
Licochalcone B, a Natural Autophagic Agent for Alleviating Oxidative Stress-Induced Cell Death in Neuronal Cells and Caenorhabditis elegans Models

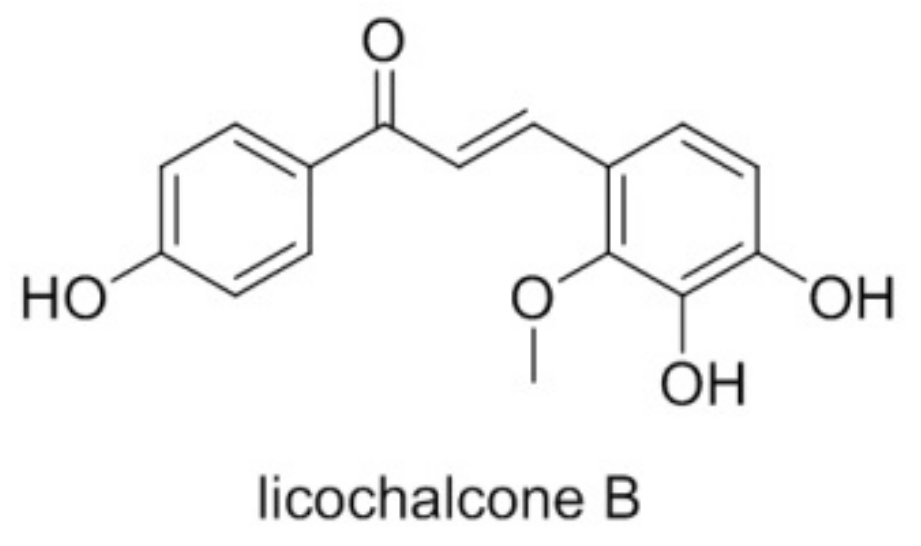{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
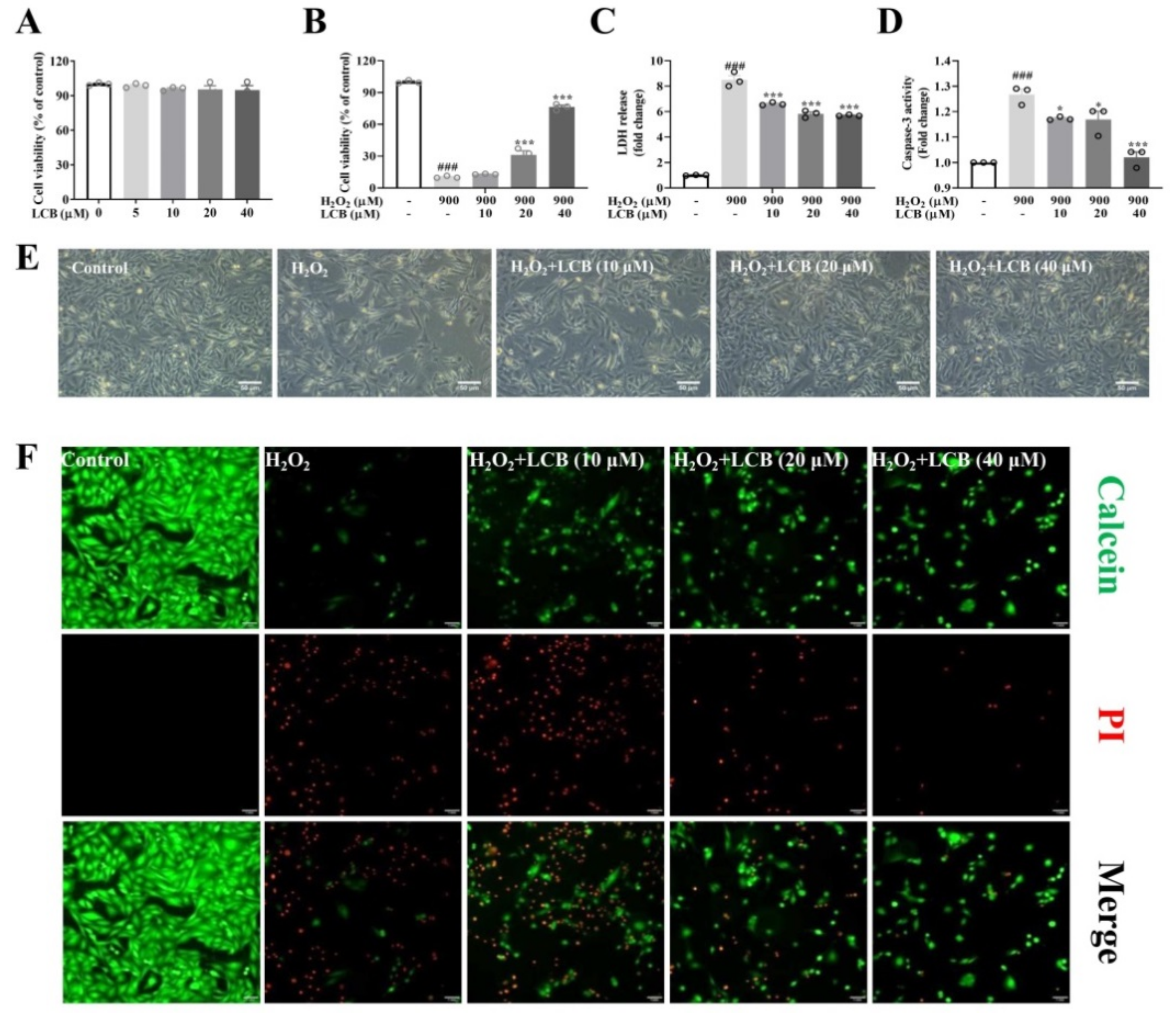
2.1. LCB Attenuated H2O2-Induced Cell Death in PC-12 Cells
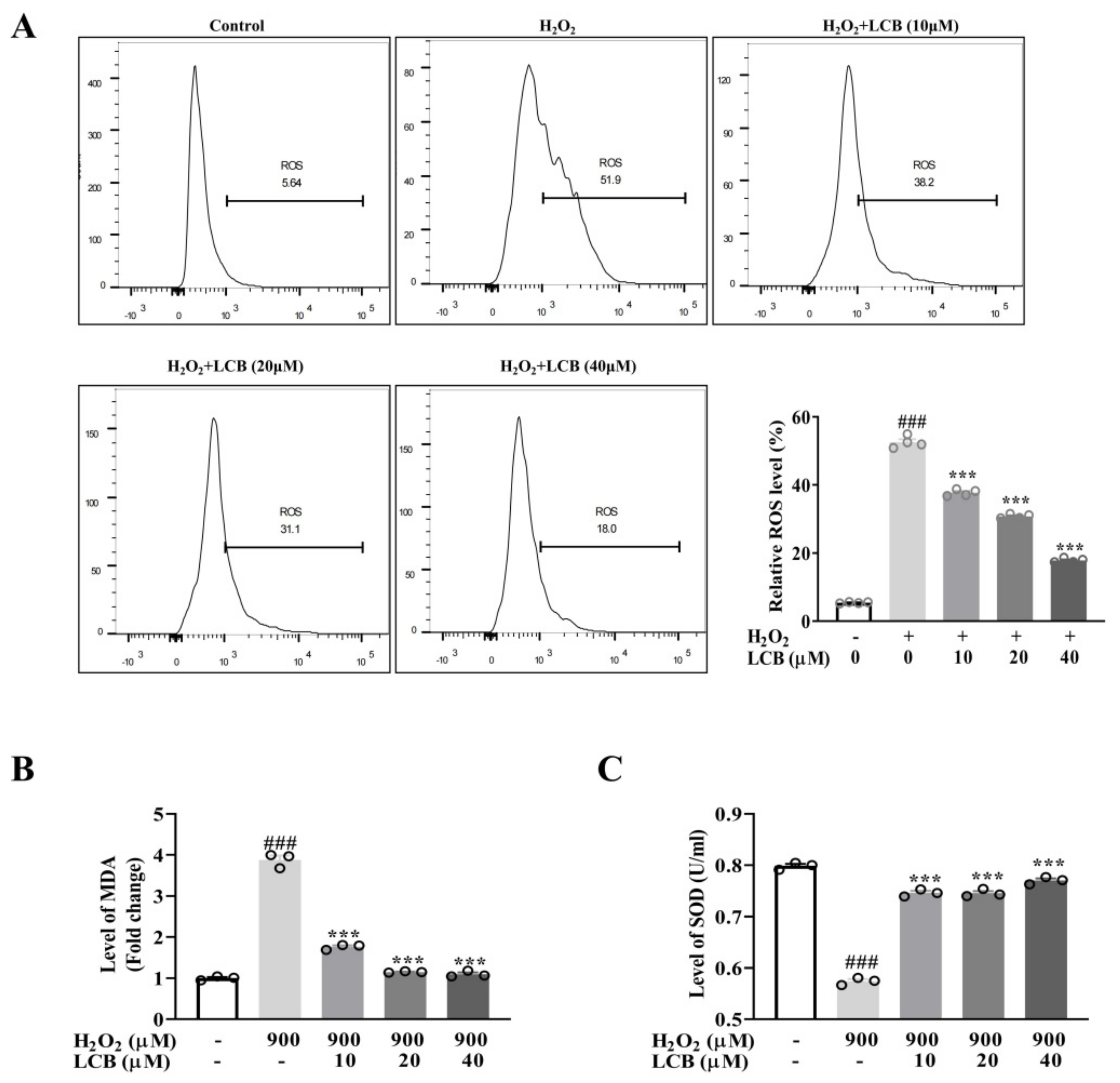
2.2. LCB Inhibited H2O2-Induced Oxidative Stress
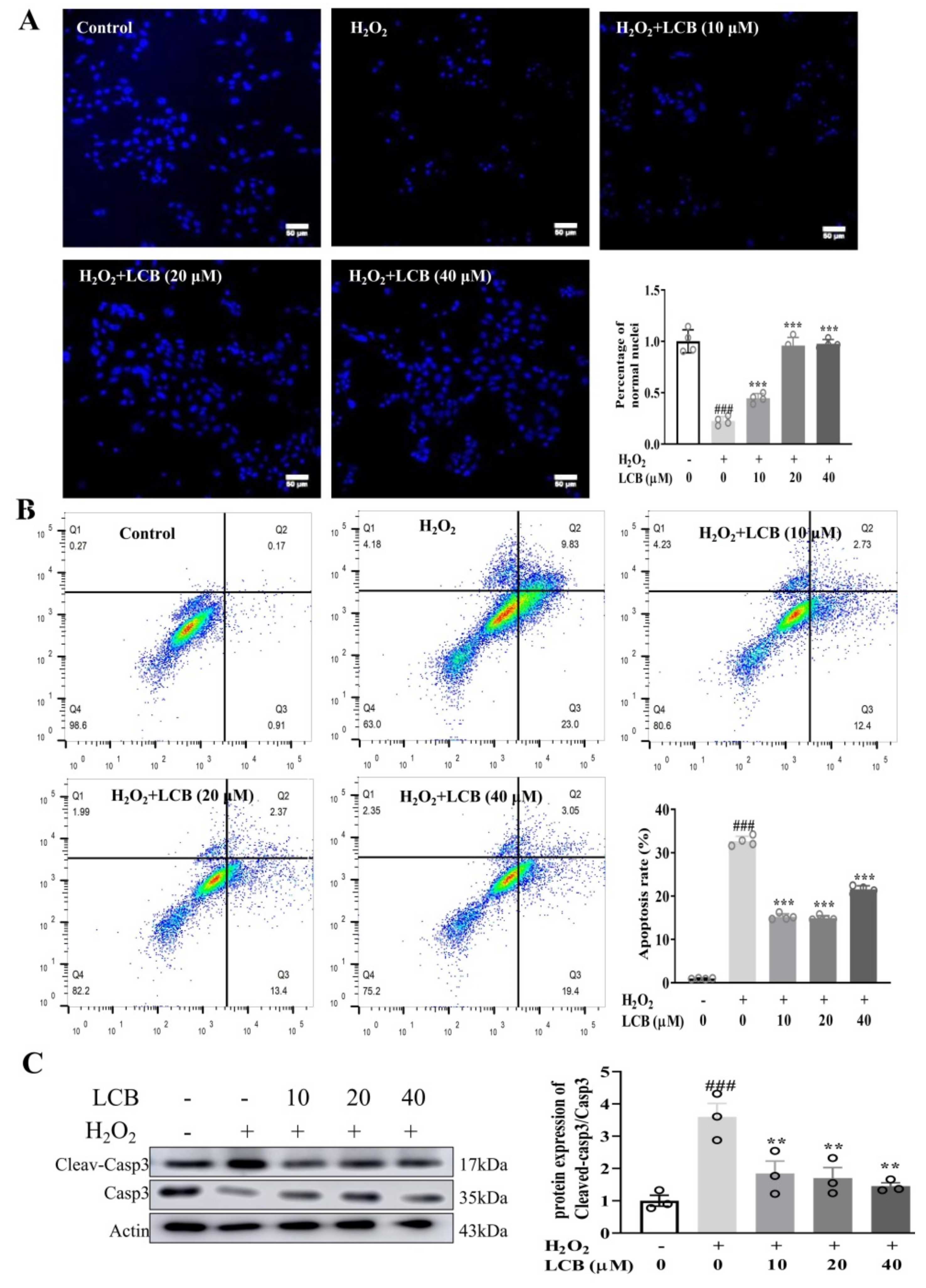
2.3. LCB Protected PC-12 Cells from H2O2-Induced Apoptotic Cell Death
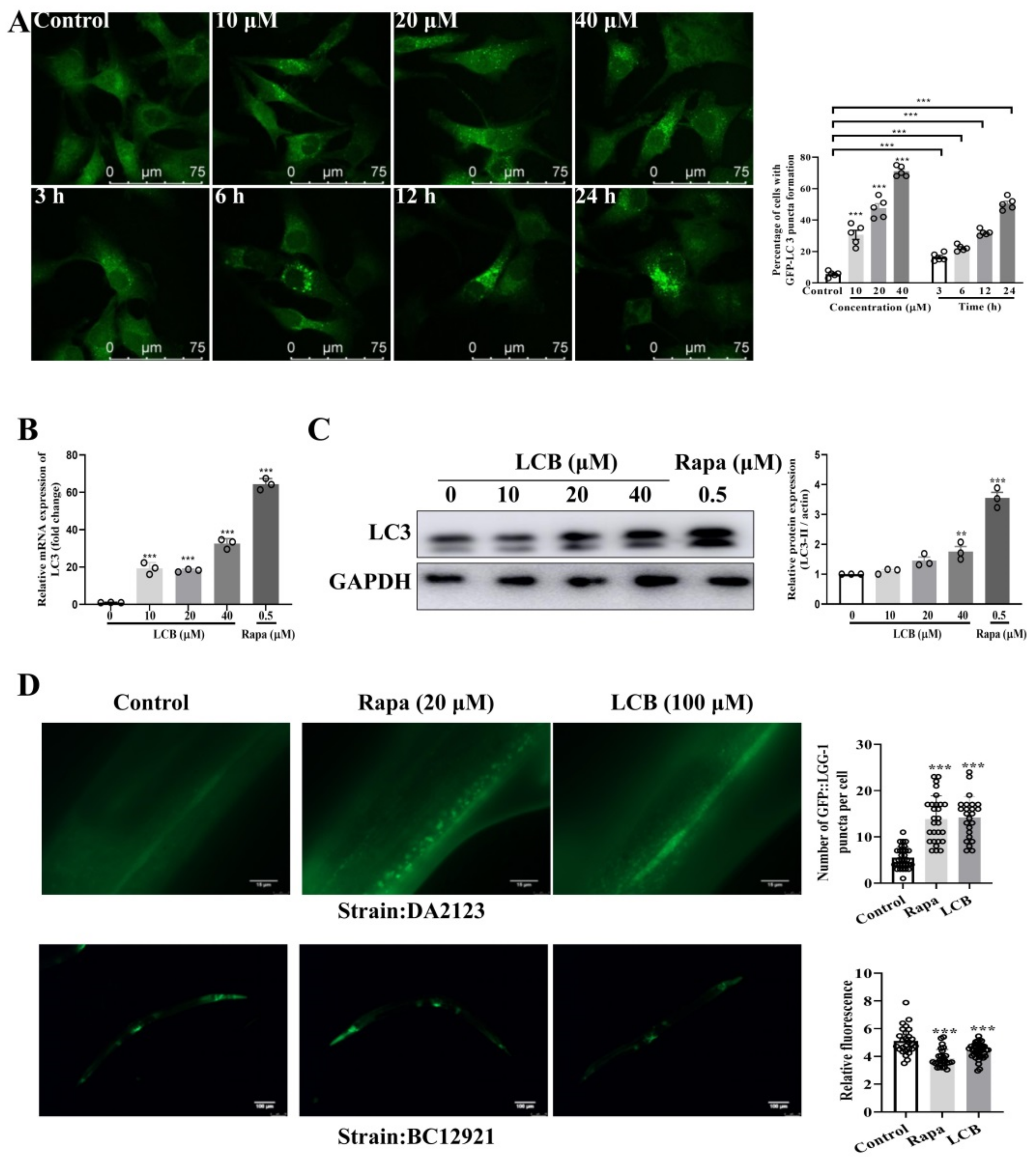
2.4. LCB Induced Autophagy in PC-12 and C. elegans Models
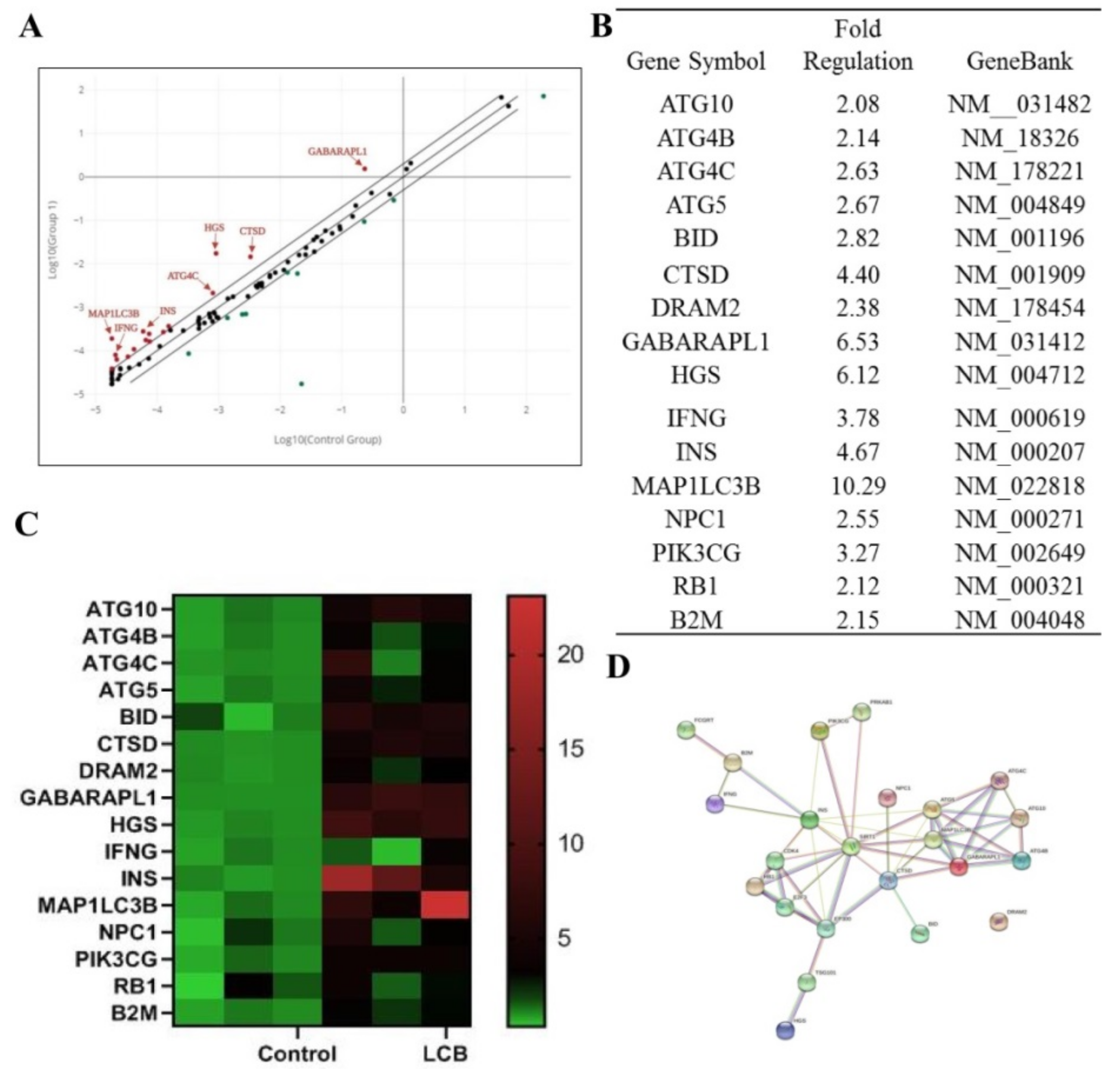
2.5. The Expression Profile of Autophagy–Related Genes in SH–SY5Y Human Cells after Treatment of LCB
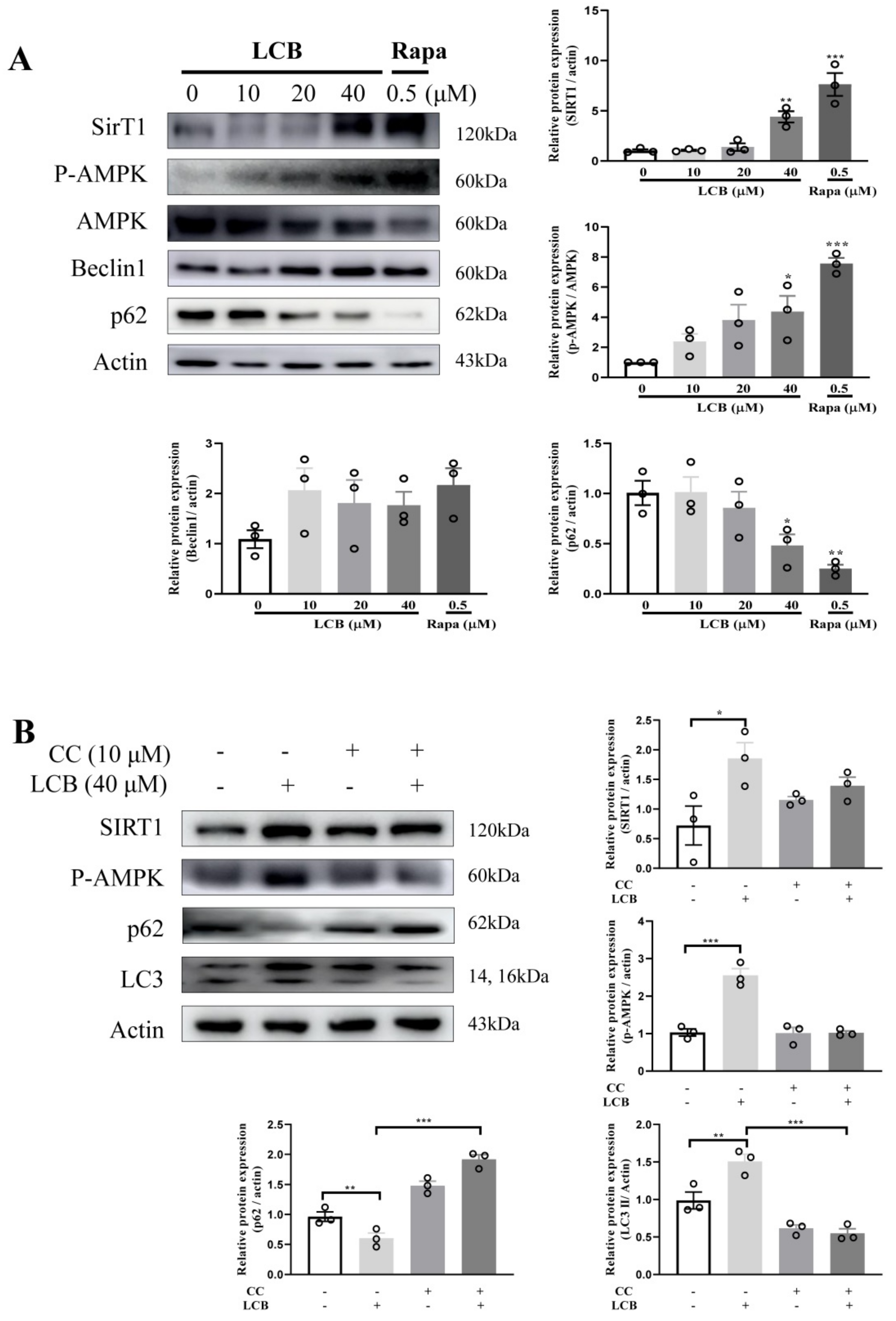
2.6. LCB Activated Autophagy via the AMPK/SIRT1 Signaling Pathway in PC-12 Cells
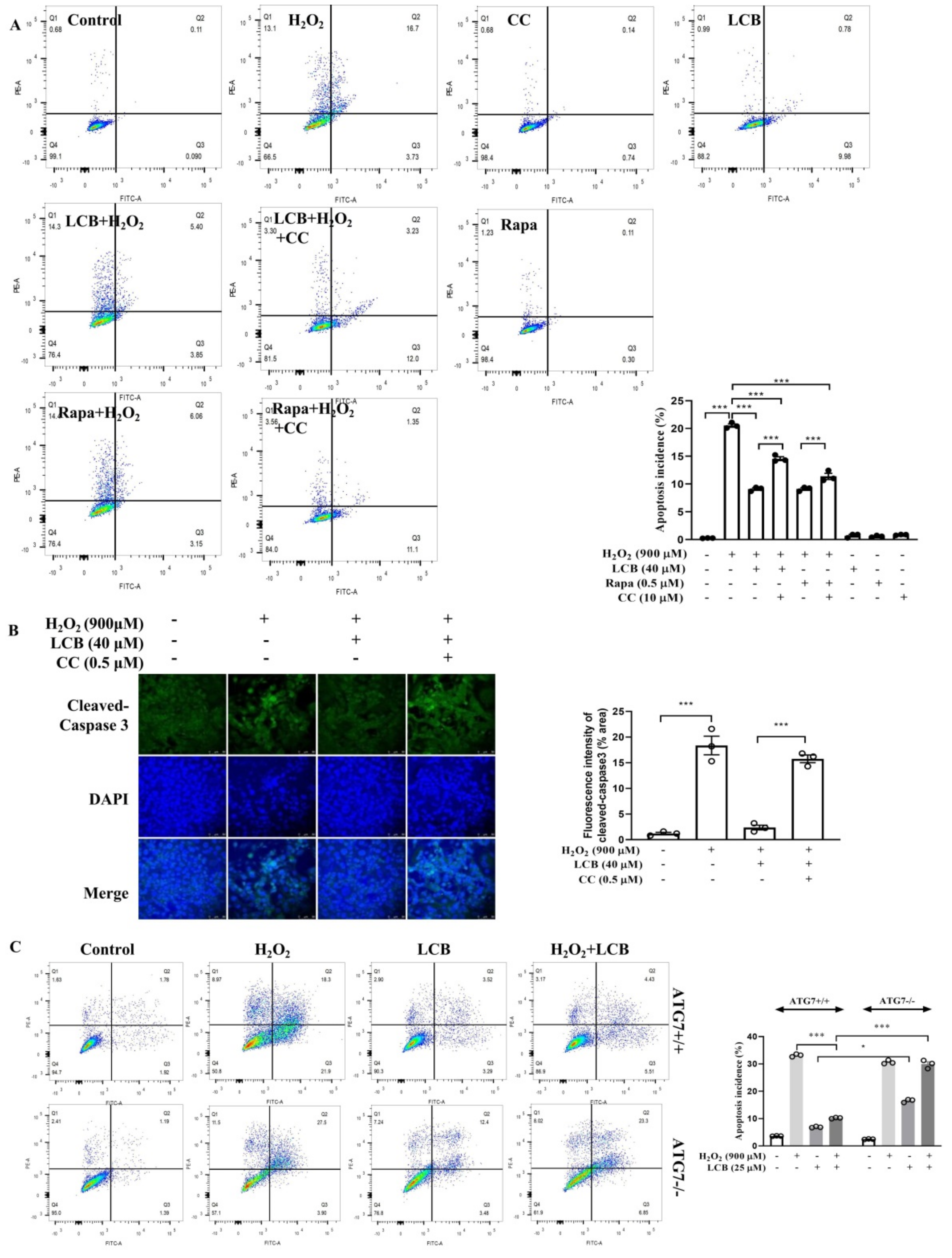
2.7. LCB Reduced H2O2-Induced Apoptosis through AMPK/SIRT1–Mediated Autophagy
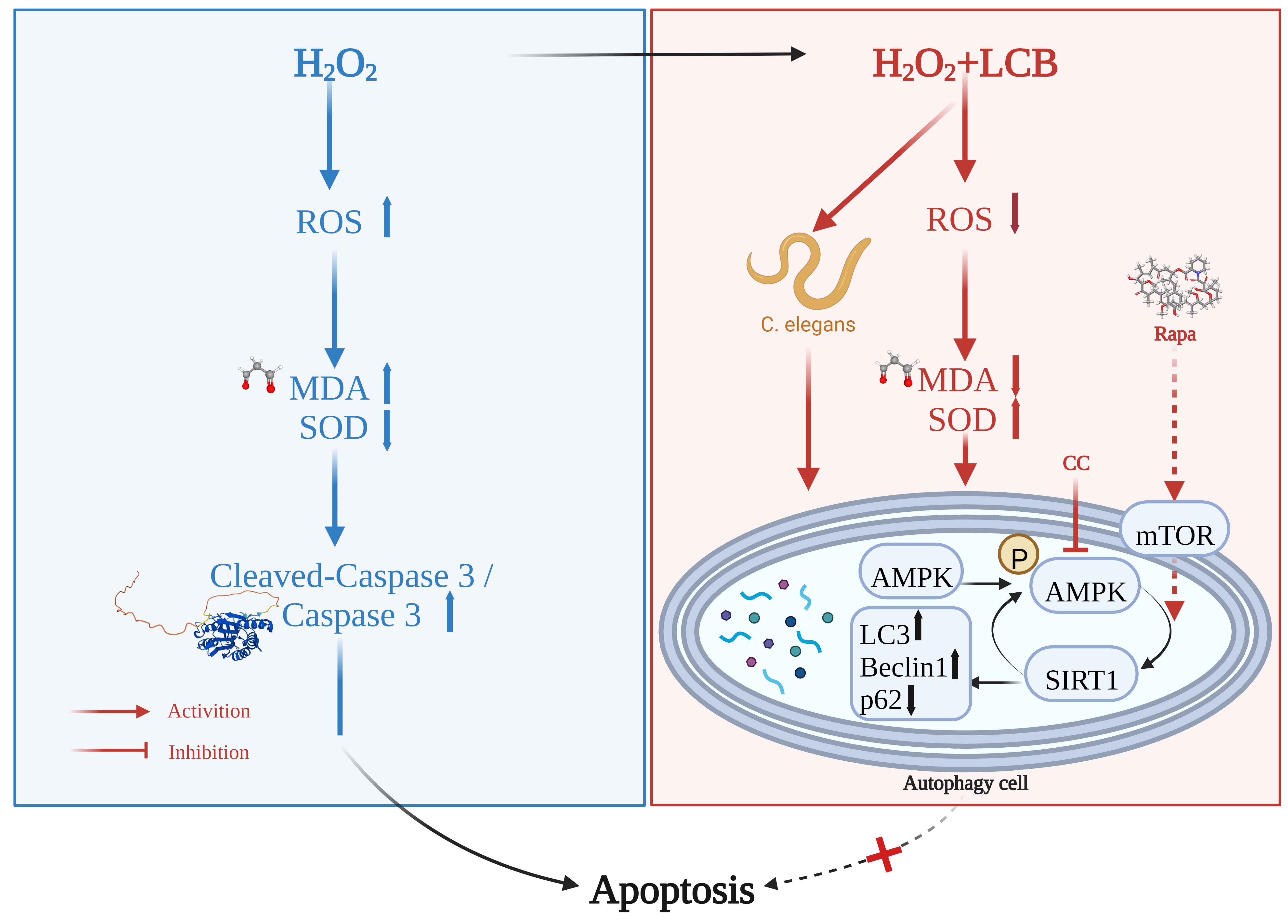
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Establishment of Cellular Oxidative Stress Model
4.4. Cell Viability Assay
4.5. Lactate Dehydrogenase Release Assay
4.6. Caspase–3 Activity Assay
4.7. Cellular Autophagy Staining Kit
4.8. Hoechst 33342 Staining
4.9. Quantification of Superoxide Dismutase (SOD) and Malondialdehyde (MDA)
4.10. Quantitation of Cellular Apoptosis
4.11. Quantitation of ROS Production
4.12. Quantitative Real–Time Polymerase Chain Reaction (PCR)
4.13. RT2 Profiler PCR Array
4.14. Western Blot Analysis
4.15. Gene Enrichment Analysis
4.16. Autophagy Assays in C. elegans
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharm. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R. What causes neurodegenerative disease? Folia Neuropathol. 2020, 58, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, F.G. In vivo detection of apoptosis. J. Nucl. Med. 2008, 49 (Suppl. S2), 81S–95S. [Google Scholar] [CrossRef]
- Schultz, D.R.; Harrington, W.J. Apoptosis: Programmed cell death at a molecular level. Semin. Arthritis Rheum. 2003, 32, 345–369. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef]
- Tian, Y.; Bustos, V.; Flajolet, M.; Greengard, P. A small-molecule enhancer of autophagy decreases levels of Aβ and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011, 25, 1934–1942. [Google Scholar] [CrossRef]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef]
- Huang, B.; Liu, J.; Ju, C.; Yang, D.; Chen, G.; Xu, S.; Zeng, Y.; Yan, X.; Wang, W.; Liu, D.; et al. Licochalcone A Prevents the Loss of Dopaminergic Neurons by Inhibiting Microglial Activation in Lipopolysaccharide (LPS)-Induced Parkinson’s Disease Models. Int. J. Mol. Sci. 2017, 18, 2043. [Google Scholar] [CrossRef] [Green Version]
- Maharajan, N.; Ganesan, C.; Moon, C.; Jang, C.-H.; Oh, W.-K.; Cho, G.-W. Licochalcone D Ameliorates Oxidative Stress-Induced Senescence via AMPK Activation. Int. J. Mol. Sci. 2021, 22, 7324. [Google Scholar] [CrossRef]
- Kim, S.S.; Lim, J.; Bang, Y.; Gal, J.; Lee, S.; Cho, Y.; Yoon, G.; Kang, B.Y.; Cheon, S.H.; Choi, H.J. Licochalcone E activates Nrf2/antioxidant response element signaling pathway in both neuronal and microglial cells: Therapeutic relevance to neurodegenerative disease. J. Nutr. Biochem. 2012, 23, 1314–1323. [Google Scholar] [CrossRef]
- Cao, Y.; Xu, W.; Huang, Y.; Zeng, X. Licochalcone B, a chalcone derivative from Glycyrrhiza inflata, as a multifunctional agent for the treatment of Alzheimer’s disease. Nat. Prod. Res. 2020, 34, 736–739. [Google Scholar] [CrossRef]
- Gao, X.-P.; Qian, D.-W.; Xie, Z.; Hui, H. Protective role of licochalcone B against ethanol-induced hepatotoxicity through regulation of Erk signaling. Iran. J. Basic Med. Sci. 2017, 20, 131–137. [Google Scholar]
- Chen, S.-Y.; Gao, Y.; Sun, J.; Meng, X.; Yang, D.; Fan, L.; Xiang, L.; Wang, P. Traditional Chinese medicine: Role in reducing β-amyloid, apoptosis, autophagy, neuroinflammation, oxidative stress, and mitochondrial dysfunction of Alzheimer’s disease. Front. Pharmacol. 2020, 11, 497. [Google Scholar] [CrossRef]
- Fang, J.; Zhu, Y.; Wang, H.; Cao, B.; Fei, M.; Niu, W.; Zhou, Y.; Wang, X.; Li, X.; Zhou, M.-L. Baicalin Protects Mice Brain From Apoptosis in Traumatic Brain Injury Model Through Activation of Autophagy. Front. Neurosci. 2019, 12, 1006. [Google Scholar] [CrossRef]
- Kumar, G.K.; Overholt, J.L.; Bright, G.R.; Hui, K.Y.; Lu, H.; Gratzl, M.; Prabhakar, N.R. Release of dopamine and norepinephrine by hypoxia from PC-12 cells. Am. J. Physiol. 1998, 274, C1592–C1600. [Google Scholar] [CrossRef]
- Ghaffari, H.; Venkataramana, M.; Ghassam, B.J.; Nayaka, S.C.; Nataraju, A.; Geetha, N.; Prakash, H. Rosmarinic acid mediated neuroprotective effects against H2O2-induced neuronal cell damage in N2A cells. Life Sci. 2014, 113, 7–13. [Google Scholar] [CrossRef]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and autophagy in Photoreceptors exposed to oxidative stress. Autophagy 2007, 3, 433–441. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, A.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Napolitano, M.; Tedesco, I.; Moccia, S.; Milito, A.; Russo, G.L. Neuroprotective Role of Natural Polyphenols. Curr. Top. Med. Chem. 2016, 16, 1943–1950. [Google Scholar] [CrossRef]
- Mamedov, N.A.; Egamberdieva, D. Phytochemical constituents and pharmacological effects of licorice: A review. Plant Hum. Health 2019, 3, 1–21. [Google Scholar]
- Jesberger, J.A.; Richardson, J.S. Oxygen Free-Radicals and Brain-Dysfunction. Int. J. Neurosci. 1991, 57, 1–17. [Google Scholar] [CrossRef]
- Muller, A.P.; Haas, C.B.; Camacho-Pereira, J.; Brochier, A.W.; Gnoatto, J.; Zimmer, E.R.; de Souza, D.O.; Galina, A.; Portela, L.V. Insulin prevents mitochondrial generation of H2O2 in rat brain. Exp. Neurol. 2013, 247, 66–72. [Google Scholar] [CrossRef]
- Picaud, J.C.; Steghens, J.P.; Auxenfans, C.; Barbieux, A.; Laborie, S.; Claris, O. Total malondialdehyde (MDA) concentrations as a marker of lipid peroxidation in all-in-one parenteral nutrition admixtures (APA) used in newborn infants. Pediatric Res. 2003, 53, 406a. [Google Scholar]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef]
- Kon, M.; Cuervo, A.M. Chaperone-mediated autophagy in health and disease. FEBS Lett. 2010, 584, 1399–1404. [Google Scholar] [CrossRef]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta-Mol. Cell Res. 2011, 1813, 238–259. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Y.; Liu, K.; Sun, X. Autophagy: A double-edged sword for neuronal survival after cerebral ischemia. Neural Regen. Res. 2014, 9, 1210–1216. [Google Scholar]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson’s Disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Feng, J.; Zhang, J.; Kang, X.; Qian, D. Quercetin modulates AMPK/SIRT1/NF-kappaB signaling to inhibit inflammatory/oxidative stress responses in diabetic high fat diet-induced atherosclerosis in the rat carotid artery. Exp. Ther. Med. 2020, 20, 280. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Wu, S. Biochemical and Structural Studies of p62/SQSTM1 and the Beclin1-UVRAG Interaction in Autophagy and Endosomal Trafficking. Ph.D. Thesis, Hong Kong Polytechnic University, Hong Kong, China, 2016. [Google Scholar]
- Kaminskyy, V.O.; Zhivotovsky, B. Free Radicals in Cross Talk Between Autophagy and Apoptosis. Antioxid. Redox Signal. 2014, 21, 86–102. [Google Scholar] [CrossRef]
- Collier, J.J.; Suomi, F.; Oláhová, M.; McWilliams, T.G.; Taylor, R.W. Emerging roles of ATG7 in human health and disease. Embo Mol. Med. 2021, 13, e14824. [Google Scholar] [CrossRef]
- Kaja, S.; Payne, A.J.; Singh, T.; Ghuman, J.K.; Sieck, E.G.; Koulen, P. An optimized lactate dehydrogenase release assay for screening of drug candidates in neuroscience. J. Pharmacol. Toxicol. Methods 2015, 73, 1–6. [Google Scholar] [CrossRef]
- Klein, A.; Hüsselmann, L.; Keyster, M.; Ludidi, N. Exogenous nitric oxide limits salt-induced oxidative damage in maize by altering superoxide dismutase activity. S. Afr. J. Bot. 2018, 115, 44–49. [Google Scholar] [CrossRef]
- Zhang, H.; Chang, J.T.; Guo, B.; Hansen, M.; Jia, K.; Kovács, A.L.; Kumsta, C.; Lapierre, L.R.; Legouis, R.; Lin, L.; et al. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 2015, 11, 9–27. [Google Scholar]
- Wang, Z.; Liu, Z.; Cao, Y.; Paudel, S.; Yoon, G.; Cheon, S.H. Short and Efficient Synthesis of Licochalcone B and D through Acid-Mediated Claisen-Schmidt Condensation. Bull. Korean Chem. Soc. 2013, 34, 3906–3908. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, L.; Wu, J.; Tang, Y.; Yun, X.; Lo, H.H.; Yu, L.; Li, W.; Wu, A.; Law, B.Y.K. Licochalcone B, a Natural Autophagic Agent for Alleviating Oxidative Stress-Induced Cell Death in Neuronal Cells and Caenorhabditis elegans Models. Pharmaceuticals 2022, 15, 1052. https://doi.org/10.3390/ph15091052
Qu L, Wu J, Tang Y, Yun X, Lo HH, Yu L, Li W, Wu A, Law BYK. Licochalcone B, a Natural Autophagic Agent for Alleviating Oxidative Stress-Induced Cell Death in Neuronal Cells and Caenorhabditis elegans Models. Pharmaceuticals. 2022; 15(9):1052. https://doi.org/10.3390/ph15091052
Chicago/Turabian StyleQu, Liqun, Jianhui Wu, Yong Tang, Xiaoyun Yun, Hang Hong Lo, Lu Yu, Wenhua Li, Anguo Wu, and Betty Yuen Kwan Law. 2022. "Licochalcone B, a Natural Autophagic Agent for Alleviating Oxidative Stress-Induced Cell Death in Neuronal Cells and Caenorhabditis elegans Models" Pharmaceuticals 15, no. 9: 1052. https://doi.org/10.3390/ph15091052
APA StyleQu, L., Wu, J., Tang, Y., Yun, X., Lo, H. H., Yu, L., Li, W., Wu, A., & Law, B. Y. K. (2022). Licochalcone B, a Natural Autophagic Agent for Alleviating Oxidative Stress-Induced Cell Death in Neuronal Cells and Caenorhabditis elegans Models. Pharmaceuticals, 15(9), 1052. https://doi.org/10.3390/ph15091052