
Identification of Immune Subtypes of Esophageal Adenocarcinoma to Predict Prognosis and Immunotherapy Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
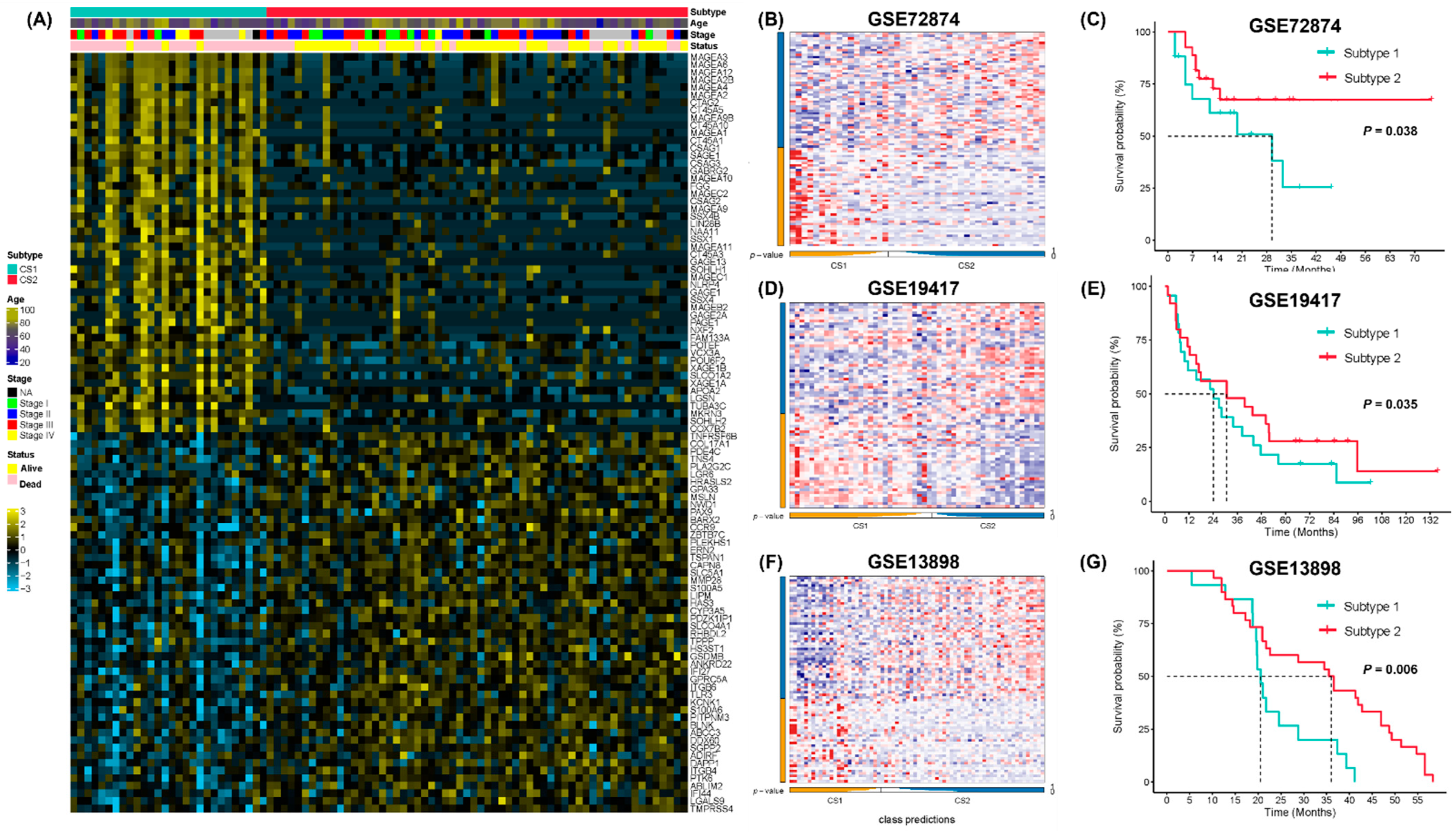{kind=link}
Abstract
:1. Introduction
2. Results
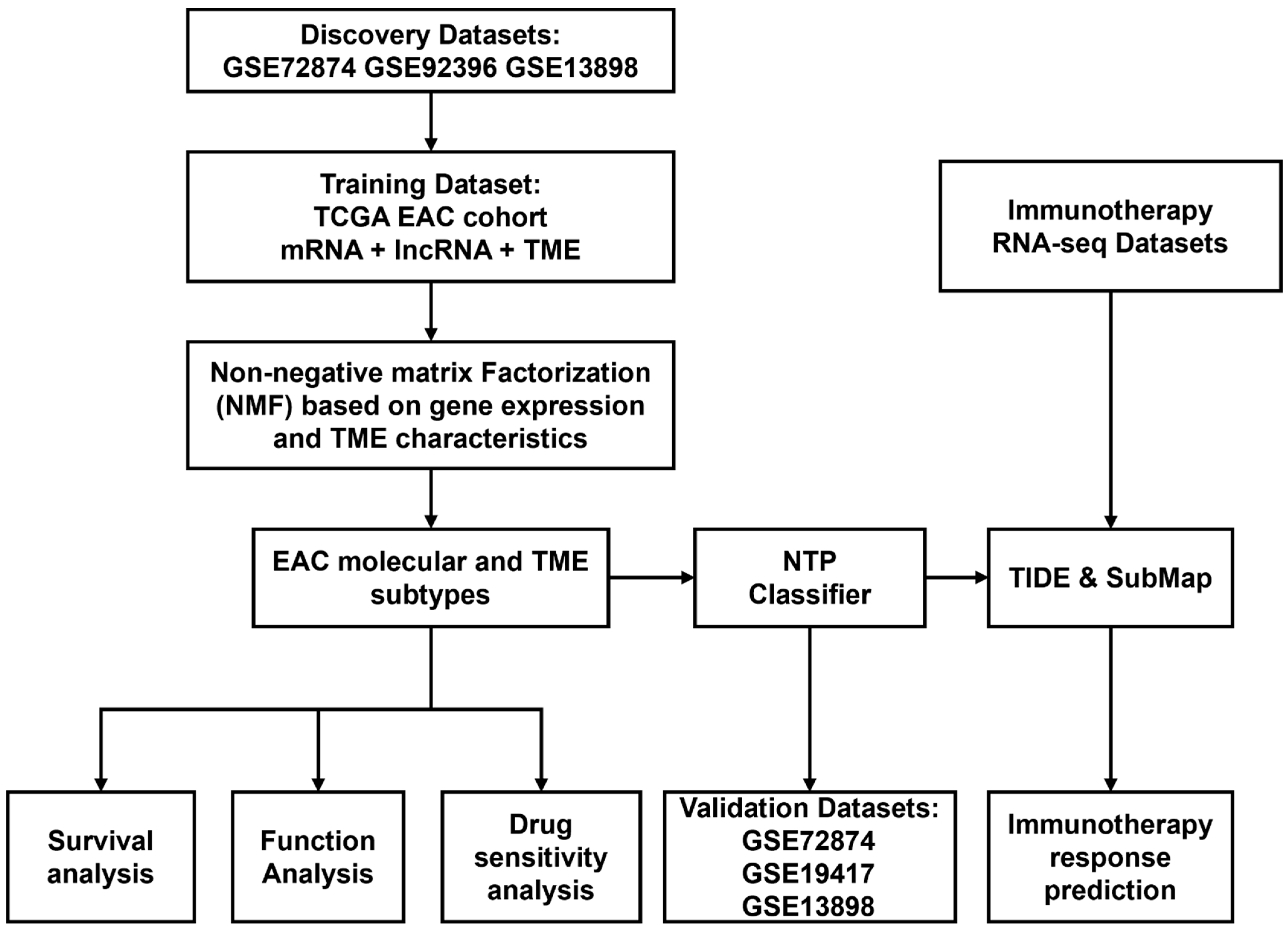
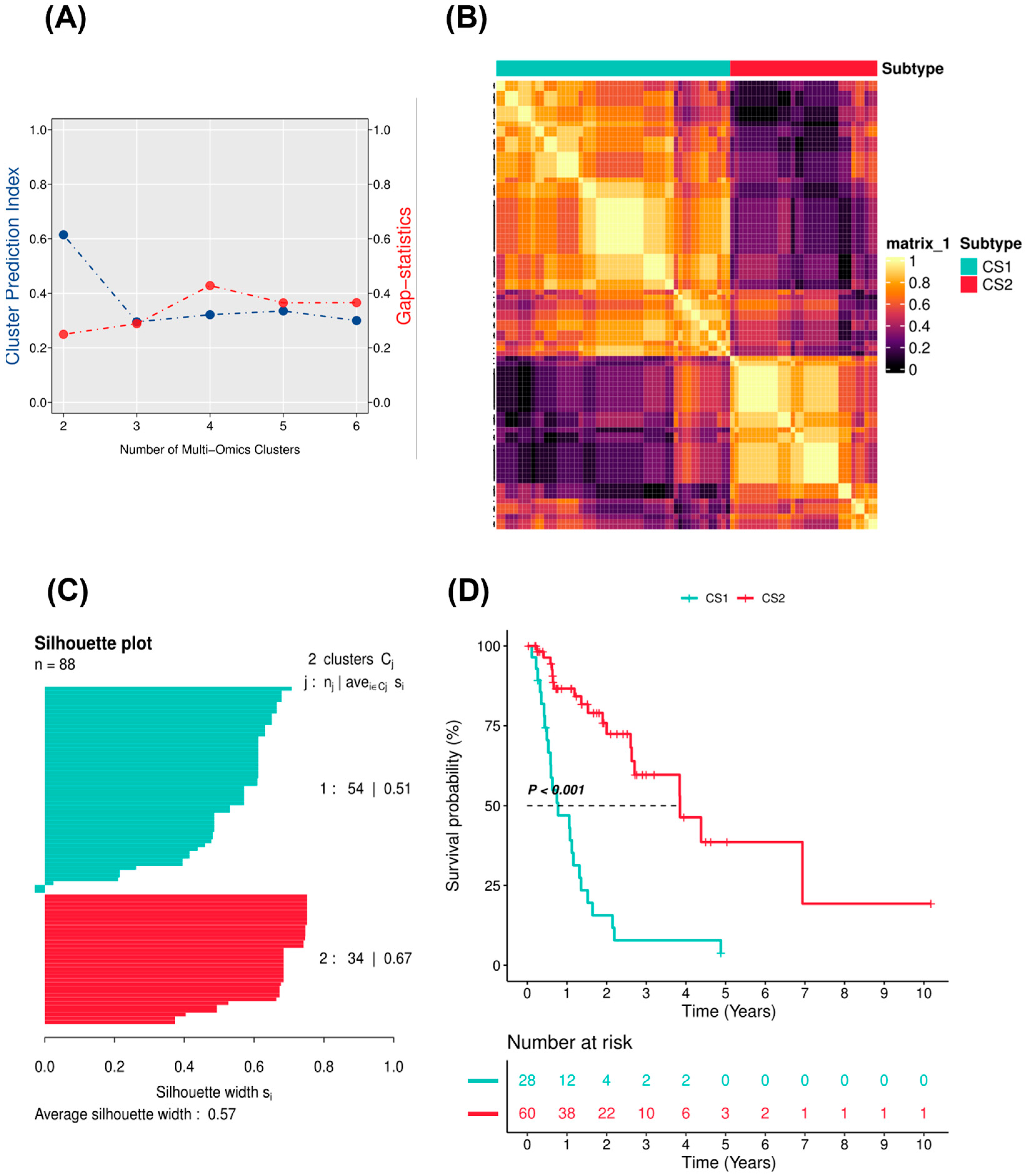
2.1. Identification of EAC Subtypes
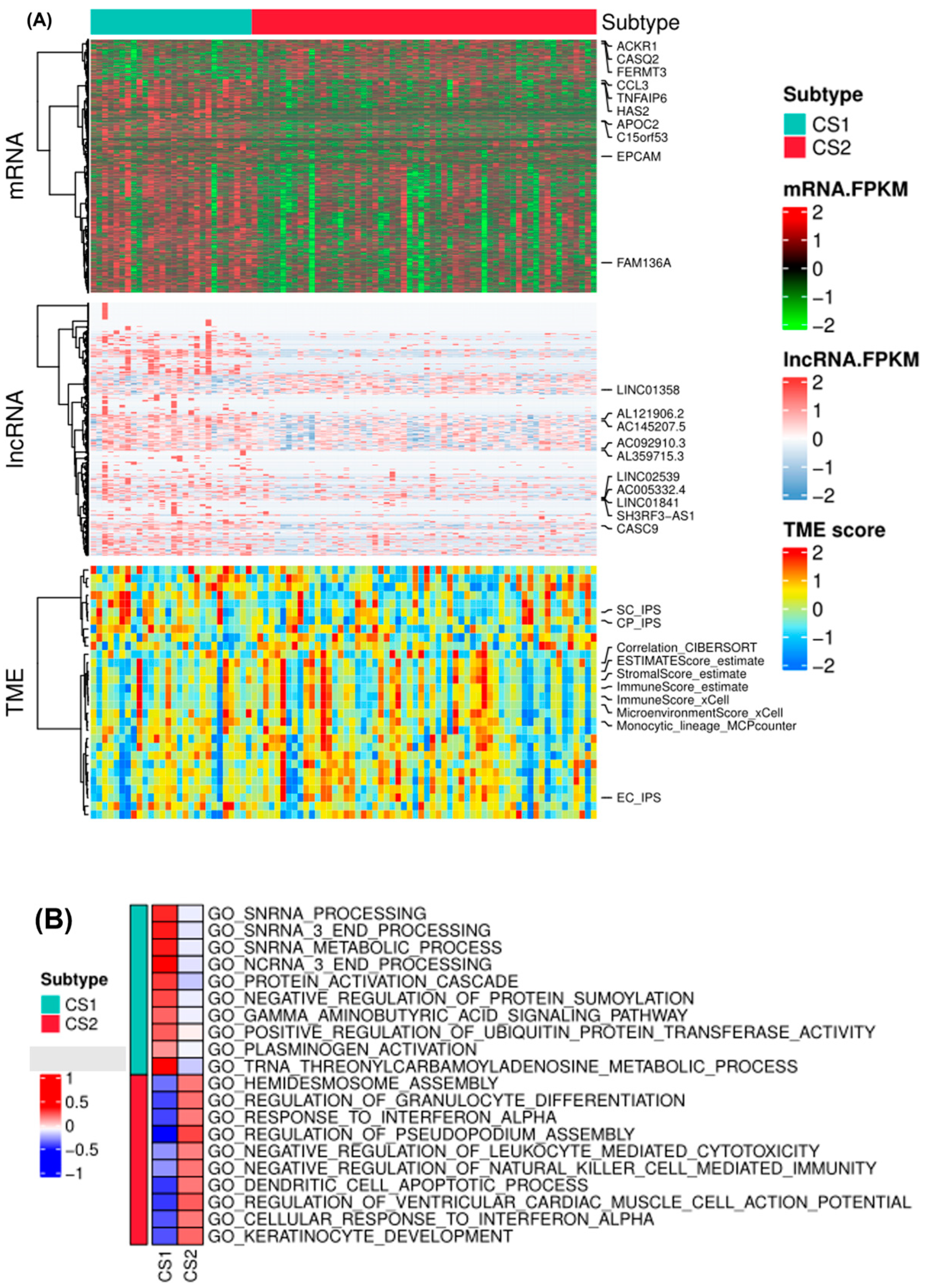
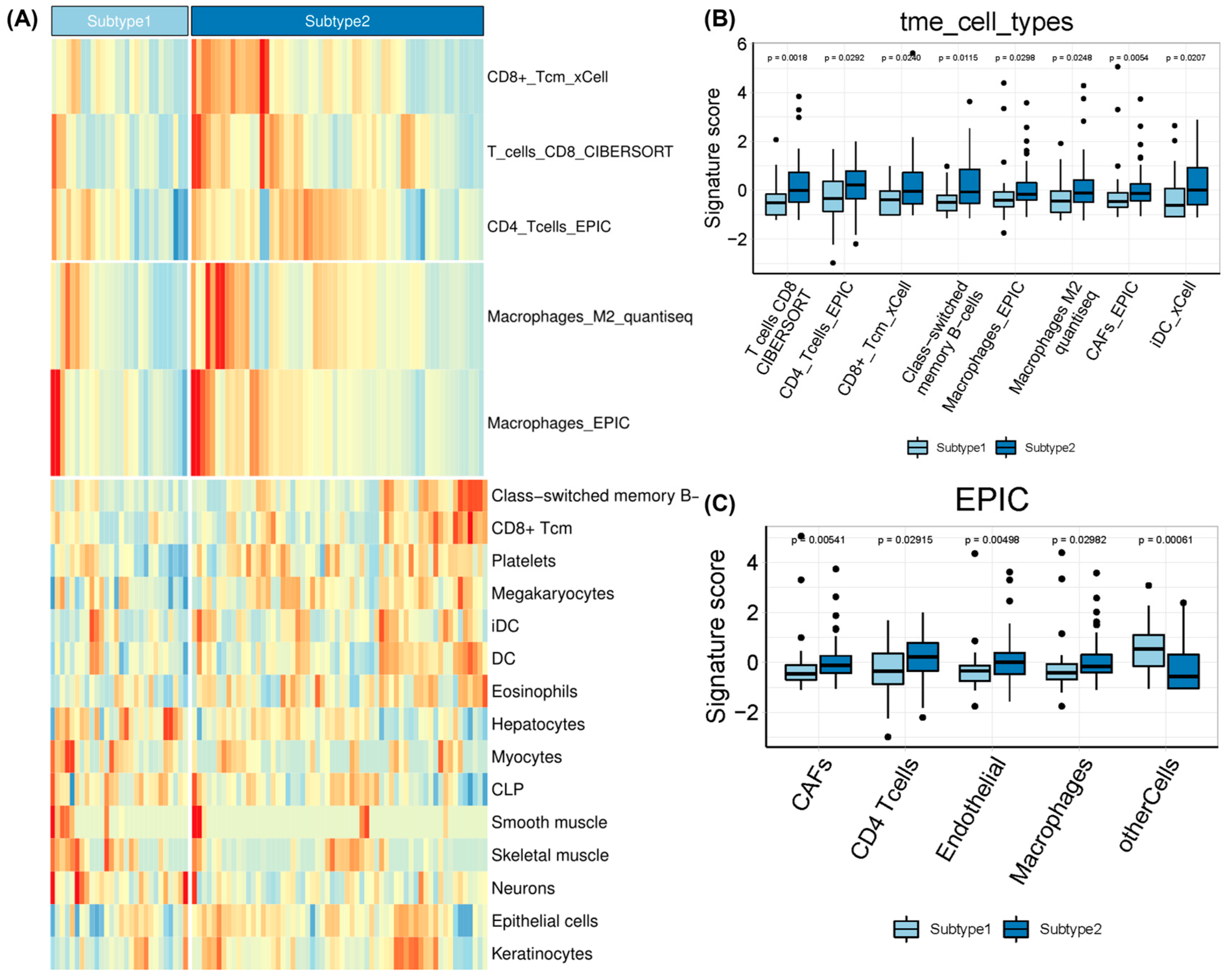
2.2. Molecular and TME Characteristics of EAC Subtypes
2.3. Therapeutic Sensitivity of EAC Subtypes
2.4. Classifier Construction and Validation
3. Discussion
4. Materials and Methods
4.1. Data Source and Processing
4.2. Selection of Differentially Expressed Genes with Prognostic Value
4.3. Estimation of Immune Infiltration
4.4. Identification and Validation of EAC Subtypes
4.5. Molecular and TME Characterization between EAC Subtypes
4.6. Therapeutic Response Prediction
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EAC | Esophageal adenocarcinoma |
| ESCC | Esophageal squamous cell carcinoma |
| CCL | Cancer cell lines |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus dataset |
| DEG | Differentially expressed gene |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| TIDE | Tumor Immune Dysfunction and Exclusion |
References
- Collaboration GBoDC. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.-L.; Yu, S.-J. Esophageal cancer: Risk factors, genetic association, and treatment. Asian J. Surg. 2018, 41, 210–215. [Google Scholar] [CrossRef]
- Offman, J.; Pesola, F.; Sasieni, P. Trends and projections in adenocarcinoma and squamous cell carcinoma of the oesophagus in England from 1971 to 2037. Br. J. Cancer 2018, 118, 1391–1398. [Google Scholar] [CrossRef]
- Eyck, B.M.; van Lanschot, J.J.B.; Hulshof, M.C.C.M.; van der Wilk, B.J.; Shapiro, J.; van Hagen, P.; Henegouwen, M.I.V.B.; Wijnhoven, B.P.L.; van Laarhoven, H.W.M.; Nieuwenhuijzen, G.A.P.; et al. Ten-Year Outcome of Neoadjuvant Chemoradiotherapy Plus Surgery for Esophageal Cancer: The Randomized Controlled CROSS Trial. J. Clin. Oncol. 2021, 39, 1995–2004. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Van Cutsem, E.; Fuchs, C.S.; Ohtsu, A.; Tabernero, J.; Ilson, D.H.; Hyung, W.J.; Strong, V.E.; Goetze, T.O.; Yoshikawa, T.; et al. KEYNOTE-585: Phase III study of perioperative chemotherapy with or without pembrolizumab for gastric cancer. Futur. Oncol. 2019, 15, 943–952. [Google Scholar] [CrossRef]
- Luo, Y.; Mao, Q.; Wang, X.; Yu, J.; Li, M. Radiotherapy for esophageal carcinoma: Dose, response and survival. Cancer Manag. Res. 2017, 10, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-M.; Hong, P.; Xu, W.W.; He, Q.-Y.; Li, B. Advances in targeted therapy for esophageal cancer. Signal Transduct. Target. Ther. 2020, 5, 229. [Google Scholar] [CrossRef]
- Schneider, B.J.; Shah, M.A.; Klute, K.; Ocean, A.; Popa, E.; Altorki, N.; Lieberman, M.; Schreiner, A.; Yantiss, R.; Christos, P.J.; et al. Phase I Study of Epigenetic Priming with Azacitidine Prior to Standard Neoadjuvant Chemotherapy for Patients with Resectable Gastric and Esophageal Adenocarcinoma: Evidence of Tumor Hypomethylation as an Indicator of Major Histopathologic Response. Clin. Cancer Res. 2016, 23, 2673–2680. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar]
- Waddell, T.; Chau, I.; Cunningham, D.; Gonzalez, D.; Okines, A.F.C.; Wotherspoon, A.; Saffery, C.; Middleton, G.; Wadsley, J.; Ferry, D.; et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial. Lancet Oncol. 2013, 14, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.; Awasthi, N.; Li, J.; Williams, F.; Schwarz, M.A.; Schwarz, R.E.; von Holzen, U. Superior Therapeutic Efficacy of Nanoparticle Albumin Bound Paclitaxel Over Cremophor-Bound Paclitaxel in Experimental Esophageal Adenocarcinoma. Transl. Oncol. 2018, 11, 426–435. [Google Scholar] [CrossRef]
- Zhan, W.; Li, H.; Guo, Y.; Du, G.; Wu, Y.; Zhang, D. Construction of Biocompatible Dual-Drug Loaded Complicated Nanoparticles for in vivo Improvement of Synergistic Chemotherapy in Esophageal Cancer. Front. Oncol. 2020, 10, 662. [Google Scholar] [CrossRef]
- Fu, Q.; Wang, J.; Liu, H. Chemo-immune synergetic therapy of esophageal carcinoma: Trastuzumab modified, cisplatin and fluorouracil co-delivered lipid–polymer hybrid nanoparticles. Drug Deliv. 2020, 27, 1535–1543. [Google Scholar] [CrossRef]
- Contino, G.; Vaughan, T.L.; Whiteman, D.; Fitzgerald, R.C. The Evolving Genomic Landscape of Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology 2017, 153, 657–673. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.J. The emerging role of immunotherapy for esophageal cancer. Curr. Opin. Gastroenterol. 2019, 35, 337–343. [Google Scholar] [CrossRef]
- Doi, T.; Piha-Paul, S.A.; Jalal, S.I.; Saraf, S.; Lunceford, J.; Koshiji, M.; Bennouna, J. Safety and Antitumor Activity of the Anti–Programmed Death-1 Antibody Pembrolizumab in Patients With Advanced Esophageal Carcinoma. J. Clin. Oncol. 2018, 36, 61–67. [Google Scholar] [CrossRef]
- Shah, M.A.; Kojima, T.; Hochhauser, D.; Enzinger, P.; Raimbourg, J.; Hollebecque, A.; Kato, K. Efficacy and Safety of Pembrolizumab for Heavily Pretreated Patients with Advanced, Metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus: The Phase 2 KEYNOTE-180 Study. JAMA Oncol. 2019, 5, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Muro, K.; Francois, E.; Hsu, C.-H.; Moriwaki, T.; Kim, S.-B.; Lee, S.-H.; Bennouna, J.; Kato, K.; Lin, S.; et al. Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: Phase III KEYNOTE-181 study. J. Clin. Oncol. 2019, 37 (Suppl. S4), 2. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Yoon, H.H. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Imamura, Y.; Watanabe, M.; Toihata, T.; Takamatsu, M.; Kawachi, H.; Haraguchi, I.; Ogata, Y.; Yoshida, N.; Saeki, H.; Oki, E.; et al. Recent Incidence Trend of Surgically Resected Esophagogastric Junction Adenocarcinoma and Microsatellite Instability Status in Japanese Patients. Digestion 2018, 99, 6–13. [Google Scholar] [CrossRef]
- Greally, M.; Chou, J.F.; Chatila, W.K.; Margolis, M.; Capanu, M.; Hechtman, J.F.; Tuvy, Y.; Kundra, R.; Daian, F.; Ladanyi, M.; et al. Clinical and Molecular Predictors of Response to Immune Checkpoint Inhibitors in Patients with Advanced Esophagogastric Cancer. Clin. Cancer Res. 2019, 25, 6160–6169. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2016, 387, 61–68. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Combes, A.J.; Samad, B.; Tsui, J.; Chew, N.W.; Yan, P.; Reeder, G.C.; Kushnoor, D.; Shen, A.; Davidson, B.; Barczak, A.J.; et al. Discovering dominant tumor immune archetypes in a pan-cancer census. Cell 2021, 185, 184–203.e19. [Google Scholar] [CrossRef]
- Ho, J.W.; Stefani, M.; Dos Remedios, C.G.; Charleston, M.A. Differential variability analysis of gene expression and its application to human diseases. Bioinformatics 2008, 24, i390–i398. [Google Scholar] [CrossRef] [Green Version]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.-L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitrodrug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Castelli, G.; Pelosi, E. Esophageal Cancer: Genomic and Molecular Characterization, Stem Cell Compartment and Clonal Evolution. Medicines 2017, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal cancer. Nat. Rev. Dis. Primers 2017, 3, 17048. [Google Scholar] [CrossRef]
- Huang, T.-X.; Fu, L. The immune landscape of esophageal cancer. Cancer Commun. 2019, 39, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lokeshwar, B.L.; Kallifatidis, G.; Hoy, J.J. Chapter One—Atypical chemokine receptors in tumor cell growth and metastasis. In Advances in Cancer Research; Shukla, A.K., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 1–27. [Google Scholar]
- Pruenster, M.; Mudde, L.; Bombosi, P.; Dimitrova, S.; Zsak, M.; Middleton, J.; Richmond, A.; Graham, G.J.; Segerer, S.; Nibbs, R.J.; et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat. Immunol. 2009, 10, 101–108. [Google Scholar] [CrossRef]
- Gaiser, M.R.; Lämmermann, T.; Feng, X.; Igyarto, B.Z.; Kaplan, D.H.; Tessarollo, L.; Germain, R.N.; Udey, M.C. Cancer-associated epithelial cell adhesion molecule (EpCAM; CD326) enables epidermal Langerhans cell motility and migration in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E889–E897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staversky, R.J.; Byun, D.K.; Georger, M.A.; Zaffuto, B.J.; Goodman, A.; Becker, M.W.; Calvi, L.M.; Frisch, B.J. The Chemokine CCL3 Regulates Myeloid Differentiation and Hematopoietic Stem Cell Numbers. Sci. Rep. 2018, 8, 14691. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.-C.; Li, C.-F.; Ke, H.-L.; Wei, Y.-C.; Shiue, Y.-L.; Li, C.-C.; Yeh, H.-C.; Lee, H.-Y.; Huang, S.-K.; Wu, W.-J.; et al. High TNFAIP6 level is associated with poor prognosis of urothelial carcinomas. Urol. Oncol. Semin. Orig. Investig. 2018, 37, e11–e24. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Z.; Xu, E.; Shen, X.; Wang, X.; Li, Z.; Yu, H.; Chen, K.; Hu, Q.; Xia, X.; et al. Apolipoprotein C-II induces EMT to promote gastric cancer peritoneal metastasis via PI3K/AKT/mTOR pathway. Clin. Transl. Med. 2021, 11, e522. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, J.; Ling, J.; Zhu, X.; Jiang, P.; Tang, X.; Zhou, H.; Li, R. Construction of a Glycolysis-related long noncoding RNA signature for predicting survival in endometrial cancer. J. Cancer 2021, 12, 1431–1444. [Google Scholar] [CrossRef]
- Xue, S.-T.; Zheng, B.; Cao, S.-Q.; Ding, J.-C.; Hu, G.-S.; Liu, W.; Chen, C. Long non-coding RNA LINC00680 functions as a ceRNA to promote esophageal squamous cell carcinoma progression through the miR-423-5p/PAK6 axis. Mol. Cancer 2022, 21, 69. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, R.; Qiu, J. A four immune-related long noncoding RNAs signature as predictors for cervical cancer. Hum. Cell 2021, 35, 348–359. [Google Scholar] [CrossRef]
- Zhang, X.; Lian, T.; Fan, W.; Zhang, G.; Chen, Z.; Gou, X.; Jha, R.K. Long-Noncoding RNA CASC9 Promotes Progression of Non-Small Cell Lung Cancer by Promoting the Expression of CDC6 Through Binding to HuR. Cancer Manag. Res. 2020, 12, 9033–9043. [Google Scholar] [CrossRef]
- Li, X.-F.; Ren, P.; Shen, W.-Z.; Jin, X.; Zhang, J. The expression, modulation and use of cancer-testis antigens as potential biomarkers for cancer immunotherapy. Am. J. Transl. Res. 2020, 12, 7002–7019. [Google Scholar]
- Mohsenzadegan, M.; Razmi, M.; Vafaei, S.; Abolhasani, M.; Madjd, Z.; Saeednejad Zanjani, L.; Sharifi, L. Co-expression of cancer-testis antigens of MAGE-A6 and MAGE-A11 is associated with tumor aggressiveness in patients with bladder cancer. Sci. Rep. 2022, 12, 599. [Google Scholar] [CrossRef]
- Weon, J.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila Cobos, F.; Alquicira-Hernandez, J.; Powell, J.E.; Mestdagh, P.; De Preter, K. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun. 2020, 11, 5650. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Severson, E.; Pignon, J.-C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Racle, J.; De Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Trajanoski, Z. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 1–20. [Google Scholar]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalise, P.; Fridley, B.L. Integrative clustering of multi-level ‘omic data based on non-negative matrix factorization algorithm. PLoS ONE 2017, 12, e0176278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantini, L.; Zakeri, P.; Hernandez, C.; Naldi, A.; Thieffry, D.; Remy, E.; Baudot, A. Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer. Nat. Commun. 2021, 12, 124. [Google Scholar] [CrossRef]
- Lu, X.; Meng, J.; Zhou, Y.; Jiang, L.; Yan, F. MOVICS: An R package for multi-omics integration and visualization in cancer subtyping. Bioinformatics 2020, 36, 5539–5541. [Google Scholar] [CrossRef]
- Hoshida, Y. Nearest Template Prediction: A Single-Sample-Based Flexible Class Prediction with Confidence Assessment. PLoS ONE 2010, 5, e15543. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Brunet, J.-P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Subclass Mapping: Identifying Common Subtypes in Independent Disease Data Sets. PLoS ONE 2007, 2, e1195. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, C.; Zhou, X.; Gao, Y.; Sui, X. Identification of Immune Subtypes of Esophageal Adenocarcinoma to Predict Prognosis and Immunotherapy Response. Pharmaceuticals 2022, 15, 605. https://doi.org/10.3390/ph15050605
Ling C, Zhou X, Gao Y, Sui X. Identification of Immune Subtypes of Esophageal Adenocarcinoma to Predict Prognosis and Immunotherapy Response. Pharmaceuticals. 2022; 15(5):605. https://doi.org/10.3390/ph15050605
Chicago/Turabian StyleLing, Chen, Xiuman Zhou, Yanfeng Gao, and Xinghua Sui. 2022. "Identification of Immune Subtypes of Esophageal Adenocarcinoma to Predict Prognosis and Immunotherapy Response" Pharmaceuticals 15, no. 5: 605. https://doi.org/10.3390/ph15050605
APA StyleLing, C., Zhou, X., Gao, Y., & Sui, X. (2022). Identification of Immune Subtypes of Esophageal Adenocarcinoma to Predict Prognosis and Immunotherapy Response. Pharmaceuticals, 15(5), 605. https://doi.org/10.3390/ph15050605