
Impact of MnTBAP and Baricitinib Treatment on Hutchinson–Gilford Progeria Fibroblasts



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
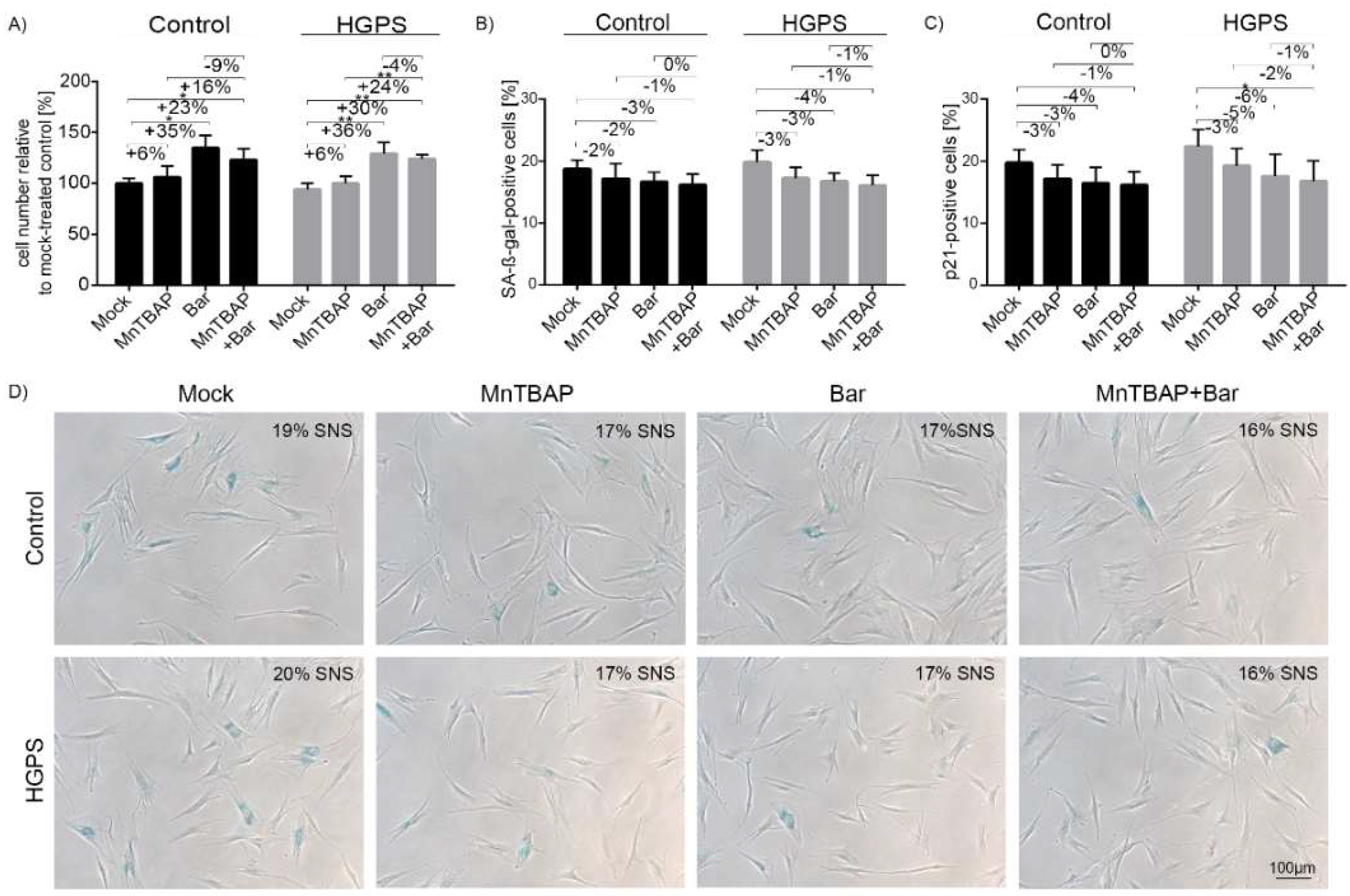
2.1. Combined MnTBAP/Bar Treatment Ameliorates HGPS Fibroblast Growth Rate and Decreases Replicative Senescence to a Range Similar to Each Drug Alone
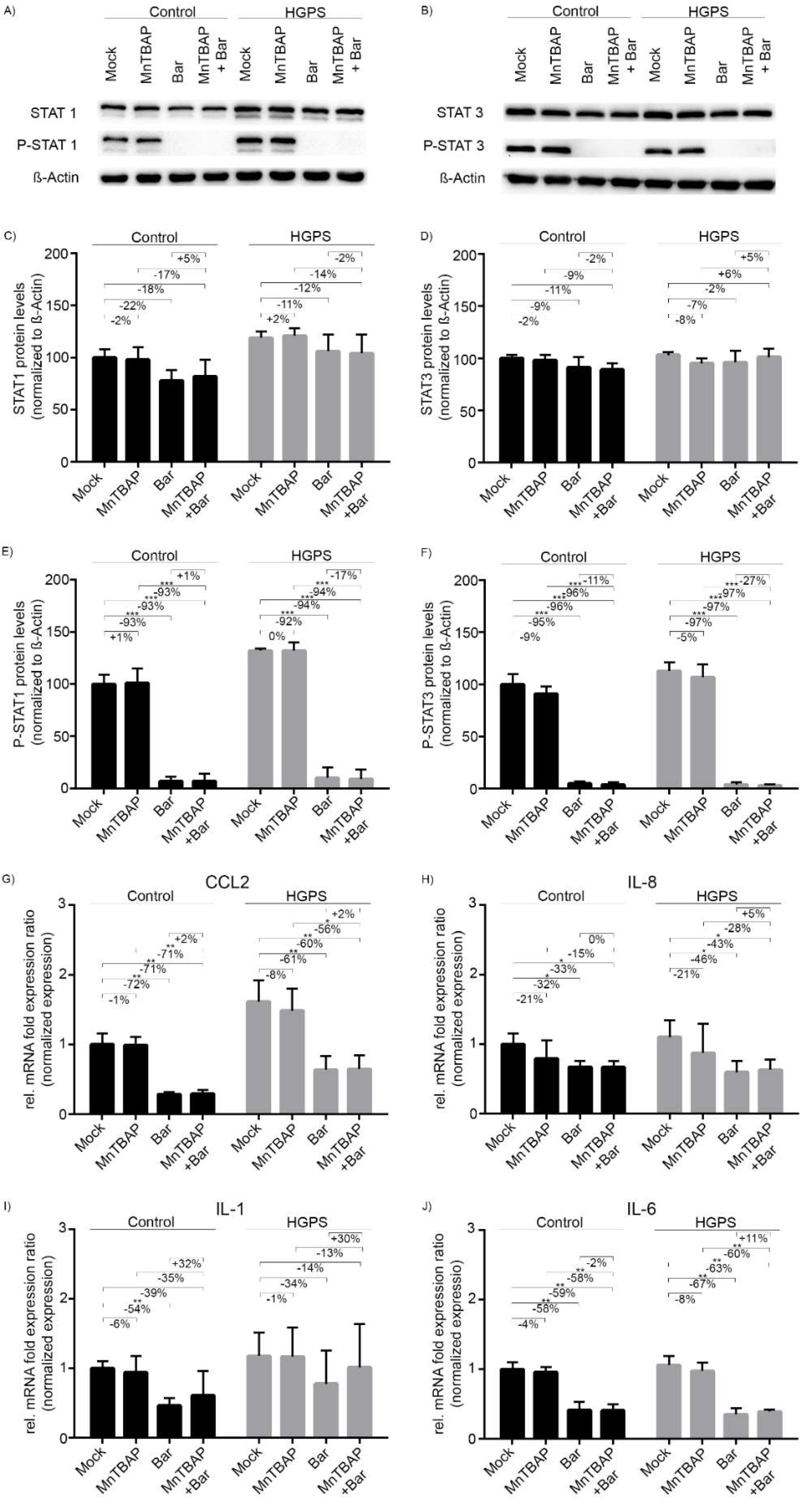
2.2. Treatment with Bar Inhibits STAT1/2 Signaling and Reduces the Expression of Pro-Inflammatory Cytokines
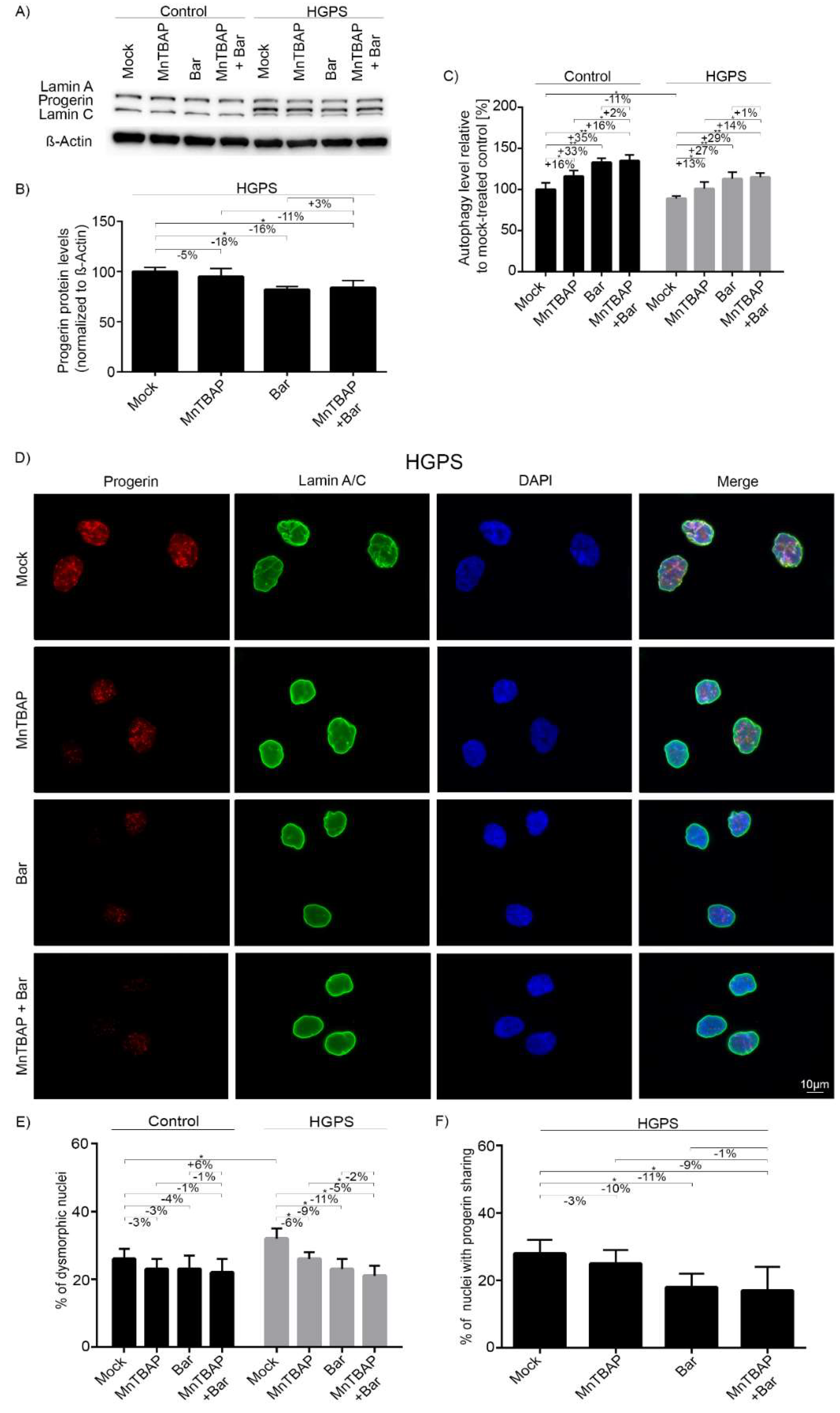
2.3. Combined MnTBAP/Bar Treatment Improves Progerin Clearance and Nuclear Morphology to a Range Similar to Bar Alone
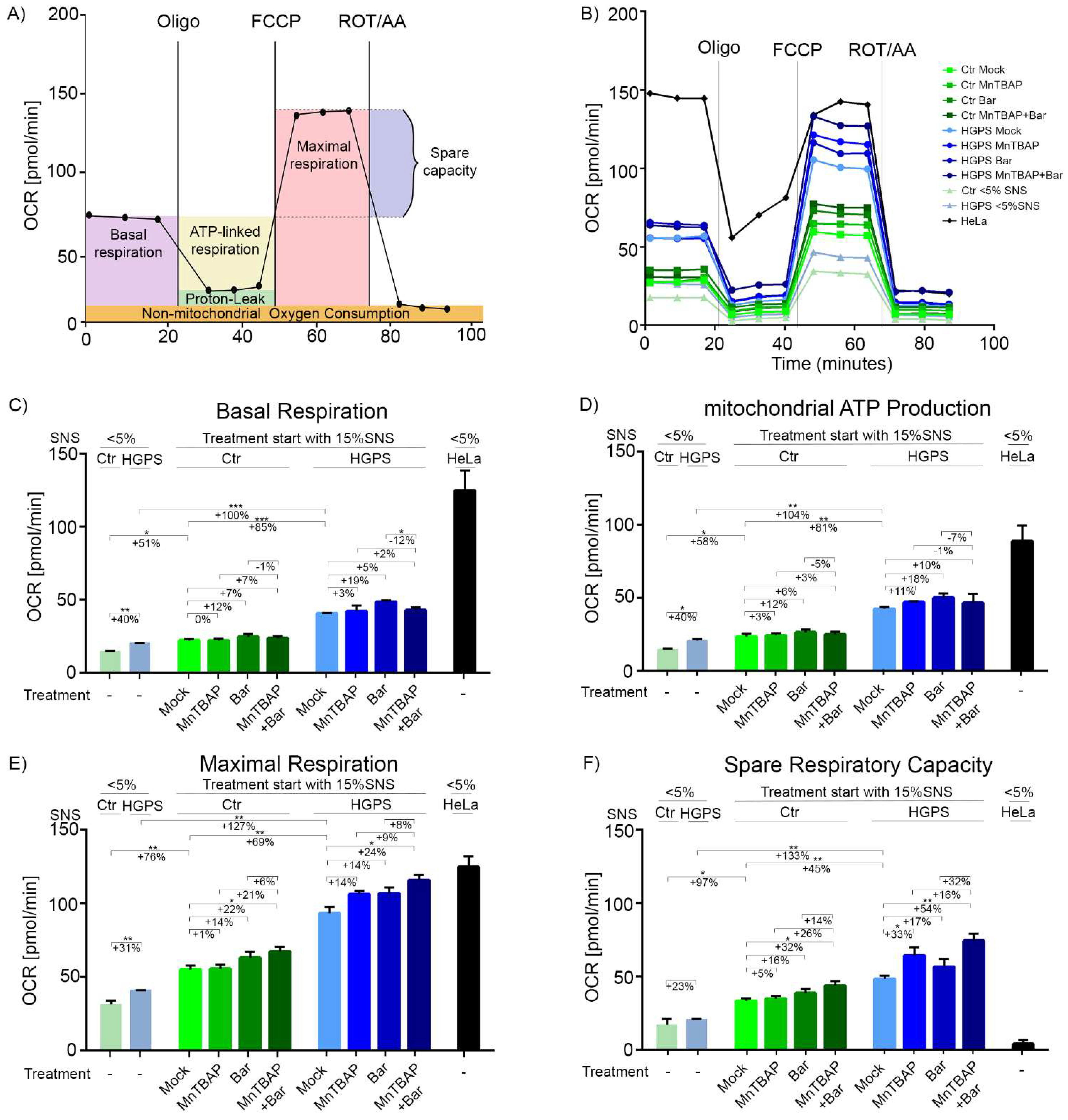
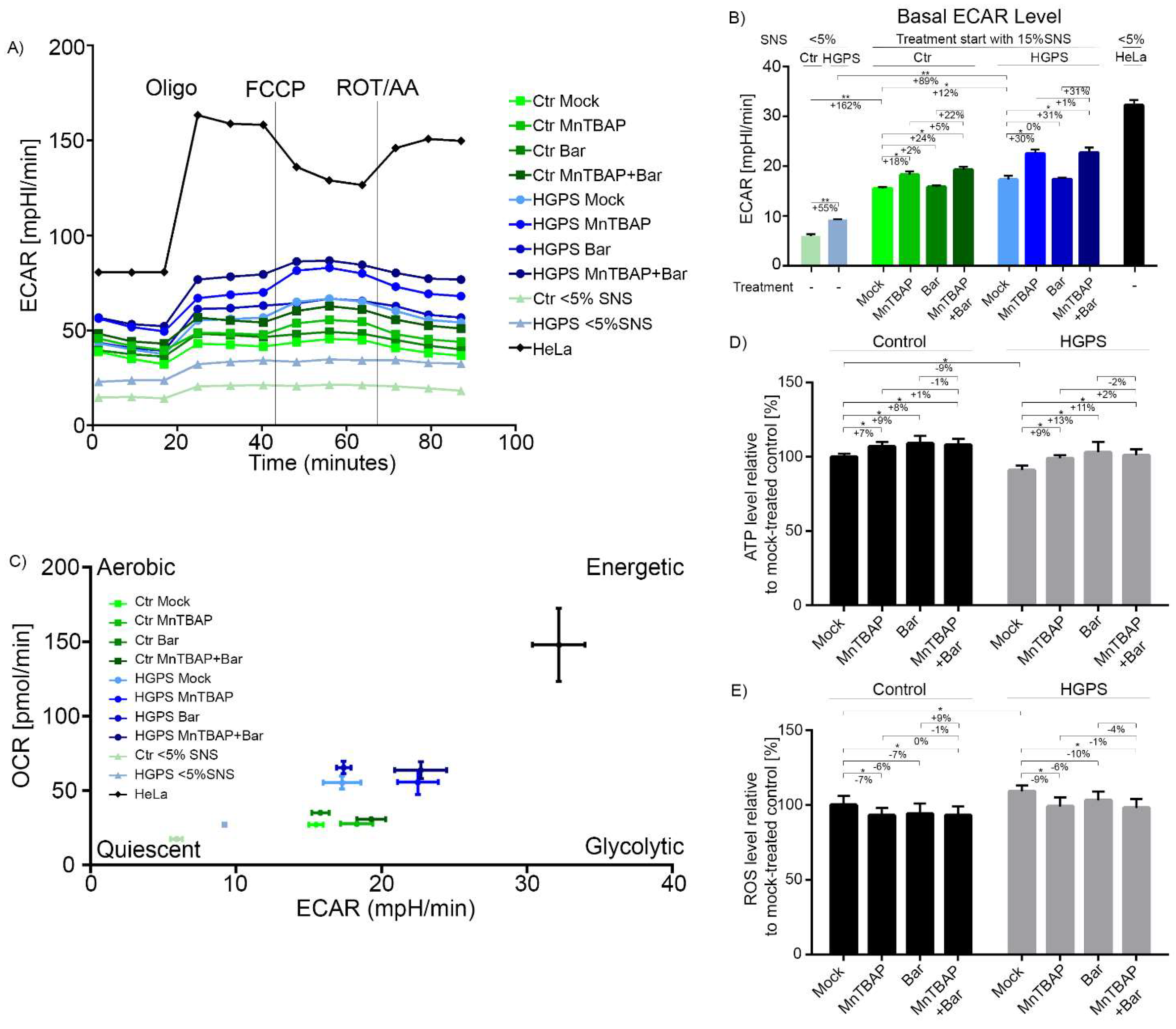
2.4. Combined MnTBAP/Bar Treatment Ameliorates Maximal Respiration and Spare Respiratory Capacity of HGPS Cells
2.5. Combined MnTBAP/Bar Treatment Reduces DNA Damage in HGPS Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Drug Treatment
4.3. Senescence-Associated Beta-Galactosidase Staining (SA-β-Gal)
4.4. Western Blot Analysis
4.5. Cell Number Determination
4.6. Respirometic Assay
4.7. Autophagy Assay
4.8. ROS Measurements
4.9. Immunohistochemistry
4.10. Measurements of Intracellular ATP Levels
4.11. Gene Expression Analysis
4.12. Toxicity Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gordon, L.B.; McCarten, K.M.; Giobbie-Hurder, A.; Machan, J.T.; Campbell, S.E.; Berns, S.D.; Kieran, M.W. Disease progression in Hutchinson-Gilford progeria syndrome: Impact on growth and development. Pediatrics 2007, 120, 824–833. [Google Scholar] [CrossRef] [PubMed]
- The Progeria Research Foundation. Available online: https://www.progeriaresearch.org/progeria-101faq/ (accessed on 3 April 2020).
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worman, H.J.; Courvalin, J.-C. How do mutations in lamins A and C cause disease? J. Clin. Investig. 2004, 113, 349–351. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Corrigan, D.P.; Kuszczak, D.; Rusinol, A.E.; Thewke, D.P.; Hrycyna, C.A.; Michaelis, S.; Sinensky, M.S. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem. J. 2005, 387, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.G.; Meta, M.; Yang, S.H.; Fong, L.G. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 2006, 281, 39741–39745. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W. Impact of Farnesylation Inhibitors on Survival in Hutchinson-Gilford Progeria Syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viteri, G.; Chung, Y.W.; Stadtman, E.R. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech. Ageing Dev. 2010, 131, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-M.; Chiang, J.-C.; Lin, Y.-C.; Lin, Y.-N.; Chuang, P.-Y.; Chang, Y.-C.; Chen, C.-C.; Wu, K.-Y.; Hsieh, J.-C.; Chen, S.-K.; et al. Lysophosphatidic acid receptor LPA3 prevents oxidative stress and cellular senescence in Hutchinson-Gilford progeria syndrome. Aging Cell 2019, 19, e13064. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Arnold, R.; Henriques, G.; Djabali, K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells 2019, 8, 1276. [Google Scholar] [CrossRef] [Green Version]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.M.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Lévy, N. An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Bergo, M.O.; Toth, J.I.; Qiao, X.; Hu, Y.; Sandoval, S.; Meta, M.; Bendale, P.; Gelb, M.H.; Young, S.G.; et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 10291–10296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov, NCT00425607. Available online: https://clinicaltrials.gov/ct2/show/NCT00425607 (accessed on 6 April 2020).
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.S.; Morse, M.A. Lonafarnib for cancer and progeria. Expert Opin. Investig. Drugs 2012, 21, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Lonafarnib: First Approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: Preclinical characterization of INCB028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griveau, A.; Wiel, C.; Ziegler, D.V.; Bergo, M.O.; Bernard, D. The JAK1/2 inhibitor ruxolitinib delays premature aging phenotypes. Aging Cell 2020, 19, e13122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, D.; Gordon, L.B.; Djabali, K. Temsirolimus Partially Rescues the Hutchinson-Gilford Progeria Cellular Phenotype. PLoS ONE 2016, 11, e0168988. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Torres, J.; Acín-Perez, R.; Cabezas-Sánchez, P.; Osorio, F.G.; Gonzalez-Gómez, C.; Megias, D.; Cámara, C.; López-Otín, C.; Enríquez, J.A.; Luque-García, J.L.; et al. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteom. 2013, 91, 466–477. [Google Scholar] [CrossRef]
- Bi, X.; Wang, J.; Liu, Y.; Wang, Y.; Ding, W. MnTBAP treatment ameliorates aldosterone-induced renal injury by regulating mitochondrial dysfunction and NLRP3 inflammasome signalling. Am. J. Transl. Res. 2018, 10, 3504–3513. [Google Scholar] [PubMed]
- Catic, A. Cellular Metabolism and Aging. Prog. Mol. Biol. Transl. Sci. 2018, 155, 85–107. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef] [Green Version]
- Salvemini, D.; Riley, D.P.; Cuzzocrea, S. SOD mimetics are coming of age. Nat. Rev. Drug Discov. 2002, 1, 367–374. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Roy, R.S. The pathophysiology of superoxide: Roles in inflammation and ischemia. Can. J. Physiol. Pharmacol. 1982, 60, 1346–1352. [Google Scholar] [CrossRef]
- Salvemini, D.; Mazzon, E.; Dugo, L.; Riley, D.P.; Serraino, I.; Caputi, A.P.; Cuzzocrea, S. Pharmacological manipulation of the inflammatory cascade by the superoxide dismutase mimetic, M40403. Br. J. Pharmacol. 2001, 132, 815–827. [Google Scholar] [CrossRef]
- Salvemini, D.; Riley, D.P.; Lennon, P.J.; Wang, Z.Q.; Currie, M.G.; Macarthur, H.; Misko, T.P. Protective effects of a superoxide dismutase mimetic and peroxynitrite decomposition catalysts in endotoxin-induced intestinal damage. Br. J. Pharmacol. 1999, 127, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Bharti, P.; Thaivalappil, A.; Cao, K. Peroxisomal abnormalities and catalase deficiency in Hutchinson-Gilford Progeria Syndrome. Aging 2020, 12, 5195–5208. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.-M.; Choi, J.Y.; Wang, K.; Zhang, H.; Tariq, Z.; Wu, D.; Ko, E.; LaDana, C.; Sesaki, H.; Cao, K. Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell 2016, 15, 279–290. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Dietary magnesium supplementation improves lifespan in a mouse model of progeria. EMBO Mol. Med. 2020, 12, e12423. [Google Scholar] [CrossRef]
- Richards, S.A.; Muter, J.; Ritchie, P.; Lattanzi, G.; Hutchison, C.J. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum. Mol. Genet. 2011, 20, 3997–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, C.S.; Bikkul, M.U.; Ofosu, W.; Eskiw, C.; Tree, D.; Makarov, E.; Kill, I.R.; Bridger, J.M. Presence and distribution of progerin in HGPS cells is ameliorated by drugs that impact on the mevalonate and mTOR pathways. Biogerontology 2019, 20, 337–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, D.; Shafry, D.D.; Gordon, L.B.; Djabali, K. Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in Hutchinson-Gilford progeria fibroblasts. Oncotarget 2017, 8, 64809–64826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.S.; Vincent, R.; Dey, M.S.; Slot, J.W.; Crapo, J.D. Transgenic models for the study of lung antioxidant defense: Enhanced manganese-containing superoxide dismutase activity gives partial protection to B6C3 hybrid mice exposed to hyperoxia. Am. J. Respir. Cell Mol. Biol. 1998, 18, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, K.M.; Liochev, S.I.; Fridovich, I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. J. Biol. Chem. 1994, 269, 23471–23476. [Google Scholar] [CrossRef]
- Zingarelli, B.; Day, B.J.; Crapo, J.D.; Salzman, A.L.; Szabó, C. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br. J. Pharmacol. 1997, 120, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Tumurkhuu, G.; Koide, N.; Dagvadorj, J.; Hassan, F.; Islam, S.; Naiki, Y.; Mori, I.; Yoshida, T.; Yokochi, T. MnTBAP, a synthetic metalloporphyrin, inhibits production of tumor necrosis factor-alpha in lipopolysaccharide-stimulated RAW 264.7 macrophages cells via inhibiting oxidative stress-mediating p38 and SAPK/JNK signaling. FEMS Immunol. Med. Microbiol. 2007, 49, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, S.; Nakayamada, S.; Sakata, K.; Kitanaga, Y.; Ma, X.; Lee, S.; Ishii, A.; Yamagata, K.; Nakano, K.; Tanaka, Y. Janus Kinase Inhibitor Baricitinib Modulates Human Innate and Adaptive Immune System. Front. Immunol. 2018, 9, 1510. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Scott, L.J. Baricitinib: A Review in Rheumatoid Arthritis. Drugs 2018, 78, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; de Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Gensch, C.; Keller, C.; Schmitt, H.; Esser, J.; Moser, M.; Liao, J.K. MnTBAP stimulates angiogenic functions in endothelial cells through mitofusin-1. Vascul. Pharmacol. 2015, 72, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signaling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisch, V.; Lu, X.; Gabriel, D.; Djabali, K. Progerin impairs chromosome maintenance by depleting CENP-F from metaphase kinetochores in Hutchinson-Gilford progeria fibroblasts. Oncotarget 2016, 7, 24700–24718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef]
- Lopez-Mejia, I.C.; de Toledo, M.; Chavey, C.; Lapasset, L.; Cavelier, P.; Lopez-Herrera, C.; Chebli, K.; Fort, P.; Beranger, G.; Fajas, L.; et al. Antagonistic functions of LMNA isoforms in energy expenditure and lifespan. EMBO Rep. 2014, 15, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin-Perez, R.; Benador, I.Y.; Petcherski, A.; Veliova, M.; Benavides, G.A.; Lagarrigue, S.; Caudal, A.; Vergnes, L.; Murphy, A.N.; Karamanlidis, G.; et al. A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J. 2020, 39, e104073. [Google Scholar] [CrossRef]
- Lardy, H.A.; Johnson, D.; McMURRAY, W.C. Antibiotics as tools for metabolic studies. I. A survey of toxic antibiotics in respiratory, phosphorylative and glycolytic systems. Arch. Biochem. Biophys. 1958, 78, 587–597. [Google Scholar] [CrossRef]
- Benz, R.; McLaughlin, S. The molecular mechanism of action of the proton ionophore FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazone). Biophys. J. 1983, 41, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Palmer, G.; Horgan, D.J.; Tisdale, H.; Singer, T.P.; Beinert, H. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. XIV. Location of the sites of inhibition of rotenone, barbiturates, and piericidin by means of electron paramagnetic resonance spectroscopy. J. Biol. Chem. 1968, 243, 844–847. [Google Scholar] [CrossRef]
- Labs, M.; Rühle, T.; Leister, D. The antimycin A-sensitive pathway of cyclic electron flow: From 1963 to 2015. Photosynth. Res. 2016, 129, 231–238. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Nacarelli, T.; Azar, A.; Altinok, O.; Orynbayeva, Z.; Sell, C. Rapamycin increases oxidative metabolism and enhances metabolic flexibility in human cardiac fibroblasts. Geroscience 2018, 40, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Yadava, N.; Nicholls, D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Wang, J.; Chan, K.M.; Tjia, W.M.; Deng, W.; Guan, X.; Huang, J.; Li, K.M.; Chau, P.Y.; Chen, D.J.; et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005, 11, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2016, 33, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef] [PubMed]
- Son, J.M.; Sarsour, E.H.; Kakkerla Balaraju, A.; Fussell, J.; Kalen, A.L.; Wagner, B.A.; Buettner, G.R.; Goswami, P.C. Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell 2017, 16, 1136–1145. [Google Scholar] [CrossRef] [Green Version]
- Kikis, E.A.; Gidalevitz, T.; Morimoto, R.I. Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 2010, 694, 138–159. [Google Scholar]
- Peth, A.; Nathan, J.A.; Goldberg, A.L. The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J. Biol. Chem. 2013, 288, 29215–29222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Meth. Enzymol. 2014, 547, 309–354. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.P.; López-Otín, C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, S.; Bigot, J.; Onodi, F.; Cosette, J.; Corre, G.; Poupiot, J.; Fenard, D.; Gjata, B.; Galy, A.; Neildez-Nguyen, T.M.A. Temporary Reduction of Membrane CD4 with the Antioxidant MnTBAP Is Sufficient to Prevent Immune Responses Induced by Gene Transfer. Mol. Ther. Methods Clin. Dev. 2019, 14, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Norell, H.; Da Martins Palma, T.; Lesher, A.; Kaur, N.; Mehrotra, M.; Naga, O.S.; Spivey, N.; Olafimihan, S.; Chakraborty, N.G.; Voelkel-Johnson, C.; et al. Inhibition of superoxide generation upon T-cell receptor engagement rescues Mart-1(27-35)-reactive T cells from activation-induced cell death. Cancer Res. 2009, 69, 6282–6289. [Google Scholar] [CrossRef] [Green Version]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.-G.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Lévy, N. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol. Med. 2017, 9, 1294–1313. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrl, J.M.; Arnold, R.; Djabali, K. Nuclear Pore Complexes Cluster in Dysmorphic Nuclei of Normal and Progeria Cells during Replicative Senescence. Cells 2021, 10, 153. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vehns, E.; Arnold, R.; Djabali, K. Impact of MnTBAP and Baricitinib Treatment on Hutchinson–Gilford Progeria Fibroblasts. Pharmaceuticals 2022, 15, 945. https://doi.org/10.3390/ph15080945
Vehns E, Arnold R, Djabali K. Impact of MnTBAP and Baricitinib Treatment on Hutchinson–Gilford Progeria Fibroblasts. Pharmaceuticals. 2022; 15(8):945. https://doi.org/10.3390/ph15080945
Chicago/Turabian StyleVehns, Elena, Rouven Arnold, and Karima Djabali. 2022. "Impact of MnTBAP and Baricitinib Treatment on Hutchinson–Gilford Progeria Fibroblasts" Pharmaceuticals 15, no. 8: 945. https://doi.org/10.3390/ph15080945
APA StyleVehns, E., Arnold, R., & Djabali, K. (2022). Impact of MnTBAP and Baricitinib Treatment on Hutchinson–Gilford Progeria Fibroblasts. Pharmaceuticals, 15(8), 945. https://doi.org/10.3390/ph15080945