

GCase Enhancers: A Potential Therapeutic Option for Gaucher Disease and Other Neurological Disorders

 ,
,  ,
,  and
and 
Abstract
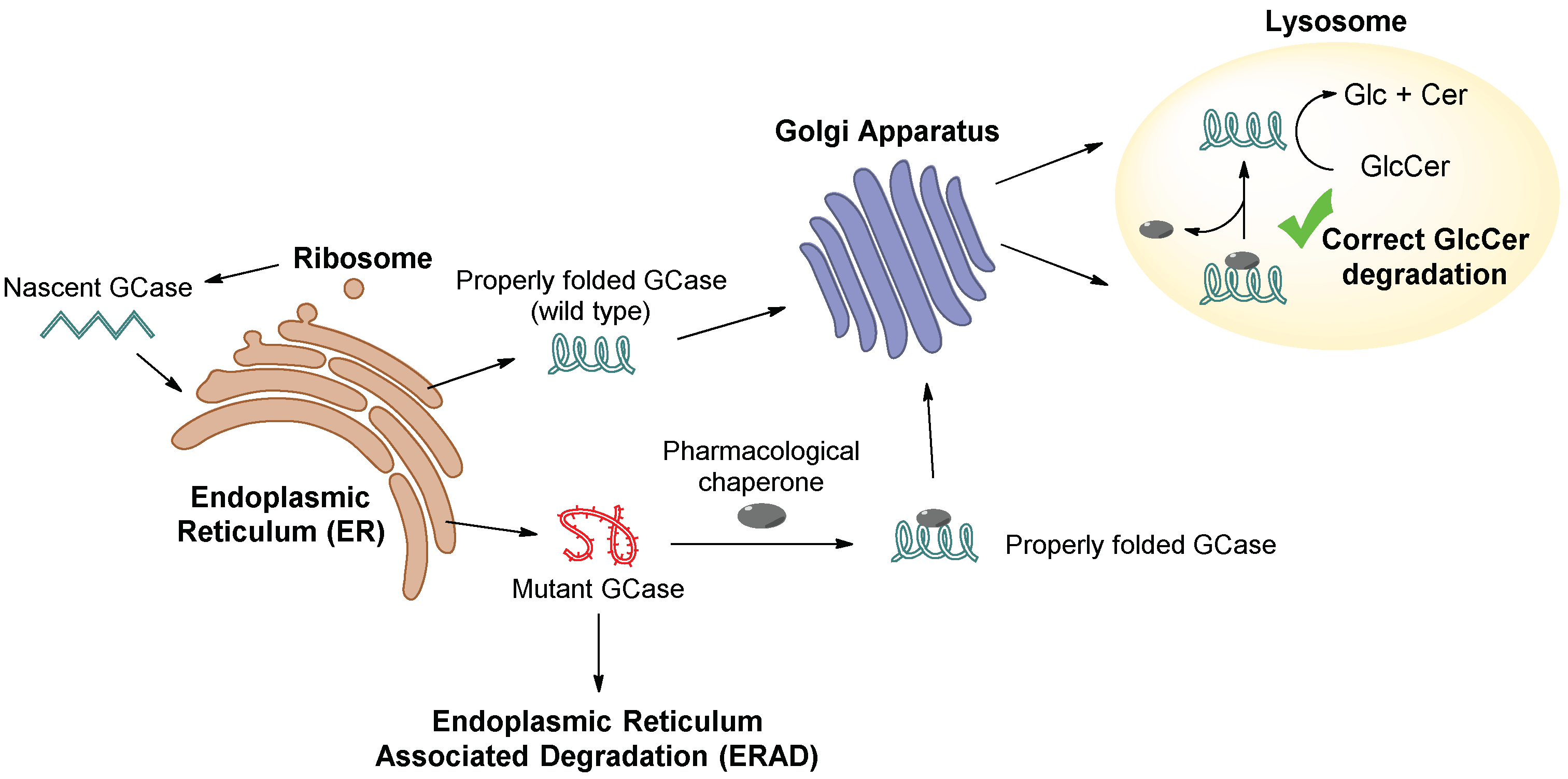
:1. Introduction
2. Inhibitory Chaperones
2.1. Iminosugars
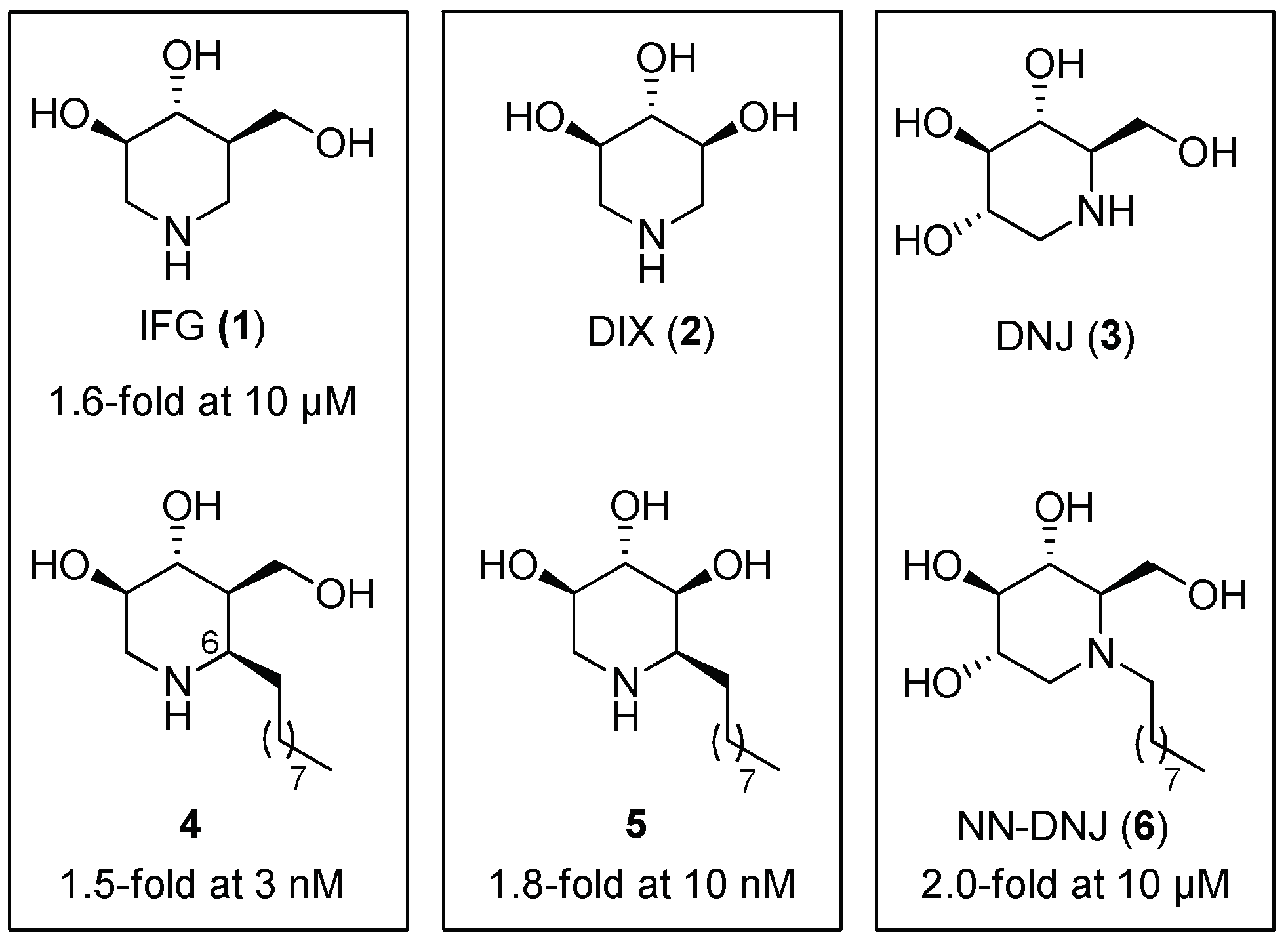
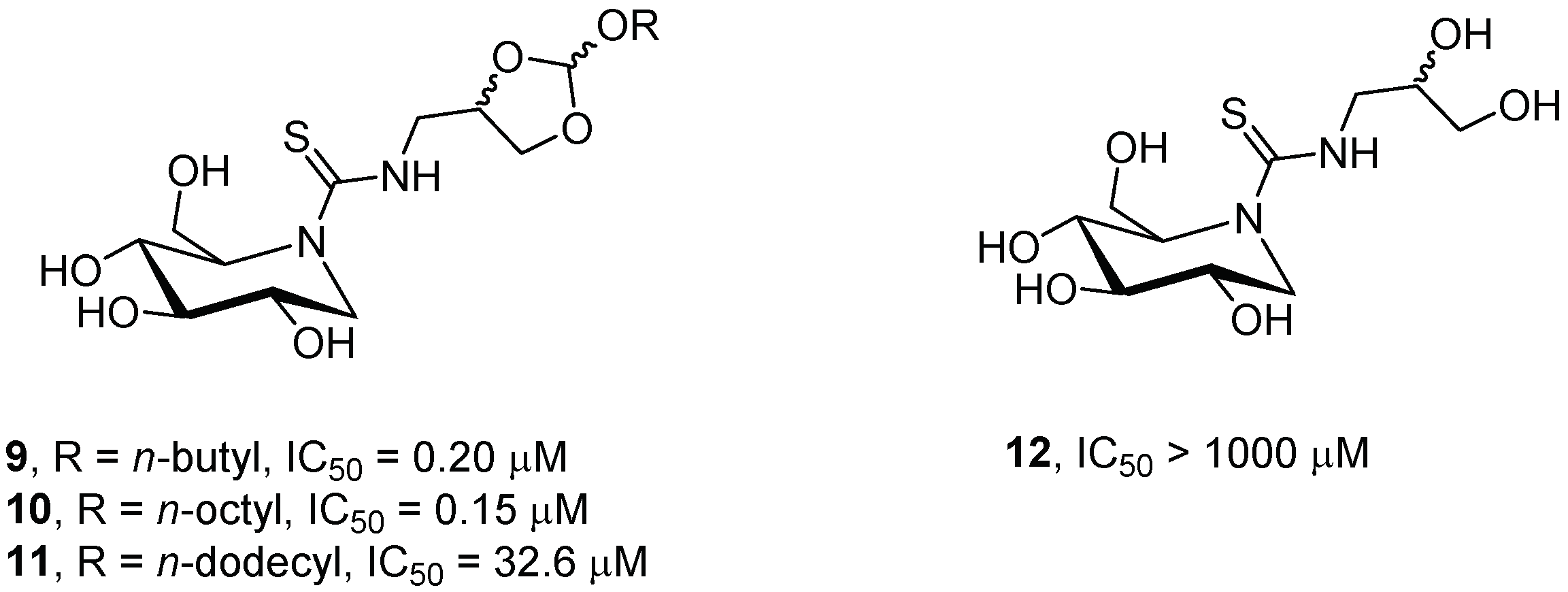
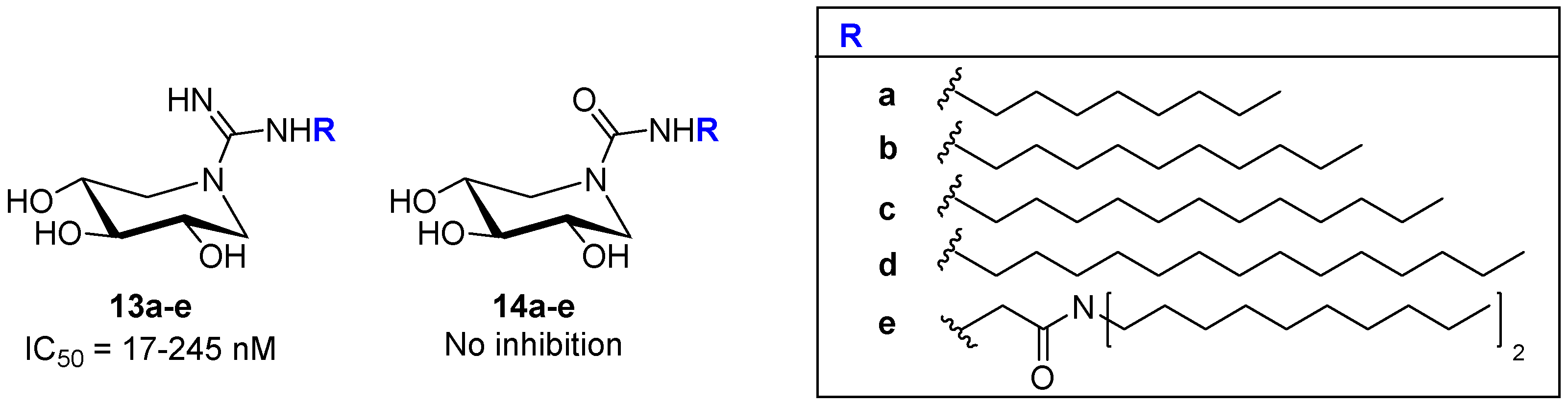
2.1.1. Six-Membered Iminosugars
2.1.2. Five-Membered Iminosugars
2.1.3. Bicyclic Iminosugars
2.1.4. Multivalent Iminosugars
2.2. Sugar Analogues
2.3. Miscellaneous Examples
3. Allosteric Enhancers
4. Studies in Parkinson’s Animal Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Valentão, P.; Andrade, P.B. Tuning protein folding in lysosomal storage diseases: The chemistry behind pharmacological chaperones. Chem. Sci. 2018, 9, 1740–1752. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, G.A. Advances in Gaucher Disease: Basic and Clinical Perspectives; Future Medicine Ltd.: London, UK, 2013. [Google Scholar]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanz, J.; Saftig, P. Parkinson’s disease: Acid-glucocerebrosidase activity and alpha-synuclein clearance. J. Neurochem. 2016, 139 (Suppl. 1), 198–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.-H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Sawkar, A.R.; Kelly, J.W. Pharmacologic chaperoning as a strategy to treat Gaucher disease. FEBS J. 2007, 274, 4944–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009, 1, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Benito, J.M.; García Fernández, J.M.; Ortiz Mellet, C. Pharmacological chaperone therapy for Gaucher disease: A patent review. Expert Opin. Ther. Pat. 2011, 21, 885–903. [Google Scholar] [CrossRef] [Green Version]
- Trapero, A.; Llebaria, A. Glucocerebrosidase inhibitors for the treatment of Gaucher disease. Future Med. Chem. 2013, 5, 573–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, G.; Pignata, C.; Vajro, P.; Salerno, M. New strategies for the treatment of lysosomal storage diseases (Review). Int. J. Mol. Med. 2013, 31, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aymami, J.; Barril, X.; Rodríguez-Pascau, L.; Martinell, M. Pharmacological chaperones for enzyme enhancement therapy in genetic diseases. Pharm. Pat. Anal. 2013, 2, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Moracci, M.; Fecarotta, S.; Andria, G. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med. Chem. 2014, 6, 1031–1045. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; García Fernández, J.M.; Ortiz Mellet, C. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from Gaucher, GM1-gangliosidosis and Fabry diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.L.; Génisson, Y.; Ballereau, S.; Dehoux, C. Second-Generation Pharmacological Chaperones: Beyond Inhibitors. Molecules 2020, 25, 3145. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Kelly, J.W. Small molecule protein binding to correct cellular folding or stabilize the native state against misfolding and aggregation. Curr. Opin. Struct. Biol. 2022, 72, 267–278. [Google Scholar] [CrossRef]
- Compain, P.; Martin, O.R. Iminosugars: From Synthesis to Therapeutic Applications; Wiley VCH: New York, NY, USA, 2007. [Google Scholar]
- Kuriyama, C.; Kamiyama, O.; Ikeda, K.; Sanae, F.; Kato, A.; Adachi, I.; Imahori, T.; Takahata, H.; Okamoto, T.; Asano, N. In vitro inhibition of glycogen-degrading enzymes and glycosidases by six-membered sugar mimics and their evaluation in cell cultures. Bioorg. Med. Chem. 2008, 16, 7330–7336. [Google Scholar] [CrossRef]
- Chang, H.-H.; Asano, N.; Ishii, S.; Ichikawa, Y.; Fan, J.-Q. Hydrophilic iminosugar active-site-specific chaperones increase residual glucocerebrosidase activity in fibroblasts from Gaucher patients. FEBS J. 2006, 273, 4082–4092. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Sheth, K.A.; Li, S.; Chang, H.-H.; Fan, J.-Q. Rational Design and Synthesis of Highly Potent β-Glucocerebrosidase Inhibitors. Angew. Chem. Int. Ed. 2005, 44, 7450–7453. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.-Q.; Zhu, X.; Sheth, K. Hydroxy Piperidine Derivatives to Treat Gaucher Disease. U.S. Patent 7,741,340, 22 June 2010. [Google Scholar]
- Compain, P.; Martin, O.R.; Boucheron, C.; Godin, G.; Yu, L.; Ikeda, K.; Asano, N. Design and Synthesis of Highly Potent and Selective Pharmacological Chaperones for the Treatment of Gaucher’s disease. ChemBiochem 2006, 7, 1356–1359. [Google Scholar] [CrossRef]
- Sawkar, A.R.; Cheng, W.-C.; Beutler, E.; Wong, C.-H.; Balch, W.E.; Kelly, J.W. Chemical chaperones increase the cellular activity of N370S β-glucosidase: A therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15428–15433. [Google Scholar] [CrossRef] [Green Version]
- Brumshtein, B.; Greenblatt, H.M.; Butters, T.D.; Shaaltiel, Y.; Aviezer, D.; Silman, I.; Futerman, A.H.; Sussman, J.L. Crystal Structures of Complexes of N-Butyl- and N-Nonyl-Deoxynojirimycin Bound to Acid β-Glucosidase: Insights into the mechanism of chemical chaperone action in Gaucher disease. J. Biol. Chem. 2007, 282, 29052–29058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmeggiani, C.; Catarzi, S.; Matassini, C.; D’Adamio, G.; Morrone, A.; Goti, A.; Paoli, P.; Cardona, F. Human Acid β-Glucosidase Inhibition by Carbohydrate Derived Iminosugars: Towards New Pharmacological Chaperones for Gaucher Disease. ChemBiochem 2015, 16, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.; Higaki, K.; Aguilar-Moncayo, M.; Ninomiya, H.; Ohno, K.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Suzuki, Y. Chaperone Activity of Bicyclic Nojirimycin Analogues for Gaucher Mutations in Comparison with N-(n-nonyl)-Deoxynojirimycin. ChemBiochem 2009, 10, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
- Mena-Barragán, T.; Narita, A.; Matias, D.; Tiscornia, G.; Nanba, E.; Ohno, K.; Suzuki, Y.; Higaki, K.; Garcia Fernández, J.M.; Ortiz Mellet, C. pH-Responsive Pharmacological Chaperones for Rescuing Mutant Glycosidases. Angew. Chem. Int. Ed. 2015, 54, 11696–11700. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03950050 (accessed on 13 May 2022).
- Sevšek, A.; Šrot, L.; Rihter, J.; Čelan, M.; van Ufford, L.Q.; Moret, E.E.; Martin, N.I.; Pieters, R.J. N-Guanidino Derivatives of 1,5-Dideoxy-1,5-imino-D-xylitol are Potent, Selective, and Stable Inhibitors of β-Glucocerebrosidase. ChemMedChem 2017, 12, 483–486. [Google Scholar] [CrossRef]
- Baudoin-Dehoux, C.; Castellan, T.; Rodriguez, F.; Rives, A.; Stauffert, F.; Garcia, V.; Levade, T.; Compain, P.; Génisson, Y. Selective Targeting of the Interconversion between Glucosylceramide and Ceramide by Scaffold Tailoring of Iminosugar Inhibitors. Molecules 2019, 24, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, F.; Matassini, C.; Goti, A.; Morrone, A.; Paoli, P.; Cardona, F. Stereoselective Synthesis of C-2 Alkylated Trihydroxypiperidines: Novel Pharmacological Chaperones for Gaucher Disease. ACS Med. Chem. Lett. 2019, 10, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Clemente, F.; Matassini, C.; Faggi, C.; Giachetti, S.; Cresti, C.; Morrone, A.; Paoli, P.; Goti, A.; Martínez-Bailén, M.; Cardona, F. Glucocerebrosidase (GCase) activity modulation by 2-alkyl trihydroxypiperidines: Inhibition and pharmacological chaperoning. Bioorg. Chem. 2020, 98, 103740. [Google Scholar] [CrossRef]
- Suzuki, K.; Nakahara, T.; Kanie, O. 3,4-Dihydroxypyrrolidine as Glycosidase Inhibitor. Curr. Top. Med. Chem. 2009, 9, 34–57. [Google Scholar] [CrossRef]
- Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R.J.; Fleet, G.W.; Natori, Y.; Watanabe, Y.; Imahori, T.; et al. Docking study and biological evaluation of pyrrolidine-based iminosugars as pharmacological chaperones for Gaucher disease. Org. Biomol. Chem. 2016, 14, 1039–1048. [Google Scholar] [CrossRef]
- Stocker, B.L.; Dangerfield, E.M.; Win-Mason, A.L.; Haslett, G.W.; Timmer, M.S.M. Recent Developments in the Synthesis of Pyrrolidine-Containing Iminosugars. Eur. J. Org. Chem. 2010, 2010, 1615–1637. [Google Scholar] [CrossRef]
- Castellan, T.; Garcia, V.; Rodriguez, F.; Fabing, I.; Shchukin, Y.; Tran, M.L.; Ballereau, S.; Levade, T.; Génisson, Y.; Dehoux, C. Concise asymmetric synthesis of new enantiomeric C-alkyl pyrrolidines acting as pharmacological chaperones against Gaucher disease. Org. Biomol. Chem. 2020, 18, 7852–7861. [Google Scholar] [CrossRef]
- Theorell, H.; Yonetani, T. Studies on Liver Alcohol Dehydrogenase Complexes. IV. Spectrophotometric Observations on the Enzyme Complexes. Arch. Biochem. Biophys. 1964, 106, 252–258. [Google Scholar] [CrossRef]
- Diot, J.D.; Garcia Moreno, I.; Twigg, G.; Ortiz Mellet, C.; Haupt, K.; Butters, T.D.; Kovensky, J.; Gouin, S.G. Amphiphilic 1-Deoxynojirimycin Derivatives through Click Strategies for Chemical Chaperoning in N370S Gaucher Cells. J. Org. Chem. 2011, 76, 7757–7768. [Google Scholar] [CrossRef]
- Martínez-Bailén, M.; Carmona, A.T.; Patterson-Orazem, A.C.; Lieberman, R.L.; Ide, D.; Kubo, M.; Kato, A.; Robina, I.; Moreno-Vargas, A.J. Exploring substituent diversity on pyrrolidine-aryltriazole iminosugars: Structural basis of β-glucocerebrosidase inhibition. Bioorg. Chem. 2019, 86, 652–664. [Google Scholar] [CrossRef]
- Kato, A.; Nakagome, I.; Nakagawa, S.; Koike, Y.; Nash, R.J.; Adachi, I.; Hirono, S. Docking and SAR studies of calystegines: Binding orientation and influence on pharmacological chaperone effects for Gaucher’s disease. Bioorg. Med. Chem. 2014, 22, 2435–2441. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; de la Mata, M.; Sánchez-Fernández, E.M.; Benito, J.M.; Díaz-Quintana, A.; Fustero, S.; Nanba, E.; Higaki, K.; Sánchez-Alcázar, J.A.; García Fernández, J.M.; et al. Fluorinated Chaperone-β-Cyclodextrin Formulations for β-Glucocerebrosidase Activity Enhancement in Neuronopathic Gaucher Disease. J. Med. Chem. 2017, 60, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Bassetto, M.; Ferla, S.; Pertusati, F. Polyfluorinated groups in medicinal chemistry. Future Med. Chem. 2015, 7, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Palchevskyy, S.S.; Posokhov, Y.O.; Olivier, B.; Popot, J.-L.; Pucci, B.; Ladokhin, A.S. Chaperoning of Insertion of Membrane Proteins into Lipid Bilayers by Hemifluorinated Surfactants: Application to Diphtheria Toxin. Biochemistry 2006, 45, 2629–2635. [Google Scholar] [CrossRef]
- Robertson, J.; Stevens, K. Pyrrolizidine alkaloids: Occurrence, biology, and chemical synthesis. Nat. Prod. Rep. 2017, 34, 62–89. [Google Scholar] [CrossRef]
- Mena-Barragán, T.; García-Moreno, M.I.; Nanba, E.; Higaki, K.; Concia, A.L.; Clapés, P.; García Fernández, J.M.; Ortiz Mellet, C. Inhibitor versus chaperone behaviour of D-fagomine, DAB and LAB sp2-iminosugar conjugates against glycosidases: A structure-activity relationship study in Gaucher fibroblasts. Eur. J. Med. Chem. 2016, 121, 880–891. [Google Scholar] [CrossRef]
- Mena-Barragán, T.; García-Moreno, M.I.; Sevšek, A.; Okazaki, T.; Nanba, E.; Higaki, K.; Martin, N.I.; Pieters, R.J.; García Fernández, J.M.; Ortiz Mellet, C. Probing the Inhibitor versus Chaperone Properties of sp2-Iminosugars towards Human β-Glucocerebrosidase: A Picomolar Chaperone for Gaucher Disease. Molecules 2018, 23, 927. [Google Scholar] [CrossRef] [Green Version]
- Sevšek, A.; Čelan, M.; Erjavec, B.; Quarles van Ufford, L.; Sastre Toraño, J.; Moret, E.E.; Pieters, R.J.; Martin, N.I. Bicyclic isoureas derived from 1-deoxynojirimycin are potent inhibitors of β-glucocerebrosidase. Org. Biomol. Chem. 2016, 14, 8670–8673. [Google Scholar] [CrossRef]
- Rodríguez-Lavado, J.; de la Mata, M.; Jiménez-Blanco, J.L.; García-Moreno, M.I.; Benito, J.M.; Díaz-Quintana, A.; Sánchez-Alcázar, J.A.; Higaki, K.; Nanba, E.; Ohno, K.; et al. Targeted delivery of pharmacological chaperones for Gaucher disease to macrophages by a mannosylated cyclodextrin carrier. Org. Biomol. Chem. 2014, 12, 2289–2301. [Google Scholar] [CrossRef]
- Compain, P.; Bodlenner, A. The Multivalent Effect in Glycosidase Inhibition: A New, Rapidly Emerging Topic in Glycoscience. ChemBiochem 2014, 15, 1239–1251. [Google Scholar] [CrossRef]
- Gouin, S.G. Multivalent Inhibitors for Carbohydrate-Processing Enzymes: Beyond the “Lock-and-Key” Concept. Chem. Eur. J. 2014, 20, 11616–11628. [Google Scholar] [CrossRef]
- Zelli, R.; Longevial, J.-F.; Dumy, P.; Marra, A. Synthesis and biological properties of multivalent iminosugars. New J. Chem. 2015, 39, 5050–5074. [Google Scholar] [CrossRef]
- Matassini, C.; Parmeggiani, C.; Cardona, F.; Goti, A. Are enzymes sensitive to the multivalent effect? Emerging evidence with glycosidases. Tetrahedron Lett. 2016, 57, 5407–5415. [Google Scholar] [CrossRef]
- Compain, P. Multivalent Effect in Glycosidase Inhibition: The End of the Beginning. Chem. Rec. 2020, 20, 10–22. [Google Scholar] [CrossRef]
- González-Cuesta, M.; Ortiz Mellet, C.; García Fernández, J.M. Carbohydrate supramolecular chemistry: Beyond the multivalent effect. Chem. Commun. 2020, 56, 5207–5222. [Google Scholar] [CrossRef] [Green Version]
- Della Sala, P.; Vanni, C.; Talotta, C.; Di Marino, L.; Matassini, C.; Goti, A.; Neri, P.; Šesták, S.; Cardona, F.; Gaeta, C. Multivalent resorcinarene clusters decorated with DAB-1 inhitopes: Targeting Golgi α-mannosidase from Drosoph. Melanogaster Org. Chem. Front. 2021, 8, 6648–6656. [Google Scholar] [CrossRef]
- Vanni, C.; Bodlenner, A.; Marradi, M.; Schneider, J.P.; Ramirez, M.d.l.A.; Moya, S.; Goti, A.; Cardona, F.; Compain, P.; Matassini, C. Hybrid Multivalent Jack Bean α-Mannosidase Inhibitors: The First Example of Gold Nanoparticles Decorated with Deoxynojirimycin Inhitopes. Molecules 2021, 26, 5864. [Google Scholar] [CrossRef]
- Alvarez-Dorta, D.; King, D.T.; Legigan, T.; Ide, D.; Adachi, I.; Deniaud, D.; Désiré, J.; Kato, A.; Vocadlo, D.; Gouin, S.G.; et al. Multivalency To Inhibit and Discriminate Hexosaminidases. Chem. Eur. J. 2017, 23, 9022–9025. [Google Scholar] [CrossRef]
- Matassini, C.; Vanni, C.; Goti, A.; Morrone, A.; Marradi, M.; Cardona, F. Multimerization of DAB-1 onto Au GNPs affords new potent and selective N-acetylgalactosamine-6-sulfatase (GALNS) inhibitors. Org. Biomol. Chem. 2018, 16, 8604–8612. [Google Scholar] [CrossRef]
- D’Adamio, G.; Matassini, C.; Parmeggiani, C.; Catarzi, S.; Morrone, A.; Goti, A.; Paoli, P.; Cardona, F. Evidence for a multivalent effect in inhibition of sulfatases involved in lysosomal storage disorders (LSDs). RSC Adv. 2016, 6, 64847–64851. [Google Scholar] [CrossRef]
- Martínez-Bailén, M.; Carmona, A.T.; Cardona, F.; Matassini, C.; Goti, A.; Kubo, M.; Kato, A.; Robina, I.; Moreno-Vargas, A.J. Synthesis of multimeric pyrrolidine iminosugar inhibitors of human β-glucocerebrosidase and α-galactosidase A: First example of a multivalent enzyme activity enhancer for Fabry disease. Eur. J. Med. Chem. 2020, 192, 112173. [Google Scholar] [CrossRef]
- Decroocq, C.; Rodríguez-Lucena, D.; Ikeda, K.; Asano, N.; Compain, P. Cyclodextrin-Based Iminosugar Click Clusters: The First Examples of Multivalent Pharmacological Chaperones for the Treatment of Lysosomal Storage Disorders. ChemBiochem 2012, 13, 661–664. [Google Scholar] [CrossRef]
- Joosten, A.; Decroocq, C.; de Sousa, J.; Schneider, J.P.; Etamé, E.; Bodlenner, A.; Butters, T.D.; Compain, P. A Systematic Investigation of Iminosugar Click Clusters as Pharmacological Chaperones for the Treatment of Gaucher Disease. ChemBiochem 2014, 15, 309–319. [Google Scholar] [CrossRef]
- Laigre, E.; Hazelard, D.; Casas, J.; Serra-Vinardell, J.; Michelakakis, H.; Mavridou, I.; Aerts, J.M.F.G.; Delgado, A.; Compain, P. Investigation of original multivalent iminosugars as pharmacological chaperones for the treatment of Gaucher disease. Carbohydr. Res. 2016, 429, 98–104. [Google Scholar] [CrossRef]
- Jing, X.; Yu, F.; Chen, L. Visualization of nitroxyl (HNO) in vivo via a lysosome-targetable near-infrared fluorescent probe. Chem. Commun. 2014, 50, 14253–14256. [Google Scholar] [CrossRef]
- Stauffert, F.; Serra-Vinardell, J.; Gómez-Grau, M.; Michelakakis, H.; Mavridou, I.; Grinberg, D.; Vilageliu, L.; Casas, J.; Bodlenner, A.; Delgado, A.; et al. Stereodivergent synthesis of right- and left-handed iminoxylitol heterodimers and monomers. Study of their impact on β-glucocerebrosidase activity. Org. Biomol. Chem. 2017, 15, 3681–3705. [Google Scholar] [CrossRef]
- Vanni, C.; Clemente, F.; Paoli, P.; Morrone, A.; Matassini, C.; Goti, A.; Cardona, F. 3,4,5-Trihydroxypiperidine Based Multivalent Glucocerebrosidase (GCase) Enhancers. ChemBiochem 2022, 23, e202200077. [Google Scholar] [CrossRef]
- Castilla, J.; Rísquez, R.; Higaki, K.; Nanba, E.; Ohno, K.; Suzuki, Y.; Díaz, Y.; Ortiz Mellet, C.; García Fernández, J.M.; Castillón, S. Conformationally-locked N-glycosides: Exploiting long-range non-glycone interactions in the design of pharmacological chaperones for Gaucher disease. Eur. J. Med. Chem. 2015, 90, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Castilla, J.; Rísquez, R.; Cruz, D.; Higaki, K.; Nanba, E.; Ohno, K.; Suzuki, Y.; Díaz, Y.; Ortiz Mellet, C.; García Fernández, J.M.; et al. Conformationally-Locked N-Glycosides with Selective β-Glucosidase Inhibitory Activity: Identification of a New Non-Iminosugar-Type Pharmacological Chaperone for Gaucher Disease. J. Med. Chem. 2012, 55, 6857–6865. [Google Scholar] [CrossRef]
- Whitworth, G.E.; Macauley, M.S.; Stubbs, K.A.; Dennis, R.J.; Taylor, E.J.; Davies, G.J.; Greig, I.R.; Vocadlo, D.J. Analysis of PUGNAc and NAG-thiazoline as Transition State Analogues for Human O-GlcNAcase: Mechanistic and Structural Insights into Inhibitor Selectivity and Transition State Poise. J. Am. Chem. Soc. 2007, 129, 635–644. [Google Scholar] [CrossRef]
- Navo, C.D.; Corzana, F.; Sánchez-Fernández, E.M.; Busto, J.H.; Avenoza, A.; Zurbano, M.M.; Nanba, E.; Higaki, K.; Ortiz Mellet, C.; García Fernández, J.M.; et al. Conformationally-locked C-glycosides: Tuning aglycone interactions for optimal chaperone behaviour in Gaucher fibroblasts. Org. Biomol. Chem. 2016, 14, 1473–1484. [Google Scholar] [CrossRef]
- Aydillo, C.; Navo, C.D.; Busto, J.H.; Corzana, F.; Zurbano, M.M.; Avenoza, A.; Peregrina, J.M. A Double Diastereoselective Michael-Type Addition as an Entry to Conformationally Restricted Tn Antigen Mimics. J. Org. Chem. 2013, 78, 10968–10977. [Google Scholar] [CrossRef]
- Ben Bdira, F.; Kallemeijn, W.W.; Oussoren, S.V.; Scheij, S.; Bleijlevens, B.; Florea, B.I.; van Roomen, C.P.A.A.; Ottenhoff, R.; van Kooten, M.J.F.M.; Walvoort, M.T.C.; et al. Stabilization of Glucocerebrosidase by Active Site Occupancy. ACS Chem. Biol. 2017, 12, 1830–1841. [Google Scholar] [CrossRef] [Green Version]
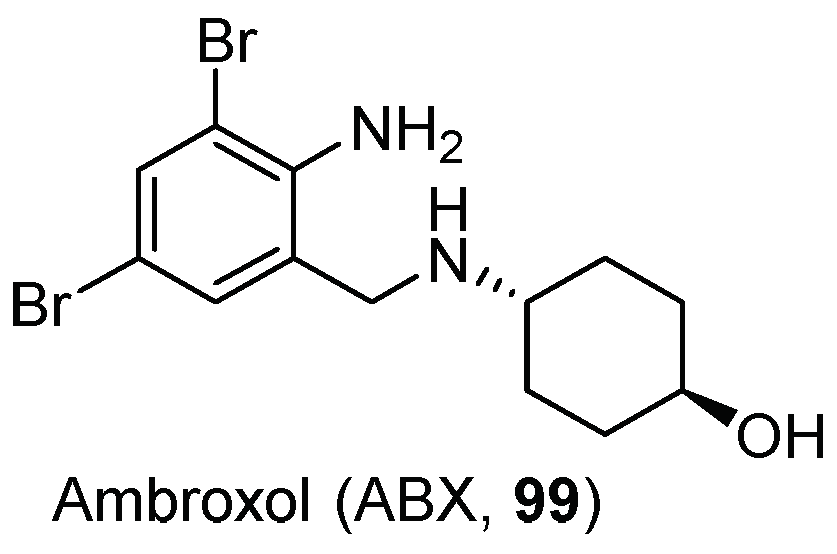
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sam, R.; Sharma, P.; Chen, L.; Do, J.; Sidransky, E. Glucocerebrosidase as a therapeutic target for Parkinson’s disease. Expert Opin. Ther. Targets 2020, 24, 287–294. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Luan, Z.; Li, L.; Higaki, K.; Nanba, E.; Suzuki, Y.; Ohno, K. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. 2013, 35, 317–322. [Google Scholar] [CrossRef]
- Zimran, A.; Altarescu, G.; Elstein, D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 134–137. [Google Scholar] [CrossRef]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [Green Version]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Han, T.-U.; Sam, R.; Sidransky, E. Small Molecule Chaperones for the Treatment of Gaucher Disease and GBA1-Associated Parkinson Disease. Front. Cell Dev. Biol. 2020, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Goldin, E.; Zheng, W.; Motabar, O.; Southall, N.; Choi, J.H.; Marugan, J.; Austin, C.P.; Sidransky, E. High Throughput Screening for Small Molecule Therapy for Gaucher Disease Using Patient Tissue as the Source of Mutant Glucocerebrosidase. PLoS ONE 2012, 7, e29861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Z. CNS-Accessible Pharmacological Chaperones for Treatment of Acid Beta-Glucosidase-Related Disease States. U.S. Patent 10,588,888, 17 March 2020. [Google Scholar]
- Zheng, J.; Chen, L.; Skinner, O.S.; Ysselstein, D.; Remis, J.; Lansbury, P.; Skerlj, R.; Mrosek, M.; Heunisch, U.; Krapp, S.; et al. β-Glucocerebrosidase Modulators Promote Dimerization of β-Glucocerebrosidase and Reveal an Allosteric Binding Site. J. Am. Chem. Soc. 2018, 140, 5914–5924. [Google Scholar] [CrossRef]
- Gruschus, J.M.; Jiang, Z.; Yap, T.L.; Hill, S.A.; Grishaev, A.; Piszczek, G.; Sidransky, E.; Lee, J.C. Dissociation of glucocerebrosidase dimer in solution by its co-factor, saposin C. Biochem. Biophys. Res. Commun. 2015, 457, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Benz, J.; Rufer, A.C.; Huber, S.; Ehler, A.; Hug, M.; Topp, A.; Guba, W.; Hofmann, E.C.; Jagasia, R.; Rodríguez Sarmiento, R.M. Novel β-Glucocerebrosidase Activators That Bind to a New Pocket at a Dimer Interface and Induce Dimerization. Angew. Chem. Int. Ed. 2021, 60, 5436–5442. [Google Scholar] [CrossRef]
- García Collazo, A.M.; Barril Alonso, X.; Cubero Jordà, E.; Revés Vilaplana, M.; Roberts, R.S. Heteroaryl Compounds and Their Use. WO Patent 2018122775, 5 July 2018. [Google Scholar]
- de Lau, L.M.L.; Breteler, M.M.B. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- García-Sanz, P.; Aerts, J.M.F.G.; Moratalla, R. The Role of Cholesterol in α-Synuclein and Lewy Body Pathology in GBA1 Parkinson’s Disease. Mov. Disord. 2021, 36, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Westbroek, W.; Sidransky, E. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron 2017, 93, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.-Y.; Whitworth, A.J.; Schapira, A.H.V. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef] [Green Version]
- Migdalska-Richards, A.; Daly, L.; Bezard, E.; Schapira, A.H.V. Ambroxol Effects in Glucocerebrosidase and α-Synuclein Transgenic Mice. Ann. Neurol. 2016, 80, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Richter, F.; Fleming, S.M.; Watson, M.; Lemesre, V.; Pellegrino, L.; Ranes, B.; Zhu, C.; Mortazavi, F.; Mulligan, C.K.; Sioshansi, P.C.; et al. A GCase Chaperone Improves Motor Function in a Mouse Model of Synucleinopathy. Neurotherapeutics 2014, 11, 840–856. [Google Scholar] [CrossRef] [Green Version]
- Migdalska-Richards, A.; Ko, W.K.D.; Li, Q.; Bezard, E.; Schapira, A.H.V. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 2017, 71, e21967. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med. 2019, 11, eaau6870. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
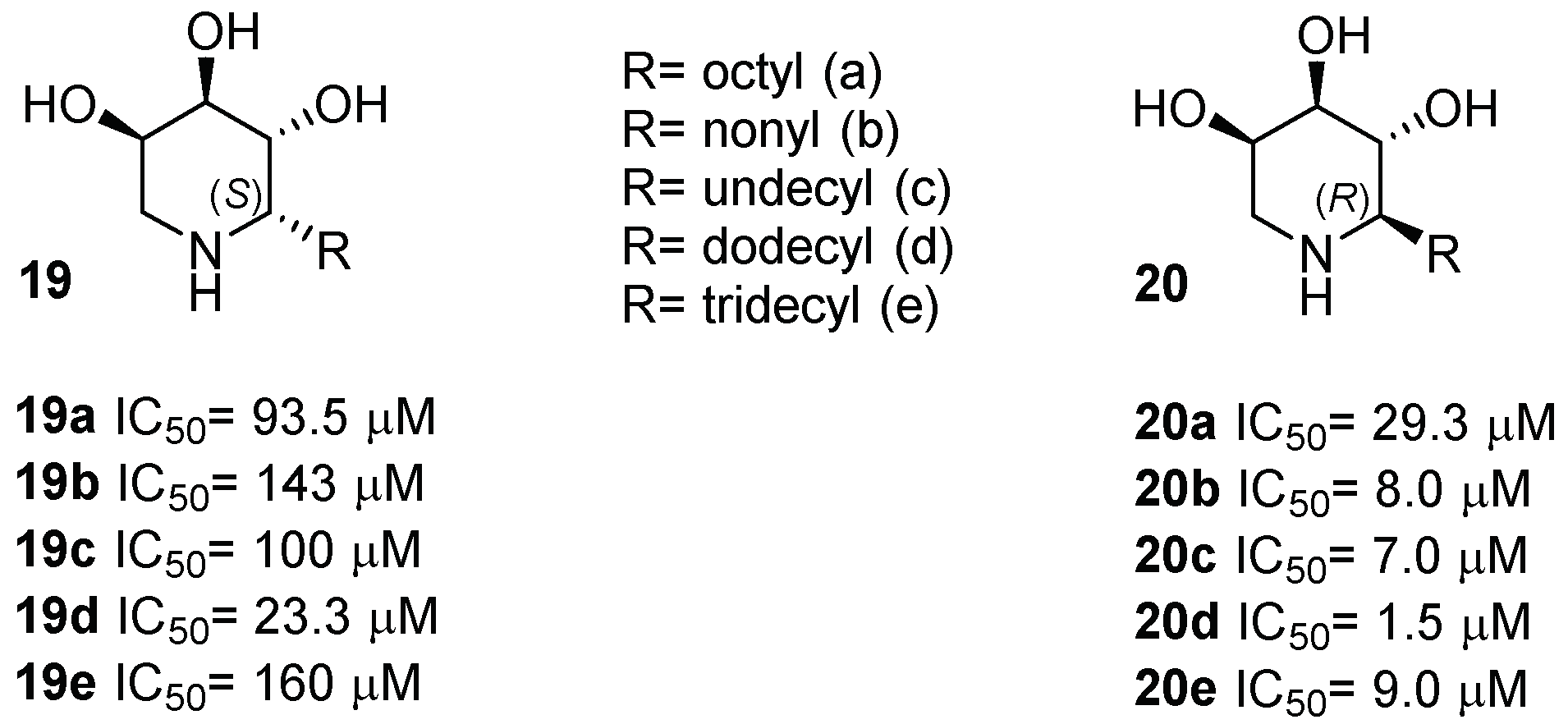{kind=link}
{kind=link}
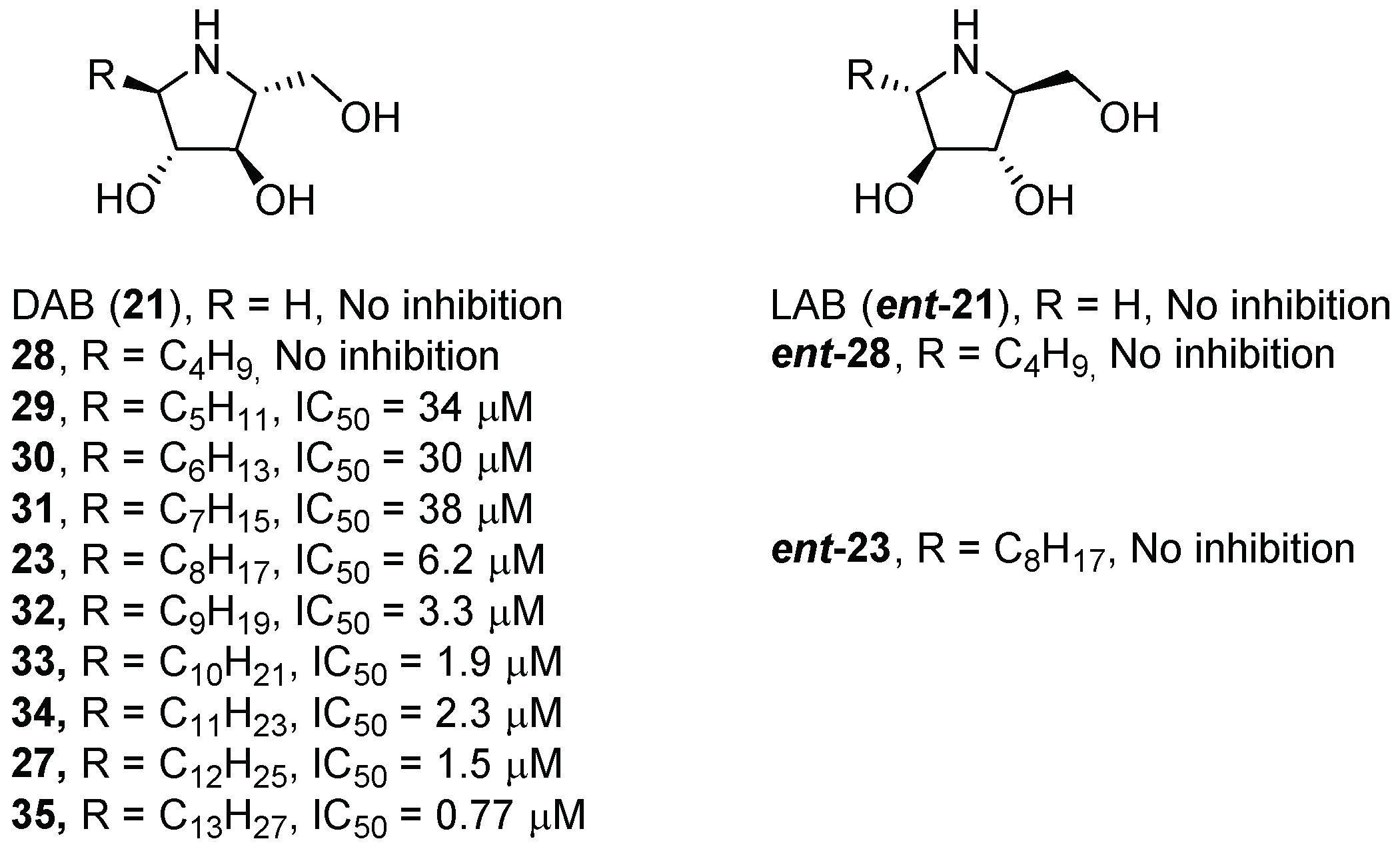{kind=link}
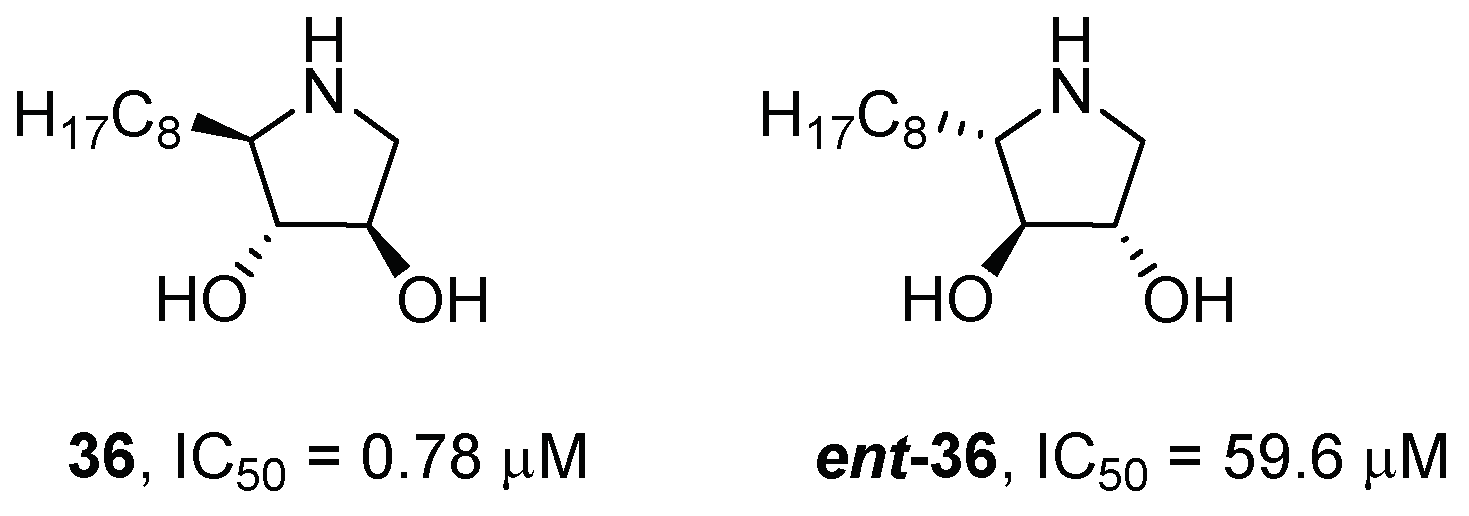{kind=link}
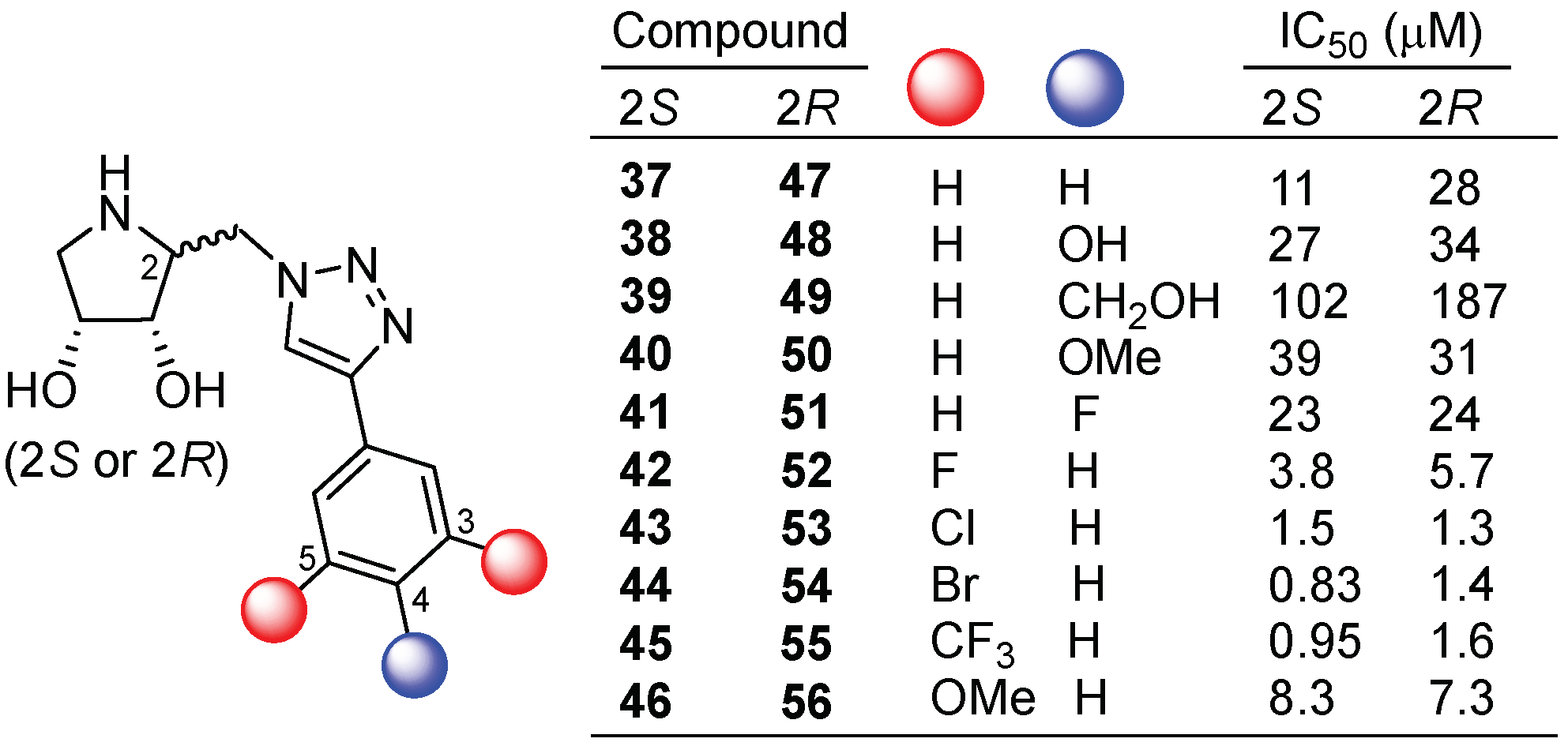{kind=link}
{kind=link}
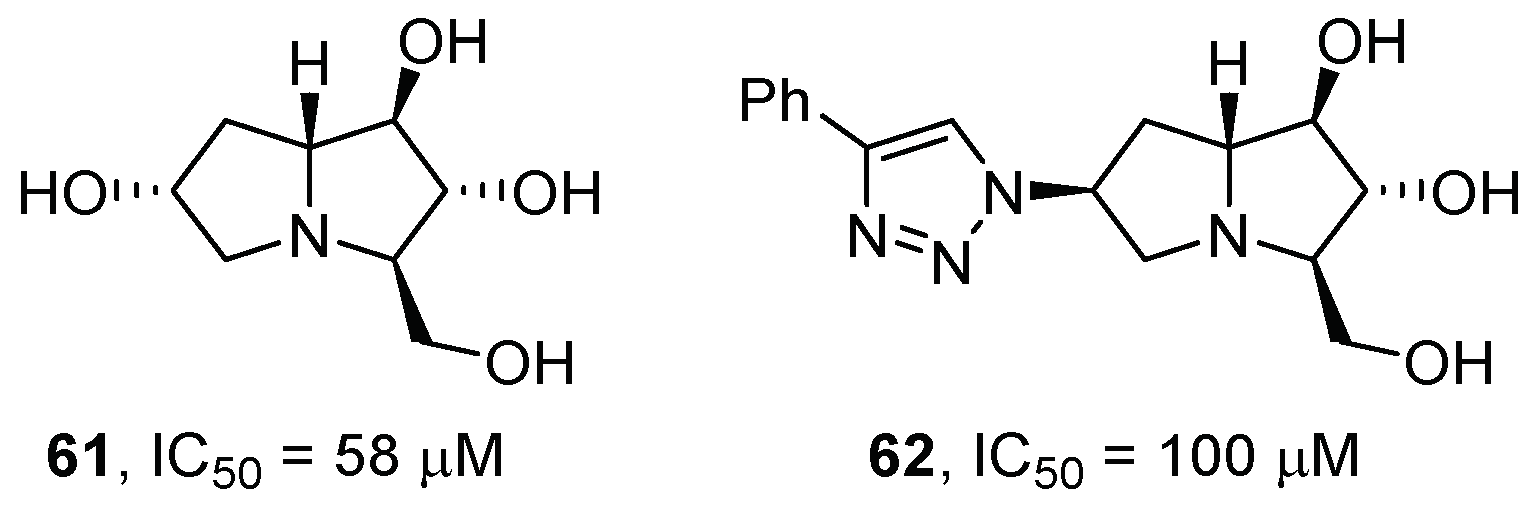{kind=link}
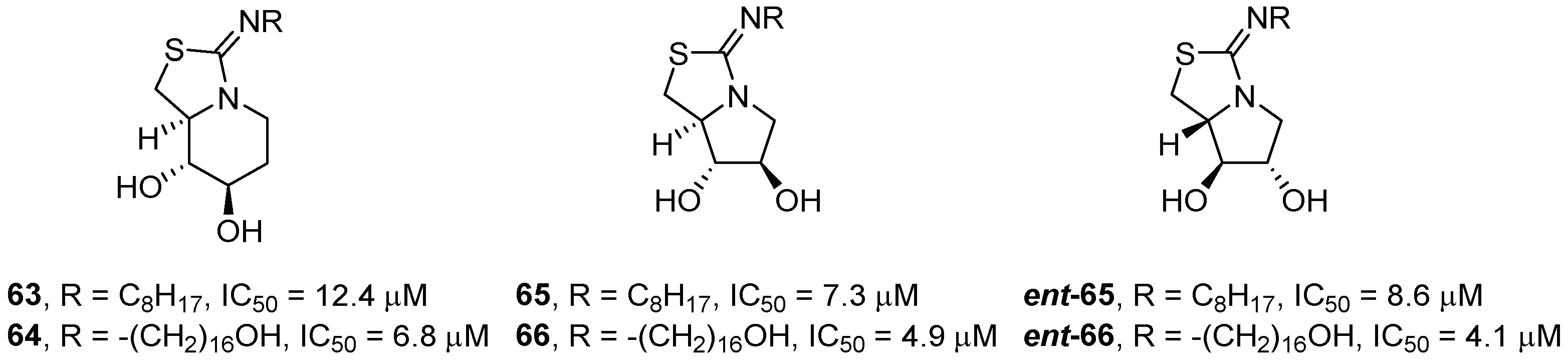{kind=link}
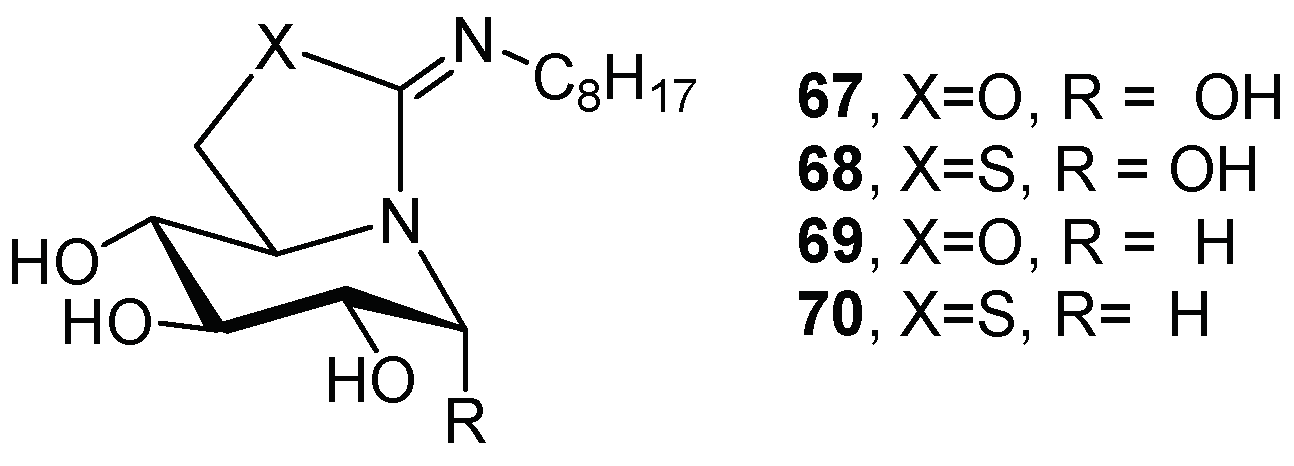{kind=link}
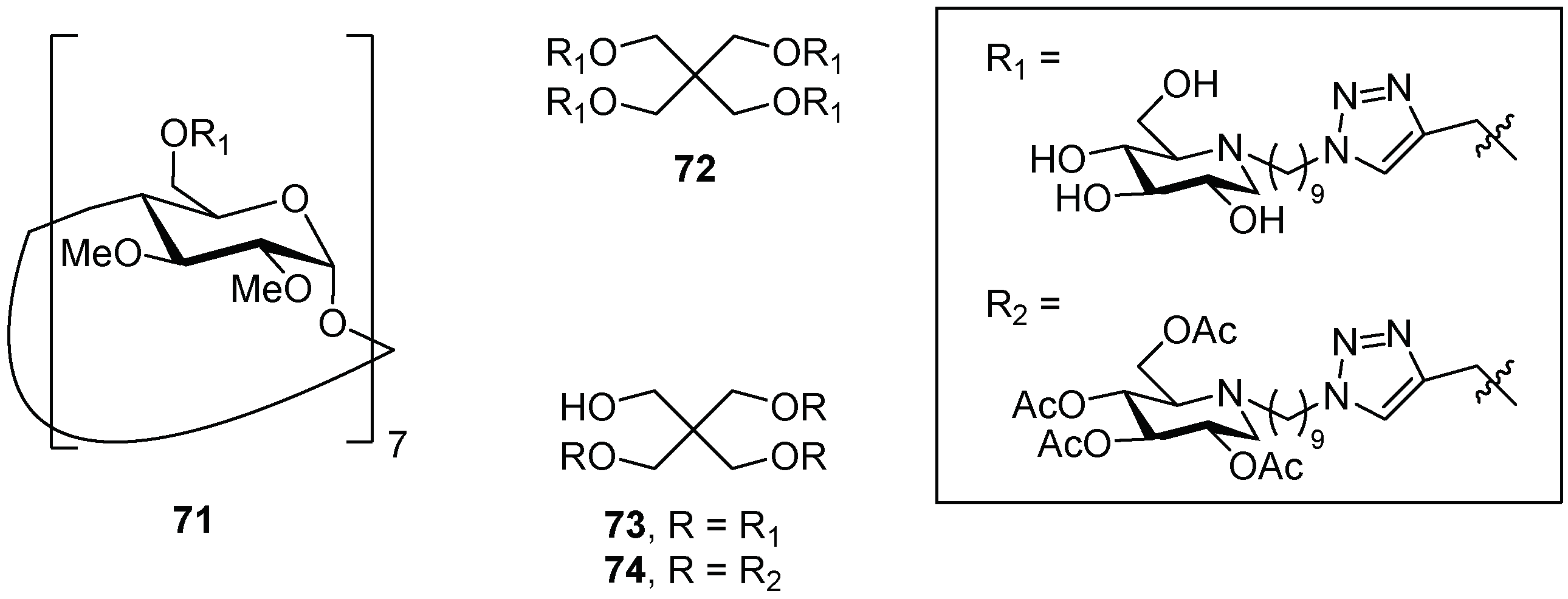{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
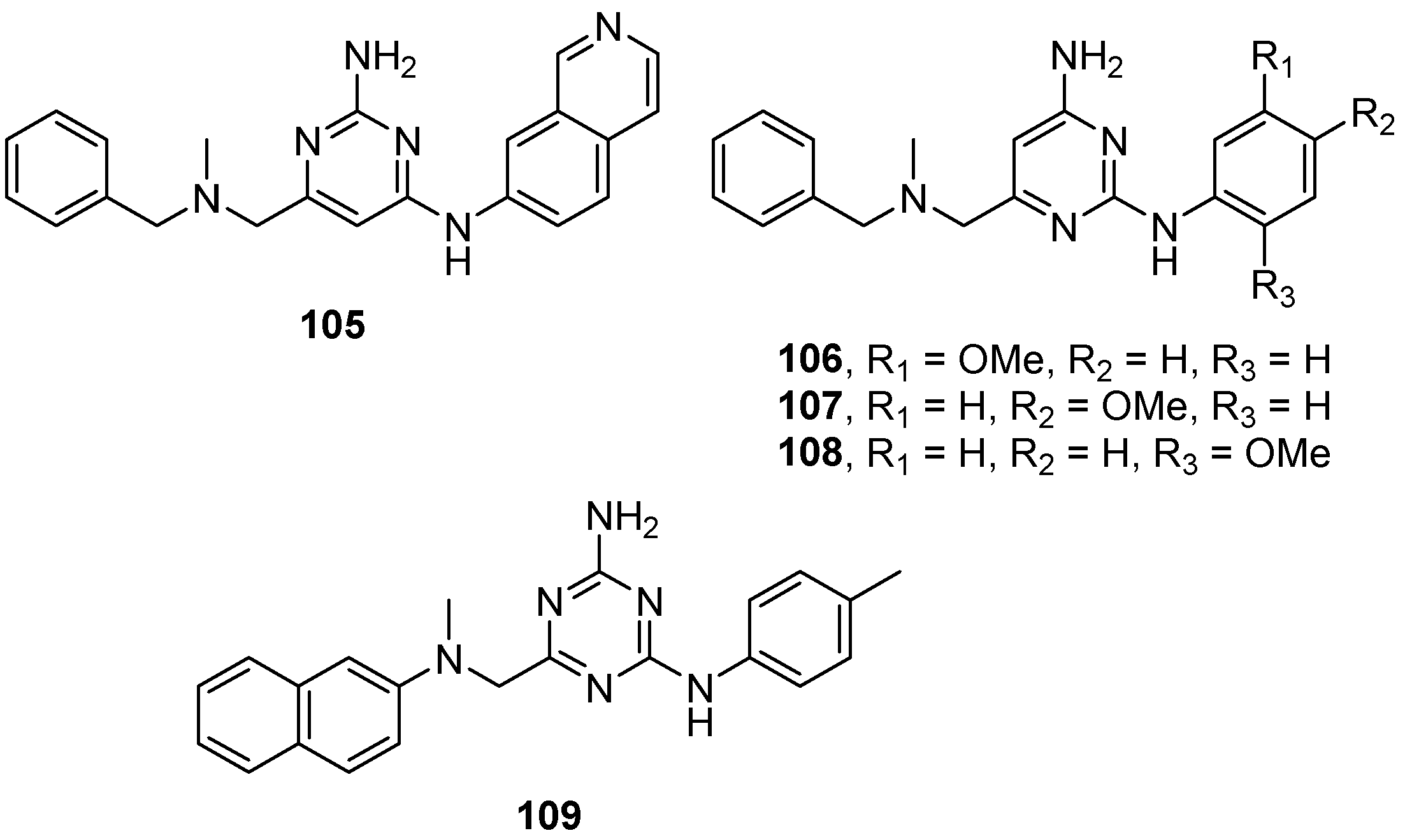{kind=link}
| Entry | GCase Variant | Severity | Best Enhancement | Concentration | Complex |
|---|---|---|---|---|---|
| 1 | N370S/N370S | Type I | 1.5-fold | 2 μM | 58:βCD |
| 2 | N370S/84GG | 2.8-fold | 20 μM | 58:βCD | |
| 3 | V230G/R296X | Type II | 2.7-fold | 20 μM | 57:βCD |
| 4 | L444P/P415R | 2.9-fold | 20 μM | 58:βCD | |
| 5 | N188S/G183W | Type III | 1.8-fold | 20 μM | 57:βCD and 58:βCD |
| 6 | L444P/L444P | 2.8-fold | 20 μM | 58:βCD |
| Entry | Compound | Ki (µM) a | Enhancement (N188S/G183W) | |
|---|---|---|---|---|
| pH 7 | pH 5 | |||
| 1 | 67 | 15.1 | 54.4 | 3-fold at 20 µM |
| 2 | 68 | 0.26 | 1.05 | 3.2-fold at 2 µM |
| 3 | 69 | 1.7 | 6.3 | 3.2-fold at 200 nM |
| 4 | 70 | 0.013 | 0.059 | 3-fold at 2 nM |
| Entry | Compound | Enhancement |
|---|---|---|
| 1 | 71 | 2.4-fold at 10 µM |
| 2 | 72 | 3.3-fold at 10 µM |
| 3 | 73 | 2.4-fold at 10 µM |
| 4 | 74 | 3.0-fold at 1 µM |
| Entry | Compound | IC50 (nM) | Enhancement |
|---|---|---|---|
| 1 | 72 | 500 | 2.5-fold |
| 2 | 75 | n.i. | 1.8-fold |
| 3 | 76 | 109 | 2.0-fold |
| 4 | 77 | n.i. | 2.1-fold |
| Entry | Compound | Valence | IC50 (nM) | Enhancement | Concentration (nM) | |
|---|---|---|---|---|---|---|
| pH 5.2 | pH 7.0 | |||||
| 1 | 78a | 2 | 1800 | 223 | 1.5-fold | 2000 |
| 2 | 78b | 2 | 23.6 | 32.5 | 1.7-fold | 2000 |
| 3 | 78c | 2 | 57.1 | 28.5 | 2.2-fold | 2000 |
| 4 | 78d | 2 | 1100 | 362 | 1.6-fold | 2000 |
| 5 | 78e | 2 | 46.6 | 17.0 | 2.2-fold | 100 |
| 6 | 79 | 2 | 3.6 | 3.3 | 2.5-fold | 100 |
| 7 | ent-79 | 2 | 162,000 | 94,000 | - | - |
| 8 | 80 a | 1 | 4.3 | 5.7 | 1.8-fold | 1 |
| 9 | 81 | 1 | 292 | 182 | 1.6-fold | 2000 |
| 10 | 82 a | 1 | 24.3 | 19.9 | 3.6-fold | 100 |
| 11 | 83 | 1 | 4.8 | 4.3 | 2.2-fold | 10 |
| 12 | ent-80 | 1 | 75,000 | 42,000 | - | 300 |
| 13 | ent-83 | 1 | 58,000 | 19,000 | - | 2000 |
| Compound | Disease Models | Animal Model | Effect of Chaperone | Refs. |
|---|---|---|---|---|
 | GBA-PD | Drosophila with mutant GBA (N370S and L444P) | - reduce ER stress - reverse locomotor deficits | [99] |
| PD | Transgenic mice overexpressing human WT α-syn | - improved motor function - reduced α-syn immunoreactivity - reduced α-syn aggregates | [102] | |
 | GBA-PD | Drosophila with mutant GBA (N370S and L444P) | - reduce ER stress - reverse locomotor deficits | [99,100] |
| GBA-PD | Gba1L444P/+ mice | - increased GCase activity in brains | [101] | |
| PD | Transgenic mice overexpressing human WT α-syn | - reduced α-syn levels | [101] | |
 | GBA-PD | Gba1D409V/+ mice | - reduced GlcCer levels in brain - reduced α-syn levels in brain | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Bailén, M.; Clemente, F.; Matassini, C.; Cardona, F. GCase Enhancers: A Potential Therapeutic Option for Gaucher Disease and Other Neurological Disorders. Pharmaceuticals 2022, 15, 823. https://doi.org/10.3390/ph15070823
Martínez-Bailén M, Clemente F, Matassini C, Cardona F. GCase Enhancers: A Potential Therapeutic Option for Gaucher Disease and Other Neurological Disorders. Pharmaceuticals. 2022; 15(7):823. https://doi.org/10.3390/ph15070823
Chicago/Turabian StyleMartínez-Bailén, Macarena, Francesca Clemente, Camilla Matassini, and Francesca Cardona. 2022. "GCase Enhancers: A Potential Therapeutic Option for Gaucher Disease and Other Neurological Disorders" Pharmaceuticals 15, no. 7: 823. https://doi.org/10.3390/ph15070823
APA StyleMartínez-Bailén, M., Clemente, F., Matassini, C., & Cardona, F. (2022). GCase Enhancers: A Potential Therapeutic Option for Gaucher Disease and Other Neurological Disorders. Pharmaceuticals, 15(7), 823. https://doi.org/10.3390/ph15070823