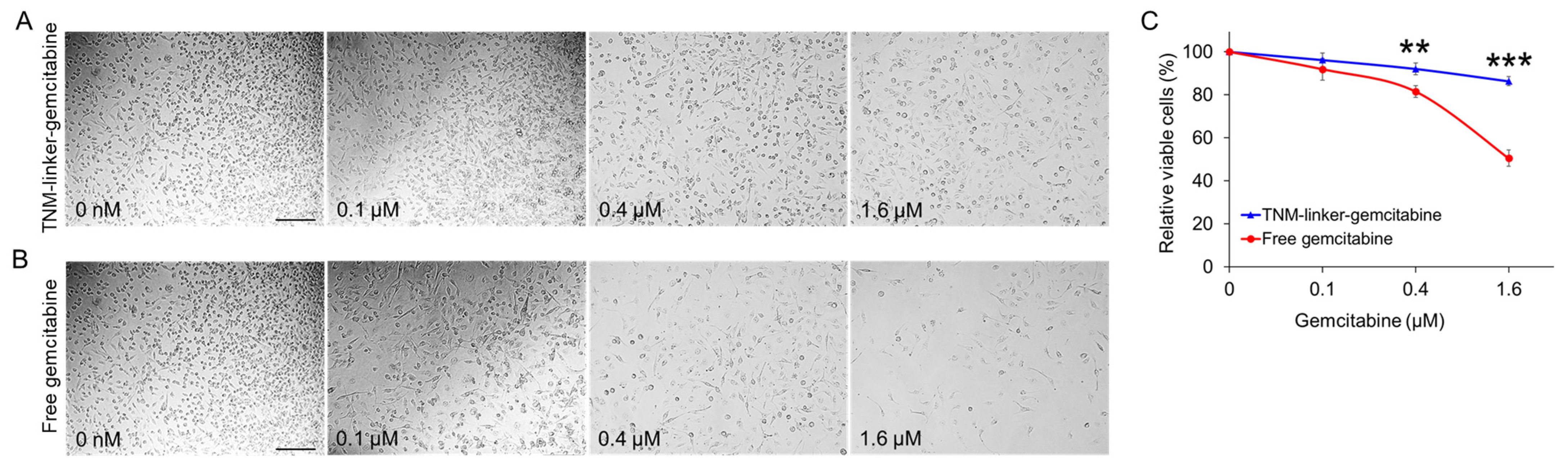
The clinical utility of chemotherapeutics is constrained by the tolerability of their side effects. Due to the fact that side effects typically arise from off-target cytotoxicity, therapeutic strategies that minimize off-target activity can facilitate higher dosing and, by extension, greater therapeutic effect. Our central objective was to develop a novel chemotherapeutic with specific anti-tumor activity in TNBC cells but limited activity in healthy tissue. To this end, we hypothesized that the chemotherapeutic could be stabilized with chemical moieties that prevent extracellular drug release. In the past, the clinical adoption of aptamer therapeutics has been hindered by instability and unpredictable behavior in vivo. To prevent aptamer release of gemcitabine in circulation, we incorporated cleavable peptide linkers into the TNM dendrimer vehicle. These linkers, commonly used in antibody–drug conjugate development, are enzymatically cleaved by activated cathepsin B in lysosomes. Notably, our study reveals that, by incorporating gemcitabine into the TNM vehicle using these cleavable linkers, the cytotoxic activity of gemcitabine was markedly reduced relative to free drug (
Figure 5), which may increase its drug delivery safety profile.
4. Materials and Methods
4.1. Chromatography and Mass Spectroscopy
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Tokyo Chemical Industry (Tokyo, Japan) and used without further purification. Column chromatography was performed on silica gel 60 (70–230 mesh) (Fair Lawn, NJ, USA). Analytical thin-layer chromatography was conducted on glass plates coated with silica gel 60 (F-254) (EMD-Millipore Corporation, Billerica, MA, USA). Mass spectra (ESI-MS data) were collected on an LCQ-Fleet™ Ion Trap Mass Spectrometer (Thermo, Waltham, MA, USA) and Waters Maldi SYNAPT HDMS Q-TOF Mass Spectrometer (Milford, MA, USA).
4.2. Synthesis and Characterization of Apt–cL–triGemcitabine Conjugate
Synthesis of TNM-linker–gemcitabine. The TNM–linker–gemcitabine was synthesized in two phases according to
Scheme S1. In the first phase, Boc-protected linker–gemcitabine (compound
6) was prepared using established peptide chemistry. Boc-Val-Cit-OH (compound
3) was coupled with p-aminobenzyl alcohol (PABOH) using N-Ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) to form compound
4, followed by the reaction with 4-nitrophenyl-chloroformate to form an activated carbonate moiety (compound
5). Compound 5 was conjugated to gemcitabine to create the Boc-protected cleavable linker peptide–gemcitabine (compound
6). The mass spectrum of compound
6 is shown in
Figure S4.
In the second phase, Boc-protected peptide-Gemcitabine was incorporated into a TNM. TNM (compound
10) was coupled with 6-maleimidocaproic acid in the presence of 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo [4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) to form compound
11. Compound
11 was deprotected to remove tert-butoxy, forming compound
12. Compound
12 was activated with HATU completely to form active ester compound
13. Dropwise addition of compound
13 to deprotected peptide–gemcitabine in Dimethylformamide (DMF) and N-Methylmorpholine (NMM) formed TNM–peptide–gemcitabine (compound
14). The mass spectrum of compound
14 is shown in
Figures S11 and S12.
Synthesis and characterization of Apt–cL–triGemcitabine. Detailed methods are described in the
supplementary materials.
4.3. Drug Release from TNM-Linker-Gemcitabine
Cathepsin B-mediated Release of free Gemcitabine drug: Dithiothreitol (DTT) solution (30 mM) was prepared by adding 4.5 mg of DTT to 1 mL of sodium acetate buffer (pH = 5.0, ~0.1 M). Then, 1 µL (0.45 µg) of cathepsin B solution (molecular weight: 35,000, purchased from EMD Millipore, Burlington, MA, USA) was added to 40 µL of DTT solution and the mixture was incubated at 37 °C for 15 min. Subsequently, 2 µL (6.5 µg) of linker–gemcitabineTNM–linker–gemcitabine stock solution (in DMF) was added to the activated cathepsin B solution and incubated for 1 h at 37 °C. The reaction solution was analyzed by HPLC with a reversed-phase column.
Cell Lysate Preparation: Two-day cultured MDA-MB-231 cells were trypsinized, washed twice with PBS (phosphate buffered saline), and quantified. Next, 1 × 107 cells were resuspended in an Eppendorf tube with 200 µL PBS. Cells were sonicated in a 4 °C water bath in a 30 s off/on cycle for 10 min. After sonication, cells were centrifugated at 10,000× g for 60 min. The supernatant was transferred to a clean tube and stored at −20 °C for further use.
Cell Lysate-mediated Release of Gemcitabine: A 10 µL aliquot of DTT solution was added to 30 µL of cell lysate and incubated for 15 min at 37 °C. After activation of the cell lysate solution, 1 µL (0.32 µg) of linker–gemcitabineTNM–linker–gemcitabine stock solution (diluted ×10) was added to the cell lysate and incubated for 1 h at 37 °C. The reaction solution was analyzed by HPLC with a reversed-phase column.
4.4. HPLC Analysis
HPLC Operating Conditions for Aptamer–linker–gemcitabineTNM–linker–gemcitabineCoupling: HPLC separations were performed using a PRP-1 C18 column (4.1 × 150 mm, 5 µm, Hamilton, Reno, NV, USA) and the Varian 920-LC liquid chromatograph (Walnut Creek, CA, USA). The reaction was monitored using gradient HPLC, and analytes were detected using an ultraviolet absorbance detector at a wavelength of 260 nm. The mobile phase consisted of solvent A (100 mM of triethylammonium acetate (TEAA) aqueous solution) and solvent B (5% of mobile A in acetonitrile) with a flow rate of 1.0 mL/min. The mobile phase composition was maintained at 100% solvent A for 3 min before changing linearly to 0% solvent A (3–18 min), which was held for 3 min (18–21 min). Subsequently, the mobile phase was returned to its initial conditions over 3 min (21–24 min) and maintained at 100% solvent A for 2 min (24–26 min) to equilibrate the column.
HPLC Operating Conditions for Purification: Semi-preparative HPLC was performed using a PRP-1 C18 column (10 × 250 mm, 10 µm, Hamilton, Reno, NV, USA) and the Hitachi L-710 liquid chromatograph with an L-7455 diode array detector (Chiyoda-ku, Tokyo, 100-8280, Japan). Purification was conducted using gradient HPLC and analytes were detected using an ultraviolet absorbance detector at a wavelength of 260 nm. The mobile phase consisted of solvent A (100 mM of TEAA aqueous solution) and solvent B (5% of mobile A in acetonitrile) at a flow rate of 3.0 mL/min. The mobile phase composition was maintained at 100% solvent A for 4 min before changing linearly to 0% solvent A (4–24 min), which was held for 4 min (24–28 min). Subsequently, the mobile phase was returned to its initial conditions over 3 min (28–30 min) and maintained at 100% solvent A for 2 min (30–32 min) to equilibrate the column.
HPLC Operating Conditions for Drug Release: HPLC separation was performed using a Phenomenex Luna C18 column (150 × 4.60 mm, 5 µm, Phenomenex, Torrance, CA, USA) and the HPLC quaternary solvent manager pump (Waters Corporation, Milford, MA, USA) with a 2998 PDA ultraviolet absorbance detector (Waters Corporation, Milford, MA, USA). Gemcitabine release was monitored using gradient HPLC and analytes were detected using an ultraviolet absorbance detector at a wavelength of 260 nm (gemcitabine) and 215 nm (decomposed compounds). The mobile phase consisted of solvent A (0.1% trifluoroacetic acid aqueous solution) and solvent B (0.05% trifluoroacetic acid in acetonitrile) at a flow rate of 1.0 mL/min. The mobile phase composition was maintained at 100% solvent A for 3 min before changing linearly to 0% solvent A (3–18 min), which was held for 3 min (18–21 min). Subsequently, the mobile phase was returned to its initial conditions over 3 min (21–24 min) and maintained at 100% solvent A for 2 min (24–26 min) to equilibrate the column. The injection volume was 20 µL.
4.5. Gel Electrophoresis
Thiol-PDGC21-T aptamer (0.4 µg), Apt–cL–triGemcitabine (0.5 µg), and linker–gemcitabineTNM–linker–gemcitabine (0.5 µg) were loaded onto a 10% denaturing PAGE (Polyacrylamide) gel. Separation was performed at 60 V/cm for 1.5 h in 0.5 × TBE running buffer. The gel was stained with 1× SYBR Green for 25 min and imaged using a Gel Doc™ EZ imaging system (Bio-Rad, Hercules, CA, USA).
4.6. Cell Lines and Cell Culture
Thr MDA-MB-231 (human TNBC) and T47D (human non-TNBC) cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA) and tested negative for mycoplasma. The MDA-MB-231 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Corning, Manassas, VA, USA), and T47D cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (HyClone, GE Healthcare Life Sciences, South Logan, UT, USA). Both media were supplemented with 10% FBS (Atlanta Biologicals, Inc., Flowery Branch, GA, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco, Waltham, MA, USA). Cells were maintained at 37 °C in 5% CO2 and ≥95% humidity.
4.7. PDGC21-T Aptamer Probe Synthesis
The PDGC21-T aptamer sequence was previously reported as ACACCAAAATCGTCCGTTTCGTTTTAGTCCGTCTCTTTAGGGTGT. Cy3-conjugated PDGC21-T was synthesized by Integrated DNA Technologies (Coralville, IA, USA). A 100 µM stock solution of the aptamer probe was prepared with nuclease-free water and stored at −20 °C.
4.8. Cell-Binding Assay
Flow Cytometry Analysis: Two-day cultured cells (MDA-MB-231 and T47D) were trypsinized and washed twice with PBS. The Cy3-PDGC21-T conjugate was heated at 95 °C for 5 min and then cooled on ice for 10 min. Cells were mixed with a Cy3-PDGC21-T probe in a binding buffer to a final concentration of 200 nM and incubated in the dark at RT for 30 min. After incubation, the cells were washed twice with a washing buffer and subjected to flow cytometry analysis with a BD LSR II cytometer (BD Biosciences, San Jose, CA, USA). Flow cytometry data were analyzed using FlowJo v10.0.7 software (FlowJo, Ashland, OR, USA).
Fluorescent Microscopy: MDA-MB-231 and T47D cells were cultured in black 96-well plates for 1 day and then incubated with a 200 nM of Cy3-PDGC21-T probe in s culture medium at 37 °C for 1 h. After incubation, aptamer-treated and control cells were counterstained with Hoechst 33342 dye for 5 min. The staining solution was discarded and 100 µL fresh culture medium was added to cells. Cells were examined using an Olympus IX81 fluorescence microscope (Olympus Corporation, Shinjuku City, Tokyo, Japan).
Confocal Microscopy: MDA-MB-231 cells and T47D cells were cultured in black 96-well plates for 1 day and then incubated with 200 nM of a Cy3-PDGC21-T probe in a culture medium at 37 °C for 30 min. The cells were washed once with PBS, and then cell membranes were counterstained with Vybrant™ DiO Cell-Labeling Solution (5 µL/mL) at 37 °C for 15 min, and nuclei were stained with Hoechst 33342 dye for 5 min. To evaluate aptamer internalization, the cells were examined using a Nikon A1 Confocal Imaging System (Nikon, Tokyo, Japan).
4.9. Cell Proliferation Assay
4.9.1. Toxicity Evaluation of TNM-gemcitabine
Cell treatment: 4 × 103 MDA-MB-231cells in 100 µL of DMEM culture medium were seeded into 96-well plates. Cells were incubated with synthetic TNM–gemcitabine products or free drug at equimolar amounts of gemcitabine, at final concentrations of 100 nM, 400 nM, and 1600 nM for 30 min at 37 °C. After incubation, the drug-containing supernatant was discarded, the cells were washed twice with PBS, and fresh DMEM was added. The cells were further cultured at 37 °C in 5% CO2 and ≥95% humidity for two days.
CCK-8 Proliferation Assay: Cell proliferation was quantified using the Cell Counting Kit-8 (CCK-8) assay (APExBIO Technology, Houston, TX, USA) according to the manufacturer’s instructions. Cell proliferation was evaluated one, two, or three days after drug administration. Formazan products were measured using a BioTek Synergy H4 microplate reader (Winooski, VT, USA) at a wavelength of 450 nm. All experiments were performed in triplicate, and the average absorbance standard deviation was calculated. Student’s t-tests were performed to assess different groups.
4.9.2. Effects of Apt–cL–triGemcitabine Conjugate on TNBC Cells
Cell Treatment: 4 × 103 cells in 100 µL of DMEM culture medium (MDA-MB-231 cells) or RPMI 1640 culture medium (T47D cells) were seeded into 96-well plates. Cells were incubated with the Apt–cL–triGemcitabine conjugate or the free gemcitabine drug at concentrations of 100 nM or 250 nM for 30 min at 37 °C. An untreated control group was prepared using the same number of cells. After incubation, the drug-containing supernatant was discarded, cells were washed twice with PBS, and fresh DMEM or RPMI 1640 culture medium was added.
Fluorescence Microscopy: Two days after treatment with the Apt–cL–triGemcitabine conjugate or free gemcitabine drug, MDA-MB-231 and T47D cells were stained with a mixture of PI (Invitrogen, Eugene, OR, USA), Annexin V (BD Biosciences, Franklin Lakes, NJ, USA), and Hoechst 33342 (Thermo Fischer Scientific, Waltham, MA, USA) dyes at RT for 10 min. The supernatant was discarded, cells were washed with PBS, and 100 µL of new culture medium was added. Cells were examined using an Olympus IX81 fluorescence microscope. Images of each experimental group were captured at low magnification (×100). Annexin V positive apoptosis cells and PI positive dead cells were quantified for four fields of each experiment. Mean values and standard deviations were calculated.
CCK-8 Proliferation Assay: Cell proliferation was evaluated one, two, or three days after drug administration using the methods prescribed previously. All experiments were performed in triplicate, and the average absorbance standard deviation was calculated. Student’s t-tests were performed to assess the different groups and treatment timepoints.
4.10. Data Analysis
Statistical analysis was performed using Microsoft Excel software. An unpaired Student’s t-test was used in the data analysis, and p < 0.05 was considered a statistically significant difference.
4.11. Study Design and Rationale
The goal of ligand-directed cancer therapy is to minimize the side effects related to the activity of the cytotoxic agents in normal cells while simultaneously delivering these agents at therapeutic doses to the target cells. To this end, we designed aptamer–drug conjugates to facilitate the targeted delivery of gemcitabine, an effective and well-validated chemotherapeutic, to cancer cells. To introduce a controlled drug-release switch, gemcitabine was conjugated to the carrier vehicle through an enzymatically cleavable linker (
Figure 1A). This peptide linker is highly stable in blood but readily cleaved by activated cathepsin B (present in the low pH microenvironment of cell lysosomes). In addition, the linker conjugation masks the 4-NH2 group on gemcitabine, a region that contributes to its cytotoxicity [
18]. To increase the drug payload, we incorporated three linker–gemcitabine molecules into a TNM. To increase the specificity for cancer cells, we conjugated the TNM–linker–gemcitabine products to PDGC21 aptamer sequences that selectively bind TNBC cells [
19]. The formulated Aptamer, enzymatically cleavable Linker, and tri-Gemcitabine (Apt–cL–triGemcitabine) drug conjugate allows one aptamer to carry three gemcitabine molecules, ensures stable drug delivery, and results in an enzymatically controlled lysosomal drug release.
Under aptamer guidance, the Apt–cL–triGemcitabine conjugate will bind the TNBC cells and be rapidly internalized [
20]. Within targeted cells, enzymatic cleavage of Apt–cL–triGemcitabine by lysosomal cathepsin B protease will liberate gemcitabine molecules. These freed molecules will disrupt DNA synthesis, resulting in apoptosis and the death of targeted tumor cells (
Figure 1B). This drug-delivery system should have high therapeutic efficacy as it promotes the selective accumulation of gemcitabine in targeted cancer cells and hinders the non-specific uptake of gemcitabine by healthy cells.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






