
Metabolic Action of Metformin


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
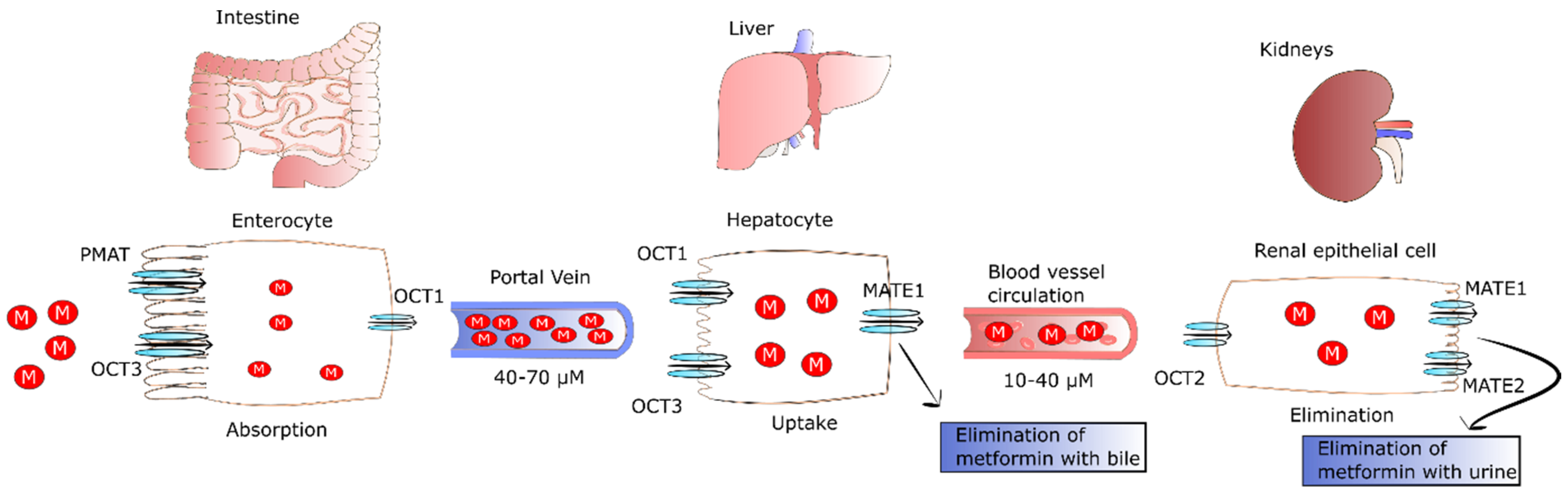
2. The Fate of Metformin in the Human Body
3. Dose-Dependent and Tissue-Specific Effect of Metformin on Cellular Respiration
3.1. Metformin Inhibits Hepatic Glucose Production (Gluconeogenesis)
3.2. The Molecular Background of Gluconeogenesis Inhibition Exerted by Metformin
3.2.1. AMPK-Dependent
3.2.2. AMPK-Independent
3.3. How Does Metformin Act in Muscles, Adipose Tissue and Intestines?
3.3.1. The Effect of Metformin Action in Intestine
3.3.2. Muscle and Adipose Tissue
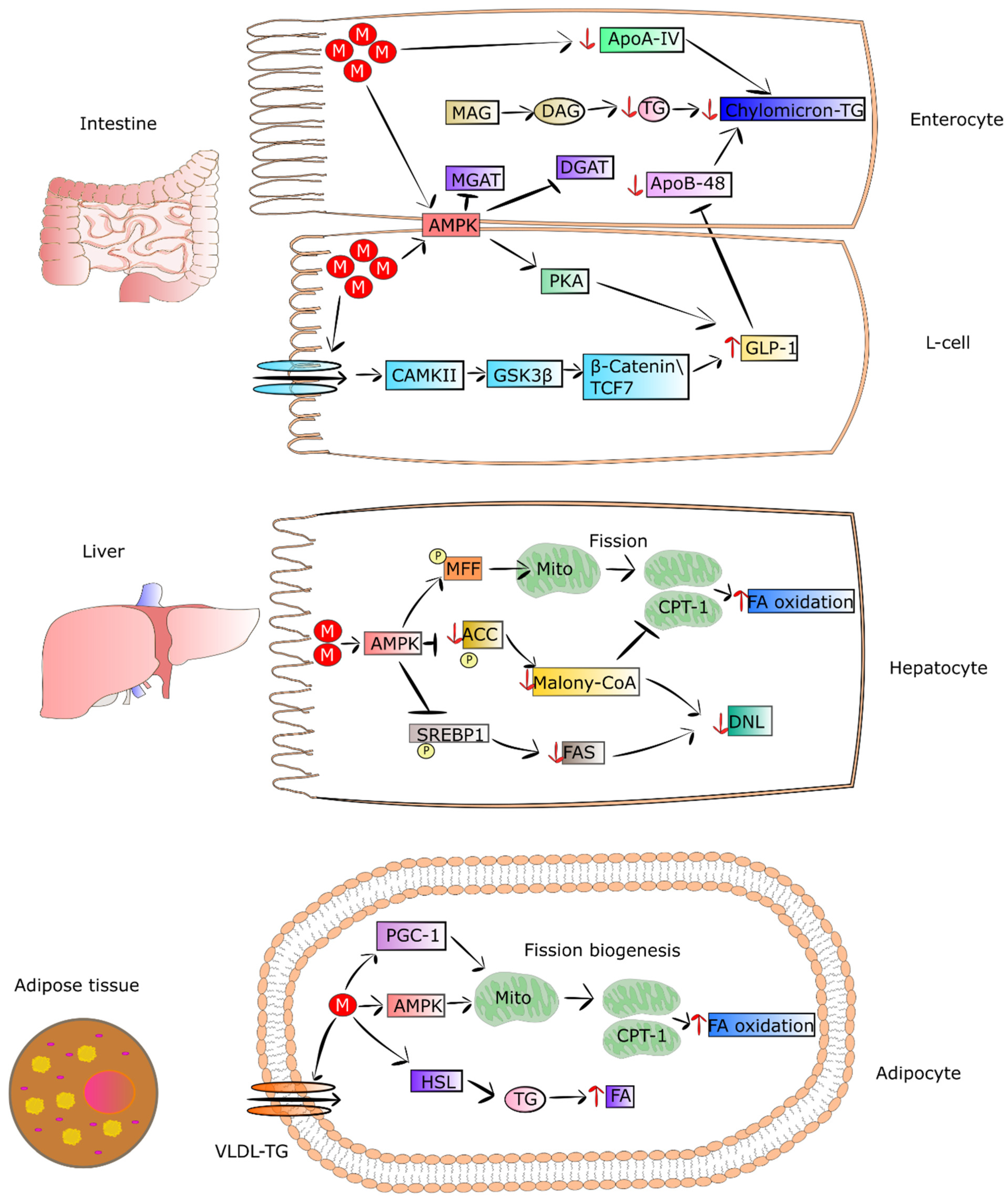
4. Metformin Regulates Lipid Metabolism
4.1. Metformin Decreases the Secretion of Lipids from Intestinal Epithelial Cells
4.2. Metformin as an Enhancer of Oxidation of Fatty Acids in Adipose Tissue and Muscles
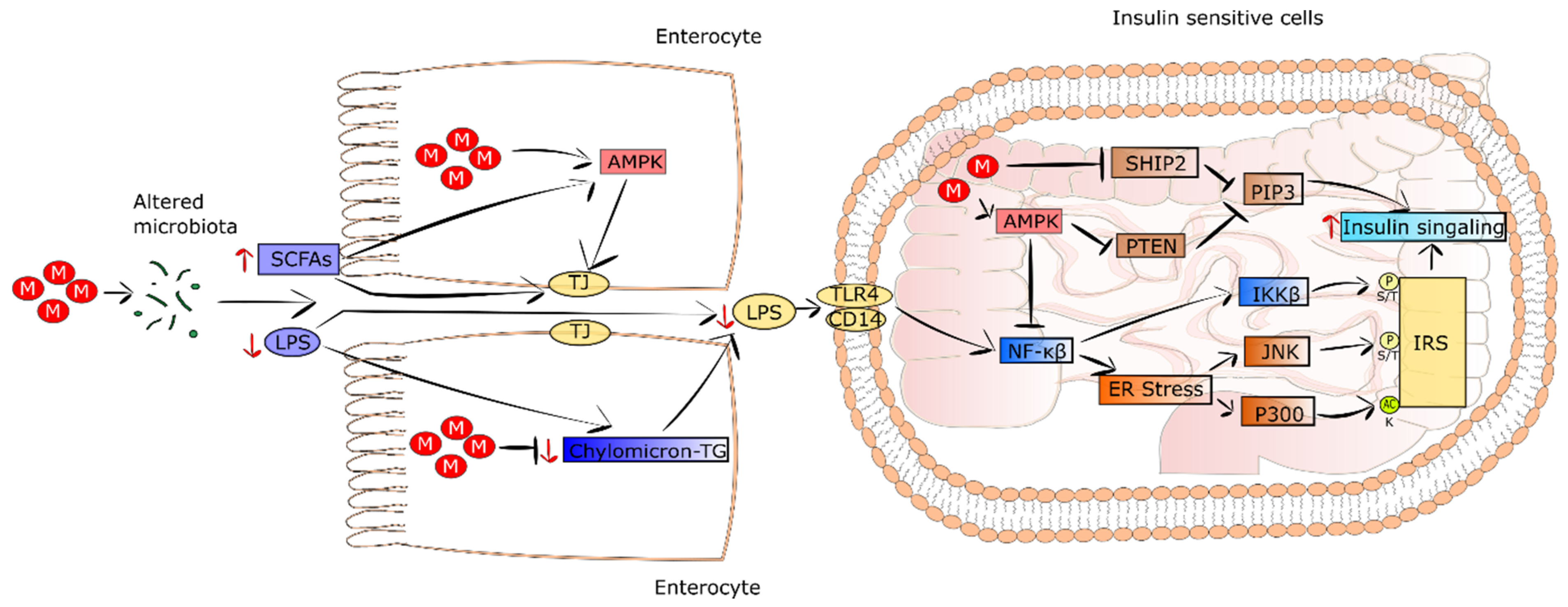
5. Other Metformin-Mediated Mechanisms Leading to Improvement of Systemic Insulin Sensitivity
5.1. Metformin Changes the Composition of Gut Microbiome and Maintains Intestinal Barrier Integrity
5.2. Metformin Decreases Systemic Low-Grade Inflammation
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S98–S110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheen, A.J.; Paquot, N. Metformin Revisited: A Critical Review of the Benefit-Risk Balance in at-Risk Patients with Type 2 Diabetes. Diabetes Metab. 2013, 39, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin Resistance: Review of the Underlying Molecular Mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- King, P.; Peacock, I.; Donnelly, R. The UK Prospective Diabetes Study (UKPDS): Clinical and Therapeutic Implications for Type 2 Diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643–648. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Goodman, A.M.; The Multicenter Metformin Study Group. Efficacy of Metformin in Patients with Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1995, 333, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Shirasaka, Y.; Seki, M.; Hatakeyama, M.; Kurokawa, Y.; Uchiyama, H.; Takemura, M.; Yasugi, Y.; Kishimoto, H.; Tamai, I.; Wang, J.; et al. Multiple Transport Mechanisms Involved in the Intestinal Absorption of Metformin: Impact on the Nonlinear Absorption Kinetics. J. Pharm. Sci. 2022, 111, 1531–1541. [Google Scholar] [CrossRef]
- Zhou, M.; Xia, L.; Wang, J. Metformin Transport by a Newly Cloned Proton-Stimulated Organic Cation Transporter (Plasma Membrane Monoamine Transporter) Expressed in Human Intestine. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.C.; Liang, X.; Yee, S.W.; Geier, E.G.; Stocker, S.L.; Chen, L.; Giacomini, K.M. Targeted Disruption of Organic Cation Transporter 3 Attenuates the Pharmacologic Response to Metformin. Mol. Pharmacol. 2015, 88, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.H.; Koepsell, H.; Brandsch, M. Drug Specificity and Intestinal Membrane Localization of Human Organic Cation Transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860. [Google Scholar] [CrossRef]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin Pathways: Pharmacokinetics and Pharmacodynamics. Pharmacogenet. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.-E.; Hong, S.-S.; Choi, M.-K.; Maeng, H.-J.; Kim, D.-D.; Chung, S.-J.; Shim, C.-K. Reduced Antidiabetic Effect of Metformin and Down-Regulation of Hepatic Oct1 in Rats with Ethynylestradiol-Induced Cholestasis. Pharm. Res. 2009, 26, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Brown, C.; Castro, R.A.; Shi, R.J.; Lin, E.T.; Owen, R.P.; Sheardown, S.A.; Yue, L.; Burchard, E.G.; Brett, C.M.; et al. Effect of Genetic Variation in the Organic Cation Transporter 1, OCT1, on Metformin Pharmacokinetics. Clin. Pharmacol. Ther. 2008, 83, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K.-I. Metformin Is a Superior Substrate for Renal Organic Cation Transporter OCT2 Rather than Hepatic OCT1. Drug Metab. Pharmacokinet. 2005, 20, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, C.; Bailey, C.J. Accumulation of Metformin by Tissues of the Normal and Diabetic Mouse. Xenobiotica Fate Foreign Compd. Biol. Syst. 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Shingaki, T.; Hume, W.E.; Takashima, T.; Katayama, Y.; Okauchi, T.; Hayashinaka, E.; Wada, Y.; Cui, Y.; Kusuhara, H.; Sugiyama, Y.; et al. Quantitative Evaluation of MMate1 Function Based on Minimally Invasive Measurement of Tissue Concentration Using PET with [(11)C]Metformin in Mouse. Pharm. Res. 2015, 32, 2538–2547. [Google Scholar] [CrossRef]
- Tucker, G.T.; Casey, C.; Phillips, P.J.; Connor, H.; Ward, J.D.; Woods, H.F. Metformin Kinetics in Healthy Subjects and in Patients with Diabetes Mellitus. Br. J. Clin. Pharmacol. 1981, 12, 235–246. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.-K.; Jin, Q.-R.; Jin, H.-E.; Shim, C.-K.; Cho, D.-Y.; Shin, J.-G.; Song, I.-S. Effects of Tetraalkylammonium Compounds with Different Affinities for Organic Cation Transporters on the Pharmacokinetics of Metformin. Biopharm. Drug Dispos. 2007, 28, 501–510. [Google Scholar] [CrossRef]
- Sato, T.; Masuda, S.; Yonezawa, A.; Tanihara, Y.; Katsura, T.; Inui, K.-I. Transcellular Transport of Organic Cations in Double-Transfected MDCK Cells Expressing Human Organic Cation Transporters HOCT1/HMATE1 and HOCT2/HMATE1. Biochem. Pharmacol. 2008, 76, 894–903. [Google Scholar] [CrossRef]
- Chen, Y.; Teranishi, K.; Li, S.; Yee, S.W.; Hesselson, S.; Stryke, D.; Johns, S.J.; Ferrin, T.E.; Kwok, P.; Giacomini, K.M. Genetic Variants in Multidrug and Toxic Compound Extrusion-1, HMATE1, Alter Transport Function. Pharm. J. 2009, 9, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K. Identification and Functional Characterization of a New Human Kidney-Specific H+/Organic Cation Antiporter, Kidney-Specific Multidrug and Toxin Extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, S.C.; Barr, J.; Arieff, A.I.; Pearl, R.G. Biguanide-Associated Lactic Acidosis. Case Report and Review of the Literature. Arch. Intern. Med. 1992, 152, 2333–2336. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; De Vivo, D. Pyruvate Carboxylase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Soeters, P.B.; Shenkin, A.; Sobotka, L.; Soeters, M.R.; de Leeuw, P.W.; Wolfe, R.R. The Anabolic Role of the Warburg, Cori-Cycle and Crabtree Effects in Health and Disease. Clin. Nutr. Edinb. Scotl. 2021, 40, 2988–2998. [Google Scholar] [CrossRef]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory Metabolism: Glycolysis, the TCA Cycle and Mitochondrial Electron Transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Rajasurya, V.; Anjum, H.; Surani, S. Metformin Use and Metformin-Associated Lactic Acidosis in Intensive Care Unit Patients with Diabetes. Cureus 2019, 11, e4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-Activated Protein Kinase Mediates Mitochondrial Fission in Response to Energy Stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, C.; Wyre, N.D.; Bailey, C.J. Subcellular Distribution of Metformin in Rat Liver. J. Pharm. Pharmacol. 1991, 43, 442–444. [Google Scholar] [CrossRef]
- Bailey, C.J.; Wilcock, C.; Scarpello, J.H.B. Metformin and the Intestine. Diabetologia 2008, 51, 1552–1553. [Google Scholar] [CrossRef] [Green Version]
- Schommers, P.; Thurau, A.; Bultmann-Mellin, I.; Guschlbauer, M.; Klatt, A.R.; Rozman, J.; Klingenspor, M.; de Angelis, M.H.; Alber, J.; Gründemann, D.; et al. Metformin Causes a Futile Intestinal-Hepatic Cycle Which Increases Energy Expenditure and Slows down Development of a Type 2 Diabetes-like State. Mol. Metab. 2017, 6, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Hollunger, G. Guanidines and Oxidative Phosphorylations. Acta Pharmacol. Toxicol. 1955, 11, 1–84. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence That Metformin Exerts Its Anti-Diabetic Effects through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Chappell, J.B. The Effect of Alkylguanidines on Mitochondrial Metabolism. J. Biol. Chem. 1963, 238, 410–417. [Google Scholar] [CrossRef]
- Davidoff, F. Effects of Guanidine Derivatives on Mitochondrial Function. I. Phenethylbiguanide Inhibition of Respiration in Mitochondria from Guinea Pig and Rat Tissues. J. Clin. Investig. 1968, 47, 2331–2343. [Google Scholar] [CrossRef]
- Evans, P.F.; King, L.J.; Parke, D.V.; Margetts, G.; Jones, W.E. The Mechanism of Action of Phenformin in Starved Rats. Biochem. Pharmacol. 1983, 32, 3459–3463. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-Induced Mitochondrial Dysfunction Yields Increased Lactate Production and Cytotoxicity of Aerobically-Poised HepG2 Cells and Human Hepatocytes In Vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e5. [Google Scholar] [CrossRef]
- Larsen, S.; Rabøl, R.; Hansen, C.N.; Madsbad, S.; Helge, J.W.; Dela, F. Metformin-Treated Patients with Type 2 Diabetes Have Normal Mitochondrial Complex I Respiration. Diabetologia 2012, 55, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Victor, V.M.; Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Castelló, R.; Falcón, R.; Gómez, M.; Rocha, M.; Hernández-Mijares, A. Effects of Metformin on Mitochondrial Function of Leukocytes from Polycystic Ovary Syndrome Patients with Insulin Resistance. Eur. J. Endocrinol. 2015, 173, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin Improves Healthspan and Lifespan in Mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Karise, I.; Bargut, T.C.; Del Sol, M.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Metformin Enhances Mitochondrial Biogenesis and Thermogenesis in Brown Adipocytes of Mice. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 111, 1156–1165. [Google Scholar] [CrossRef]
- Geerling, J.J.; Boon, M.R.; van der Zon, G.C.; van den Berg, S.A.A.; van den Hoek, A.M.; Lombès, M.; Princen, H.M.G.; Havekes, L.M.; Rensen, P.C.N.; Guigas, B. Metformin Lowers Plasma Triglycerides by Promoting VLDL-Triglyceride Clearance by Brown Adipose Tissue in Mice. Diabetes 2014, 63, 880–891. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides Suppress Hepatic Glucagon Signalling by Decreasing Production of Cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the Glucoregulatory Mechanisms of Metformin in Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [Green Version]
- Takashima, M.; Ogawa, W.; Hayashi, K.; Inoue, H.; Kinoshita, S.; Okamoto, Y.; Sakaue, H.; Wataoka, Y.; Emi, A.; Senga, Y.; et al. Role of KLF15 in Regulation of Hepatic Gluconeogenesis and Metformin Action. Diabetes 2010, 59, 1608–1615. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Andrikopoulos, S.; Filippis, C.; Thorburn, A.W.; Khan, D.; Proietto, J. Mechanism of Fat-Induced Hepatic Gluconeogenesis: Effect of Metformin. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E275–E282. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by Which Metformin Reduces Glucose Production in Type 2 Diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inzucchi, S.E.; Maggs, D.G.; Spollett, G.R.; Page, S.L.; Rife, F.S.; Walton, V.; Shulman, G.I. Efficacy and Metabolic Effects of Metformin and Troglitazone in Type II Diabetes Mellitus. N. Engl. J. Med. 1998, 338, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Consoli, A.; DeFronzo, R.A. Metabolic Effects of Metformin on Glucose and Lactate Metabolism in Noninsulin-Dependent Diabetes Mellitus. J. Clin. Endocrinol. Metab. 1996, 81, 4059–4067. [Google Scholar] [CrossRef] [Green Version]
- Gormsen, L.C.; Søndergaard, E.; Christensen, N.L.; Brøsen, K.; Jessen, N.; Nielsen, S. Metformin Increases Endogenous Glucose Production in Non-Diabetic Individuals and Individuals with Recent-Onset Type 2 Diabetes. Diabetologia 2019, 62, 1251–1256. [Google Scholar] [CrossRef] [Green Version]
- Konopka, A.R.; Esponda, R.R.; Robinson, M.M.; Johnson, M.L.; Carter, R.E.; Schiavon, M.; Cobelli, C.; Wondisford, F.E.; Lanza, I.R.; Nair, K.S. Hyperglucagonemia Mitigates the Effect of Metformin on Glucose Production in Prediabetes. Cell Rep. 2016, 15, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin Suppresses Gluconeogenesis by Inhibiting Mitochondrial Glycerophosphate Dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin Inhibits Gluconeogenesis via a Redox-Dependent Mechanism In Vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin Inhibits Hepatic Gluconeogenesis in Mice Independently of the LKB1/AMPK Pathway via a Decrease in Hepatic Energy State. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The Antidiabetic Drug Metformin Activates the AMP-Activated Protein Kinase Cascade via an Adenine Nucleotide-Independent Mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low Concentrations of Metformin Suppress Glucose Production in Hepatocytes through AMP-Activated Protein Kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin Reduces Liver Glucose Production by Inhibition of Fructose-1-6-Bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakhill, J.S.; Scott, J.W.; Kemp, B.E. AMPK Functions as an Adenylate Charge-Regulated Protein Kinase. Trends Endocrinol. Metab. 2012, 23, 125–132. [Google Scholar] [CrossRef]
- Savage, D.B.; Choi, C.S.; Samuel, V.T.; Liu, Z.-X.; Zhang, D.; Wang, A.; Zhang, X.-M.; Cline, G.W.; Yu, X.X.; Geisler, J.G.; et al. Reversal of Diet-Induced Hepatic Steatosis and Hepatic Insulin Resistance by Antisense Oligonucleotide Inhibitors of Acetyl-CoA Carboxylases 1 and 2. J. Clin. Investig. 2006, 116, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.-P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single Phosphorylation Sites in Acc1 and Acc2 Regulate Lipid Homeostasis and the Insulin-Sensitizing Effects of Metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.A.; Kim, K.H. Regulation of Hepatic Acetyl Coenzyme A Carboxylase by Phosphorylation and Dephosphorylation. J. Biol. Chem. 1973, 248, 378–380. [Google Scholar] [CrossRef]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A Common Bicyclic Protein Kinase Cascade Inactivates the Regulatory Enzymes of Fatty Acid and Cholesterol Biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine Palmitoyltransferase I. The Site of Inhibition of Hepatic Fatty Acid Oxidation by Malonyl-CoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar] [CrossRef]
- Koo, S.-H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB Coactivator TORC2 Is a Key Regulator of Fasting Glucose Metabolism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, I.; Lenzner, C.; Gourdon, L.; Vaulont, S.; Kahn, A.; Viollet, B. Hepatocyte Nuclear Factor-4α Involved in Type 1 Maturity-Onset Diabetes of the Young Is a Novel Target of AMP-Activated Protein Kinase. Diabetes 2001, 50, 1515–1521. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.-D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, S.; Cao, J.; He, Q.; Xiong, L.; Chang, E.; Radovick, S.; Wondisford, F.E.; He, L. Metformin Activates AMP-Activated Protein Kinase by Promoting Formation of the Aβγ Heterotrimeric Complex. J. Biol. Chem. 2015, 290, 3793–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.-W.; Wu, Y.-Q.; Lin, S.-Y.; Lin, S.-C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Alshawi, A.; Agius, L. Low Metformin Causes a More Oxidized Mitochondrial NADH/NAD Redox State in Hepatocytes and Inhibits Gluconeogenesis by a Redox-Independent Mechanism. J. Biol. Chem. 2019, 294, 2839–2853. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, E.B.; Vienberg, S.G.; Ørskov, C.; Hansen, H.S.; Andersen, B. Metformin Stimulates FGF21 Expression in Primary Hepatocytes. Exp. Diabetes Res. 2012, 2012, 465282. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I.; Lieber, C.S.; Beattie, D.S.; Rubin, E. Characterization of Shuttle Mechanisms for the Transport of Reducing Equivalents into Mitochondria. Arch. Biochem. Biophys. 1973, 158, 763–781. [Google Scholar] [CrossRef]
- LaNoue, K.F.; Williamson, J.R. Interrelationships between Malate-Aspartate Shuttle and Citric Acid Cycle in Rat Heart Mitochondria. Metabolism 1971, 20, 119–140. [Google Scholar] [CrossRef]
- Harding, J.W.; Pyeritz, E.A.; Copeland, E.S.; White, H.B. Role of Glycerol 3-Phosphate Dehydrogenase in Glyceride Metabolism. Effect of Diet on Enzyme Activities in Chicken Liver. Biochem. J. 1975, 146, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Radziuk, J.; Zhang, Z.; Wiernsperger, N.; Pye, S. Effects of Metformin on Lactate Uptake and Gluconeogenesis in the Perfused Rat Liver. Diabetes 1997, 46, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Calza, G.; Nyberg, E.; Mäkinen, M.; Soliymani, R.; Cascone, A.; Lindholm, D.; Barborini, E.; Baumann, M.; Lalowski, M.; Eriksson, O. Lactate-Induced Glucose Output Is Unchanged by Metformin at a Therapeutic Concentration—A Mass Spectrometry Imaging Study of the Perfused Rat Liver. Front. Pharmacol. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Magno, L.; Manni, S.; Di Pastena, F.; Coni, S.; Macone, A.; Cairoli, S.; Sambucci, M.; Infante, P.; Moretti, M.; Petroni, M.; et al. Phenformin Inhibits Hedgehog-Dependent Tumor Growth through a Complex I-Independent Redox/Corepressor Module. Cell Rep. 2020, 30, 1735–1752.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, S.; Daley, B.; Gaskins, K.; Vasko, V.V.; Boufraqech, M.; Patel, D.; Sourbier, C.; Reece, J.; Cheng, S.-Y.; Kebebew, E.; et al. Metformin Targets Mitochondrial Glycerophosphate Dehydrogenase to Control Rate of Oxidative Phosphorylation and Growth of Thyroid Cancer In Vitro and In Vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4030–4043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Chaudhari, K.; Shetty, R.; Winters, A.; Gao, X.; Hu, Z.; Ge, W.-P.; Sumien, N.; Forster, M.; Liu, R.; et al. Metformin Alters Locomotor and Cognitive Function and Brain Metabolism in Normoglycemic Mice. Aging Dis. 2019, 10, 949–963. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Ye, J.; Cai, Z.; Luo, Y.; Zhu, X.; Deng, Y.; Feng, Y.; Liang, Y.; Liu, R.; Han, Z.; et al. GPD1 Enhances the Anticancer Effects of Metformin by Synergistically Increasing Total Cellular Glycerol-3-Phosphate. Cancer Res. 2020, 80, 2150–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, D.W.; Bi, S.; Weintraub, B.D.; Reitman, M. Rat Mitochondrial Glycerol-3-Phosphate Dehydrogenase Gene: Multiple Promoters, High Levels in Brown Adipose Tissue, and Tissue-Specific Regulation by Thyroid Hormone. DNA Cell Biol. 1998, 17, 301–309. [Google Scholar] [CrossRef]
- Koza, R.A.; Kozak, U.C.; Brown, L.J.; Leiter, E.H.; MacDonald, M.J.; Kozak, L.P. Sequence and Tissue-Dependent RNA Expression of Mouse FAD-Linked Glycerol-3-Phosphate Dehydrogenase. Arch. Biochem. Biophys. 1996, 336, 97–104. [Google Scholar] [CrossRef]
- MacDonald, M.J. High Content of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Pancreatic Islets and Its Inhibition by Diazoxide. J. Biol. Chem. 1981, 256, 8287–8290. [Google Scholar] [CrossRef]
- Wang, D.-S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of Organic Cation Transporter 1 in Hepatic and Intestinal Distribution of Metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, S.; Busk, M.; Jensen, J.B.; Munk, O.L.; Zois, N.E.; Alstrup, A.K.O.; Jessen, N.; Frøkiær, J. A PET Tracer for Renal Organic Cation Transporters, 11C-Metformin: Radiosynthesis and Preclinical Proof-of-Concept Studies. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 615–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, A.B.; Horsman, M.R.; Jakobsen, S.; Jensen, J.B.; Garm, C.; Jessen, N.; Breining, P.; Frøkiær, J.; Busk, M. Results from 11C-Metformin-PET Scans, Tissue Analysis and Cellular Drug-Sensitivity Assays Questions the View That Biguanides Affects Tumor Respiration Directly. Sci. Rep. 2017, 7, 9436. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Clinical Pharmacokinetics of Metformin. Clin. Pharmacokinet. 1996, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.J.M.; Miller, J.J.J.; McCallum, C.; Rider, O.J.; Neubauer, S.; Heather, L.C.; Tyler, D.J. Assessment of Metformin-Induced Changes in Cardiac and Hepatic Redox State Using Hyperpolarized[1-13C]Pyruvate. Diabetes 2016, 65, 3544–3551. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Nielsen, P.M.; Schroeder, M.; Bertelsen, L.B.; Palm, F.; Laustsen, C. Acute Renal Metabolic Effect of Metformin Assessed with Hyperpolarised MRI in Rats. Diabetologia 2018, 61, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Rebrin, K.; Steil, G.M.; Mittelman, S.D.; Bergman, R.N. Causal Linkage between Insulin Suppression of Lipolysis and Suppression of Liver Glucose Output in Dogs. J. Clin. Investig. 1996, 98, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Puhakainen, I.; Koivisto, V.A.; Yki-Järvinen, H. Lipolysis and Gluconeogenesis from Glycerol Are Increased in Patients with Noninsulin-Dependent Diabetes Mellitus. J. Clin. Endocrinol. Metab. 1992, 75, 789–794. [Google Scholar] [CrossRef]
- Nurjhan, N.; Consoli, A.; Gerich, J. Increased Lipolysis and Its Consequences on Gluconeogenesis in Non-Insulin-Dependent Diabetes Mellitus. J. Clin. Investig. 1992, 89, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Lietz, T.; Winiarska, K.; Bryła, J. Ketone Bodies Activate Gluconeogenesis in Isolated Rabbit Renal Cortical Tubules Incubated in the Presence of Amino Acids and Glycerol. Acta Biochim. Pol. 1997, 44, 323–331. [Google Scholar] [CrossRef]
- Winiarska, K.; Bozko, P.; Lietz, T.; Bryła, J. Importance of Glutamate Dehydrogenase Stimulation for Glucose and Glutamine Synthesis in Rabbit Renal Tubules Incubated with Various Amino Acids. Acta Biochim. Pol. 1998, 45, 825–831. [Google Scholar] [CrossRef]
- Lietz, T.; Bryła, J. Glycerol and Lactate Induce Reciprocal Changes in Glucose Formation and Glutamine Production in Isolated Rabbit Kidney-Cortex Tubules Incubated with Aspartate. Arch. Biochem. Biophys. 1995, 321, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yoshida, Y.; Morita, A.; Mori, N.; Miura, S. Glycerol-3-Phosphate Dehydrogenase 1 Deficiency Induces Compensatory Amino Acid Metabolism during Fasting in Mice. Metabolism 2016, 65, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qu, H.; Xiong, X.; Wang, Y.; Liu, X.; Zhang, L.; Liao, X.; Liao, Q.; Sun, Z.; Ouyang, Q.; et al. Deficiency of Mitochondrial Glycerol 3-Phosphate Dehydrogenase Contributes to Hepatic Steatosis. Hepatol. Baltim. Md 2019, 70, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Barberà, A.; Gudayol, M.; Eto, K.; Corominola, H.; Maechler, P.; Miró, O.; Cardellach, F.; Gomis, R. A High Carbohydrate Diet Does Not Induce Hyperglycaemia in a Mitochondrial Glycerol-3-Phosphate Dehydrogenase-Deficient Mouse. Diabetologia 2003, 46, 1394–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Sevrioukova, I.F.; Denisov, I.G.; Zhang, X.; Chiu, T.-L.; Thomas, D.G.; Hanse, E.A.; Cuellar, R.A.D.; Grinkova, Y.V.; Langenfeld, V.W.; et al. Heme Binding Biguanides Target Cytochrome P450-Dependent Cancer Cell Mitochondria. Cell Chem. Biol. 2017, 24, 1259–1275.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Snyder, M.P. Metformin Affects Heme Function as a Possible Mechanism of Action. G3 Bethesda Md 2019, 9, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Repiščák, P.; Erhardt, S.; Rena, G.; Paterson, M.J. Biomolecular Mode of Action of Metformin in Relation to Its Copper Binding Properties. Biochemistry 2014, 53, 787–795. [Google Scholar] [CrossRef]
- Soty, M.; Penhoat, A.; Amigo-Correig, M.; Vinera, J.; Sardella, A.; Vullin-Bouilloux, F.; Zitoun, C.; Houberdon, I.; Mithieux, G. A Gut-Brain Neural Circuit Controlled by Intestinal Gluconeogenesis Is Crucial in Metabolic Health. Mol. Metab. 2015, 4, 106–117. [Google Scholar] [CrossRef]
- Bailey, C.J.; Mynett, K.J.; Page, T. Importance of the Intestine as a Site of Metformin-Stimulated Glucose Utilization. Br. J. Pharmacol. 1994, 112, 671–675. [Google Scholar] [CrossRef] [Green Version]
- Koffert, J.P.; Mikkola, K.; Virtanen, K.A.; Andersson, A.-M.D.; Faxius, L.; Hällsten, K.; Heglind, M.; Guiducci, L.; Pham, T.; Silvola, J.M.U.; et al. Metformin Treatment Significantly Enhances Intestinal Glucose Uptake in Patients with Type 2 Diabetes: Results from a Randomized Clinical Trial. Diabetes Res. Clin. Pract. 2017, 131, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, A.; Miller, S.; Nicholls, A.W.; Baker, D.; Van Horn, S.; Thomas, E.; Rajpal, D.; Spivak, A.; Brown, J.R.; Nunez, D.J. Novel Gut-Based Pharmacology of Metformin in Patients with Type 2 Diabetes Mellitus. PLoS ONE 2014, 9, e100778. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, E.; Tesi, F.; Bardini, G.; Ognibene, A.; Petracca, M.G.; Ciani, S.; Pezzatini, A.; Brogi, M.; Dicembrini, I.; Cremasco, F.; et al. Effects of Metformin on Glucagon-like Peptide-1 Levels in Obese Patients with and without Type 2 Diabetes. Diabetes Nutr. Metab. 2004, 17, 336–342. [Google Scholar]
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K.T. Metformin Activates a Duodenal Ampk-Dependent Pathway to Lower Hepatic Glucose Production in Rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; Jee, J.-H.; Park, S.; Lee, M.-S.; Kim, K.-W.; Lee, M.-K. Metformin Enhances Glucagon-like Peptide 1 via Cooperation between Insulin and Wnt Signaling. J. Endocrinol. 2014, 220, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Migoya, E.M.; Bergeron, R.; Miller, J.L.; Snyder, R.N.K.; Tanen, M.; Hilliard, D.; Weiss, B.; Larson, P.; Gutierrez, M.; Jiang, G.; et al. Dipeptidyl Peptidase-4 Inhibitors Administered in Combination with Metformin Result in an Additive Increase in the Plasma Concentration of Active GLP-1. Clin. Pharmacol. Ther. 2010, 88, 801–808. [Google Scholar] [CrossRef]
- Wu, T.; Thazhath, S.S.; Bound, M.J.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Mechanism of Increase in Plasma Intact GLP-1 by Metformin in Type 2 Diabetes: Stimulation of GLP-1 Secretion or Reduction in Plasma DPP-4 Activity? Diabetes Res. Clin. Pract. 2014, 106, e3–e6. [Google Scholar] [CrossRef]
- Gontier, E.; Fourme, E.; Wartski, M.; Blondet, C.; Bonardel, G.; Le Stanc, E.; Mantzarides, M.; Foehrenbach, H.; Pecking, A.-P.; Alberini, J.-L. High and Typical 18F-FDG Bowel Uptake in Patients Treated with Metformin. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 95–99. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling Type 2 Diabetes and Metformin Treatment Signatures in the Human Gut Microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin Alters the Gut Microbiome of Individuals with Treatment-Naive Type 2 Diabetes, Contributing to the Therapeutic Effects of the Drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the Gastrointestinal Tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Coll, A.P.; Chen, M.; Taskar, P.; Rimmington, D.; Patel, S.; Tadross, J.A.; Cimino, I.; Yang, M.; Welsh, P.; Virtue, S.; et al. GDF15 Mediates the Effects of Metformin on Body Weight and Energy Balance. Nature 2020, 578, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Natali, A.; Nesti, L.; Venturi, E.; Shore, A.C.; Khan, F.; Gooding, K.; Gates, P.E.; Looker, H.C.; Dove, F.; Goncalves, I.; et al. Metformin Is the Key Factor in Elevated Plasma Growth Differentiation Factor-15 Levels in Type 2 Diabetes: A Nested, Case-Control Study. Diabetes Obes. Metab. 2019, 21, 412–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, E.A.; Ford, R.J.; Smith, B.K.; Mohammadi-Shemirani, P.; Morrow, M.R.; Gutgesell, R.M.; Lu, R.; Raphenya, A.R.; Kabiri, M.; McArthur, A.G.; et al. Metformin-Induced Increases in GDF15 Are Important for Suppressing Appetite and Promoting Weight Loss. Nat. Metab. 2019, 1, 1202–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.; Morley, J.E. Metformin Decreases Food Consumption and Induces Weight Loss in Subjects with Obesity with Type II Non-Insulin-Dependent Diabetes. Obes. Res. 1998, 6, 47–53. [Google Scholar] [CrossRef]
- Kristensen, J.M.; Treebak, J.T.; Schjerling, P.; Goodyear, L.; Wojtaszewski, J.F.P. Two Weeks of Metformin Treatment Induces AMPK-Dependent Enhancement of Insulin-Stimulated Glucose Uptake in Mouse Soleus Muscle. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1099–E1109. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Hirshman, M.F.; Nygren, J.; Svanfeldt, M.; Bavenholm, P.; Rooyackers, O.; Zhou, G.; Williamson, J.M.; Ljunqvist, O.; Efendic, S.; et al. Metformin Increases AMP-Activated Protein Kinase Activity in Skeletal Muscle of Subjects with Type 2 Diabetes. Diabetes 2002, 51, 2074–2081. [Google Scholar] [CrossRef] [Green Version]
- Polianskyte-Prause, Z.; Tolvanen, T.A.; Lindfors, S.; Dumont, V.; Van, M.; Wang, H.; Dash, S.N.; Berg, M.; Naams, J.-B.; Hautala, L.C.; et al. Metformin Increases Glucose Uptake and Acts Renoprotectively by Reducing SHIP2 Activity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2858–2869. [Google Scholar] [CrossRef] [Green Version]
- Okamura, H.; Yoshida, K.; Sasaki, E.; Qiu, L.; Amorim, B.R.; Morimoto, H.; Haneji, T. Expression of PTEN and Akt Phosphorylation in Lipopolysaccharide-Treated NIH3T3 Cells. Cell Biol. Int. 2007, 31, 119–125. [Google Scholar] [CrossRef]
- Lee, J.O.; Lee, S.K.; Kim, J.H.; Kim, N.; You, G.Y.; Moon, J.W.; Kim, S.J.; Park, S.H.; Kim, H.S. Metformin Regulates Glucose Transporter 4 (GLUT4) Translocation through AMP-Activated Protein Kinase (AMPK)-Mediated Cbl/CAP Signaling in 3T3-L1 Preadipocyte Cells. J. Biol. Chem. 2012, 287, 44121–44129. [Google Scholar] [CrossRef] [Green Version]
- Grisouard, J.; Timper, K.; Radimerski, T.M.; Frey, D.M.; Peterli, R.; Kola, B.; Korbonits, M.; Herrmann, P.; Krähenbühl, S.; Zulewski, H.; et al. Mechanisms of Metformin Action on Glucose Transport and Metabolism in Human Adipocytes. Biochem. Pharmacol. 2010, 80, 1736–1745. [Google Scholar] [CrossRef]
- Fischer, M.; Timper, K.; Radimerski, T.; Dembinski, K.; Frey, D.M.; Zulewski, H.; Keller, U.; Müller, B.; Christ-Crain, M.; Grisouard, J. Metformin Induces Glucose Uptake in Human Preadipocyte-Derived Adipocytes from Various Fat Depots. Diabetes Obes. Metab. 2010, 12, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, K.A.; Hällsten, K.; Parkkola, R.; Janatuinen, T.; Lönnqvist, F.; Viljanen, T.; Rönnemaa, T.; Knuuti, J.; Huupponen, R.; Lönnroth, P.; et al. Differential Effects of Rosiglitazone and Metformin on Adipose Tissue Distribution and Glucose Uptake in Type 2 Diabetic Subjects. Diabetes 2003, 52, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeppesen, J.; Zhou, M.Y.; Chen, Y.D.; Reaven, G.M. Effect of Metformin on Postprandial Lipemia in Patients with Fairly to Poorly Controlled NIDDM. Diabetes Care 1994, 17, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Grosskopf, I.; Ringel, Y.; Charach, G.; Maharshak, N.; Mor, R.; Iaina, A.; Weintraub, M. Metformin Enhances Clearance of Chylomicrons and Chylomicron Remnants in Nondiabetic Mildly Overweight Glucose-Intolerant Subjects. Diabetes Care 1997, 20, 1598–1602. [Google Scholar] [CrossRef]
- Kim, E.K.; Lee, S.H.; Jhun, J.Y.; Byun, J.K.; Jeong, J.H.; Lee, S.-Y.; Kim, J.K.; Choi, J.Y.; Cho, M.-L. Metformin Prevents Fatty Liver and Improves Balance of White/Brown Adipose in an Obesity Mouse Model by Inducing FGF21. Mediators Inflamm. 2016, 2016, 5813030. [Google Scholar] [CrossRef]
- Wang, C.; Liu, F.; Yuan, Y.; Wu, J.; Wang, H.; Zhang, L.; Hu, P.; Li, Z.; Li, Q.; Ye, J. Metformin Suppresses Lipid Accumulation in Skeletal Muscle by Promoting Fatty Acid Oxidation. Clin. Lab. 2014, 60, 887–896. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.-R.; Lee, J.-C.; Lee, H.-Y.; Kim, M.-S.; Whon, T.W.; Lee, M.-S.; Bae, J.-W. An Increase in the Akkermansia Spp. Population Induced by Metformin Treatment Improves Glucose Homeostasis in Diet-Induced Obese Mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Hu, N. Effects of Metformin on the Gut Microbiota in Obesity and Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 5003–5014. [Google Scholar] [CrossRef] [PubMed]
- Scarpello, J.H.; Hodgson, E.; Howlett, H.C. Effect of Metformin on Bile Salt Circulation and Intestinal Motility in Type 2 Diabetes Mellitus. Diabet. Med. J. Br. Diabet. Assoc. 1998, 15, 651–656. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Pieters, H.-J.; Jonkers, D.M. Short-Chain Fatty Acids Activate AMP-Activated Protein Kinase and Ameliorate Ethanol-Induced Intestinal Barrier Dysfunction in Caco-2 Cell Monolayers. J. Nutr. 2013, 143, 1872–1881. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Peng, J.; An, H.; He, Q.; Boronina, T.; Guo, S.; White, M.F.; Cole, P.A.; He, L. Endotoxemia-Mediated Activation of Acetyltransferase P300 Impairs Insulin Signaling in Obesity. Nat. Commun. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin Inhibits Cytokine-Induced Nuclear Factor KappaB Activation via AMP-Activated Protein Kinase Activation in Vascular Endothelial Cells. Hypertens. Dallas Tex 1979 2006, 47, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin Inhibits Monocyte-to-Macrophage Differentiation via AMPK-Mediated Inhibition of STAT3 Activation: Potential Role in Atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymczak-Pajor, I.; Wenclewska, S.; Śliwińska, A. Metabolic Action of Metformin. Pharmaceuticals 2022, 15, 810. https://doi.org/10.3390/ph15070810
Szymczak-Pajor I, Wenclewska S, Śliwińska A. Metabolic Action of Metformin. Pharmaceuticals. 2022; 15(7):810. https://doi.org/10.3390/ph15070810
Chicago/Turabian StyleSzymczak-Pajor, Izabela, Sylwia Wenclewska, and Agnieszka Śliwińska. 2022. "Metabolic Action of Metformin" Pharmaceuticals 15, no. 7: 810. https://doi.org/10.3390/ph15070810
APA StyleSzymczak-Pajor, I., Wenclewska, S., & Śliwińska, A. (2022). Metabolic Action of Metformin. Pharmaceuticals, 15(7), 810. https://doi.org/10.3390/ph15070810