
Computational Investigations of Traditional Chinese Medicinal Compounds against the Omicron Variant of SARS-CoV-2 to Rescue the Host Immune System

 , and
, and
Abstract
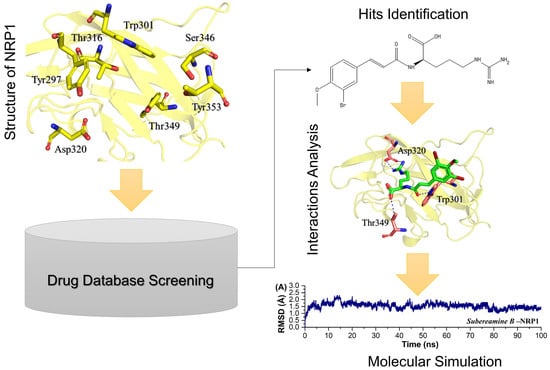
:
1. Introduction
2. Results and Discussion
2.1. Macrodomain of Omicron (B.1.1.529) and Structural Modelling
2.2. Virtual Screening and Re-Docking of TCM
2.3. Dynamic Stability Analysis of the Top Compounds
2.4. Structural Compactness Analysis
2.5. Residues Flexibility Profiling
2.6. Hydrogen Bonding Analysis
2.7. Binding Free Energy Estimation
3. Material and Methods
3.1. Modelling of the Macrodomain-I (Mac-I) of B.1.1.529 Variant
3.2. Virtual Screening of Traditional Chinese Medicine Database
3.3. Molecular Dynamics Simulation (MDS)
3.4. Trajectories Analysis Using CPPTRAJ and PTRAJ
3.5. Estimation of Post-Simulation Binding Energy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V.J.N.R.M. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D.J.T.L. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–954. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Duong, D. Alpha, Beta, Delta, Gamma: What’s important to know about SARS-CoV-2 variants of concern? Can. Med. Assoc. 2021, 193, E1059–E1060. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B. 1.1. 7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Han, P.; Su, C.; Zhang, Y.; Bai, C.; Zheng, A.; Qiao, C.; Wang, Q.; Niu, S.; Chen, Q.; Zhang, Y. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6103. [Google Scholar] [CrossRef]
- Lauring, A.S.; Malani, P.N. Variants of SARS-CoV-2. JAMA 2021, 326, 880. [Google Scholar] [CrossRef]
- Khan, A.; Waris, H.; Rafique, M.; Suleman, M.; Mohammad, A.; Ali, S.S.; Khan, T.; Waheed, Y.; Liao, C.; Wei, D.Q. The Omicron (B.1.1.529) variant of SARS-CoV-2 binds to the hACE2 receptor more strongly and escapes the antibody response: Insights from structural and simulation data. Int. J. Biol. Macromol. 2022, 200, 438–448. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S. SARS-CoV-2 B. 1.617. 2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Messali, S.; Bertelli, A.; Campisi, G.; Zani, A.; Ciccozzi, M.; Caruso, A.; Caccuri, F. A cluster of the new SARS-CoV-2 B. 1.621 lineage in Italy and sensitivity of the viral isolate to the BNT162b2 vaccine. J. Med. Virol. 2021, 93, 6468–6470. [Google Scholar] [CrossRef]
- Romero, P.E.; Dávila-Barclay, A.; Salvatierra, G.; González, L.; Cuicapuza, D.; Solís, L.; Marcos-Carbajal, P.; Huancachoque, J.; Maturrano, L.; Tsukayama, P. The emergence of SARS-CoV-2 variant lambda (C. 37) in South America. Microbiol. Spectr. 2021, 9, e00789-21. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Khan, M.T.; Saleem, S.; Junaid, M.; Ali, A.; Ali, S.S.; Khan, M.; Wei, D.-Q. Structural Insights into the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput. Struct. Biotechnol. J. 2020, 18, 2174–2184. [Google Scholar] [CrossRef]
- He, X.; Hong, W.; Pan, X.; Lu, G.; Wei, X. SARS-CoV-2 Omicron variant: Characteristics and prevention. MedComm 2021, 2, 838–845. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Wilhelm, A.; Widera, M.; Grikscheit, K.; Toptan, T.; Schenk, B.; Pallas, C.; Metzler, M.; Kohmer, N.; Hoehl, S.; Helfritz, F.A. Reduced neutralization of SARS-CoV-2 Omicron variant by vaccine sera and monoclonal antibodies. MedRxiv 2021. [Google Scholar] [CrossRef]
- Roessler, A.; Riepler, L.; Bante, D.; von Laer, D.; Kimpel, J. SARS-CoV-2 B. 1.1. 529 variant (Omicron) evades neutralization by sera from vaccinated and convalescent individuals. MedRxiv 2021. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2020, 39, 4659–4670. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Pervaiz, N.; Khan, A.; Saleem, S.; Shireen, H.; Wei, D.-Q.; Labrie, V.; Bao, Y.; Abbasi, A.A. Evolutionary and structural analysis of SARS-CoV-2 specific evasion of host immunity. Genes Immun. 2020, 21, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Brosey, C.A.; Houl, J.H.; Katsonis, P.; Balapiti-Modarage, L.P.F.; Bommagani, S.; Arvai, A.; Moiani, D.; Bacolla, A.; Link, T.; Warden, L.S.; et al. Targeting SARS-CoV-2 Nsp3 macrodomain structure with insights from human poly(ADP-ribose) glycohydrolase (PARG) structures with inhibitors. Prog. Biophys. Mol. Biol. 2021, 163, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Chang, S.-C.; Chiu, Y.-C.; Jiang, B.-C.; Wu, T.-H.; Hsu, C.-H. Structural, biophysical, and biochemical elucidation of the SARS-CoV-2 nonstructural protein 3 macro domain. ACS Infect. Dis. 2020, 6, 2970–2978. [Google Scholar] [CrossRef]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef] [Green Version]
- Hoch, N.C. Host ADP-ribosylation and the SARS-CoV-2 macrodomain. Biochem. Soc. Trans. 2021, 49, 1711–1721. [Google Scholar] [CrossRef]
- Molaei, S.; Dadkhah, M.; Asghariazar, V.; Karami, C.; Safarzadeh, E. The immune response and immune evasion characteristics in SARS-CoV, MERS-CoV, and SARS-CoV-2: Vaccine design strategies. Int. Immunopharmacol. 2021, 92, 107051. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Hosen, M.A.; Ahmad, S.; ul Qamar, M.T.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M. Synthesis, antimicrobial, anticancer activities, PASS prediction, molecular docking, molecular dynamics and pharmacokinetic studies of designed methyl α-D-glucopyranoside esters. J. Mol. Struct. 2022, 1260, 132761. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Mirza, M.U.; Song, J.-M.; Rao, M.J.; Zhu, X.; Chen, L.-L. Probing the structural basis of Citrus phytochrome B using computational modelling and molecular dynamics simulation approaches. J. Mol. Liq. 2021, 340, 116895. [Google Scholar] [CrossRef]
- Ahmad, F.; Albutti, A.; Tariq, M.H.; Din, G.; Tahir ul Qamar, M.; Ahmad, S. Discovery of Potential Antiviral Compounds against Hendra Virus by Targeting Its Receptor-Binding Protein (G) Using Computational Approaches. Molecules 2022, 27, 554. [Google Scholar] [CrossRef]
- Abbas, K.; Khan, T.; Ali, S.; Aftab, S.; Wang, Y.; Qiankun, W.; Khan, M. SARS-CoV-2 new variants: Characteristic features and impact on the efficacy of different vaccines. Biomed. Pharmacother. 2021, 143, 112176. [Google Scholar]
- Magrane, M. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Veloso, W.N.P.; Myung, Y.; Rodrigues, C.H.M.; Silk, M.; Rezende, P.M.; Silva, F.; Xavier, J.S.; Velloso, J.P.L.; da Silveira, C.H.; et al. EasyVS: A user-friendly web-based tool for molecule library selection and structure-based virtual screening. Bioinformatics 2020, 36, 4200–4202. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Muneer, I.; Ahmad, S.; Naz, A.; Abbasi, S.W.; Alblihy, A.; Aloliqi, A.A.; Alkhayl, F.F.; Alrumaihi, F.; Ahmad, S.; El Bakri, Y. Discovery of Novel Inhibitors from Medicinal Plants for V-Domain Ig Suppressor of T-Cell Activation (VISTA). Front. Mol. Biosci. 2021, 8, 716735. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- ul Qamar, M.T.; Ahmad, S.; Khan, A.; Mirza, M.U.; Ahmad, S.; Abro, A.; Chen, L.-L.; Almatroudi, A.; Wei, D.-Q. Structural probing of HapR to identify potent phytochemicals to control Vibrio cholera through integrated computational approaches. Comput. Biol. Med. 2021, 138, 104929. [Google Scholar] [CrossRef]
- Altharawi, A.; Ahmad, S.; Alamri, M.A.; ul Qamar, M.T. Structural insight into the binding pattern and interaction mechanism of chemotherapeutic agents with Sorcin by docking and molecular dynamic simulation. Colloids Surf. B Biointerfaces 2021, 208, 112098. [Google Scholar] [CrossRef] [PubMed]
- Suleman, M.; ul Qamar, M.T.; Shoaib Saleem, S.A.; Ali, S.S.; Khan, H.; Akbar, F.; Khan, W.; Alblihy, A.; Alrumaihi, F.; Waseem, M. Mutational landscape of Pirin and elucidation of the impact of most detrimental missense variants that accelerate the breast cancer pathways: A computational modelling study. Front. Mol. Biosci. 2021, 8, 692835. [Google Scholar] [CrossRef] [PubMed]
- Arif, R.; Ahmad, S.; Mustafa, G.; Mahrosh, H.S.; Ali, M.; Tahir ul Qamar, M.; Dar, H.R. Molecular Docking and Simulation Studies of Antidiabetic Agents Devised from Hypoglycemic Polypeptide-P of Momordica charantia. BioMed Res. Int. 2021, 2021, 5561129. [Google Scholar] [CrossRef]
- Mehmood, I.; Ijaz, M.; Ahmad, S.; Ahmed, T.; Bari, A.; Abro, A.; Allemailem, K.S.; Almatroudi, A.; Tahir ul Qamar, M. SARS-CoV-2: An update on genomics, risk assessment, potential therapeutics and vaccine development. Int. J. Environ. Res. Public Health 2021, 18, 1626. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; ul Qamar, M.T.; Mirza, M.U.; Alqahtani, S.M.; Froeyen, M.; Chen, L.-L. Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches. J. Pharm. Anal. 2020, 10, 546–559. [Google Scholar] [CrossRef]
- Muhseen, Z.T.; Hameed, A.R.; Al-Hasani, H.M.; ul Qamar, M.T.; Li, G. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (RBD) attachment inhibitors to the human ACE2 receptor: Integrated computational approach. J. Mol. Liq. 2020, 320, 114493. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Tahir Ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2021, 39, 4936–4948. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Li, X.; Fu, X. The macro domain protein family: Structure, functions, and their potential therapeutic implications. Mutat. Res./Rev. Mutat. Res. 2011, 727, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; McPherson, R.L.; Griffin, D.E. Macrodomain ADP-ribosylhydrolase and the pathogenesis of infectious diseases. PLoS Pathog. 2018, 14, e1006864. [Google Scholar] [CrossRef]
- Schuller, A.P.; Wu, C.C.-C.; Dever, T.E.; Buskirk, A.R.; Green, R. eIF5A functions globally in translation elongation and termination. Mol. Cell 2017, 66, 194–205.e5. [Google Scholar] [CrossRef] [Green Version]
- Babar, Z.; Khan, M.; Zahra, M.; Anwar, M.; Noor, K.; Hashmi, H.F.; Suleman, M.; Waseem, M.; Shah, A.; Ali, S. Drug similarity and structure-based screening of medicinal compounds to target macrodomain-I from SARS-CoV-2 to rescue the host immune system: A molecular dynamics study. J. Biomol. Struct. Dyn. 2020, 40, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, A.; Raki, M.; Ranki, T.; Särkioja, M.; Koponen, J.; Desmond, R.A.; Helin, A.; Stenman, U.H.; Isoniemi, H.; Höckerstedt, K.; et al. Chlorpromazine and apigenin reduce adenovirus replication and decrease replication associated toxicity. J. Gene Med. 2007, 9, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Shimu, M.S.S.; Mahmud, S.; Tallei, T.E.; Sami, S.A.; Adam, A.A.; Acharjee, U.K.; Paul, G.K.; Emran, T.B.; Zaman, S.; Uddin, M.S.; et al. Phytochemical Compound Screening to Identify Novel Small Molecules against Dengue Virus: A Docking and Dynamics Study. Molecules 2022, 27, 653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-J.; Li, W.; Li, H.-Y.; Wang, Y.-L.; Yun, T.; Song, Z.-P.; Song, Y.; Zhao, X.-W. In vivo and in vitro antiviral activity of five Tibetan medicinal plant extracts against herpes simplex virus type 2 infection. Pharm. Biol. 2009, 47, 598–607. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCM Database ID | Compound Names | 2D Structures | Docking Scores |
|---|---|---|---|
| TCM42798 | Mucic_acid_1-methyl_ester_2-O-gallate |  | −13.70 |
| TCM47007 | (2S)-5,7,2’,5’-Tetrahydroxyflavanone_7-O- -D-glucuronopyranoside |  | −13.25 |
| TCM30675 | (5S,6S,7S,8R)-5,6,7,8-Tetrahydroxy-2-[2-(3-hydroxy-4-methoxyphenyl)ethyl]-5,6,7,8-tetrahydro-4H-chromen-4-one |  | −12.49 |
| TCM27763 | 30389 |  | −11.93 |
| TCM33425 | Apigenin-bioside |  | −11.72 |
| TCM28788 | 31943 |  | −11.46 |
| TCM42159 | (-)-5’-Methoxyisolariciresinol-2-O-D-xylopyranoside_(D2) |  | −11.45 |
| TCM47184 | Tibeticanol |  | −11.36 |
| TCM31603 | 36132 |  | −11.04 |
| TCM31784 | 36381 |  | −11.02 |
| MM/GBSA | TCM42798 | TCM47007 | TCM30675 |
|---|---|---|---|
| vdW | −84.26 | −59.79 | −53.24 |
| electrostatic | −12.22 | −13.22 | −15.66 |
| ESURF | 17.45 | 14.68 | 12.25 |
| EGB | 9.25 | 8.22 | 9.01 |
| ∆G Bind | −69.78 | −50.11 | −47.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naman, Z.T.; Kadhim, S.; Al-Isawi, Z.J.K.; Butch, C.J.; Muhseen, Z.T. Computational Investigations of Traditional Chinese Medicinal Compounds against the Omicron Variant of SARS-CoV-2 to Rescue the Host Immune System. Pharmaceuticals 2022, 15, 741. https://doi.org/10.3390/ph15060741
Naman ZT, Kadhim S, Al-Isawi ZJK, Butch CJ, Muhseen ZT. Computational Investigations of Traditional Chinese Medicinal Compounds against the Omicron Variant of SARS-CoV-2 to Rescue the Host Immune System. Pharmaceuticals. 2022; 15(6):741. https://doi.org/10.3390/ph15060741
Chicago/Turabian StyleNaman, Ziad Tareq, Salim Kadhim, Zahraa J. K. Al-Isawi, Christopher J. Butch, and Ziyad Tariq Muhseen. 2022. "Computational Investigations of Traditional Chinese Medicinal Compounds against the Omicron Variant of SARS-CoV-2 to Rescue the Host Immune System" Pharmaceuticals 15, no. 6: 741. https://doi.org/10.3390/ph15060741
APA StyleNaman, Z. T., Kadhim, S., Al-Isawi, Z. J. K., Butch, C. J., & Muhseen, Z. T. (2022). Computational Investigations of Traditional Chinese Medicinal Compounds against the Omicron Variant of SARS-CoV-2 to Rescue the Host Immune System. Pharmaceuticals, 15(6), 741. https://doi.org/10.3390/ph15060741