
Applications of the Novel Quantitative Pharmacophore Activity Relationship Method QPhAR in Virtual Screening and Lead-Optimisation



Abstract
:1. Introduction
2. Results and Discussion
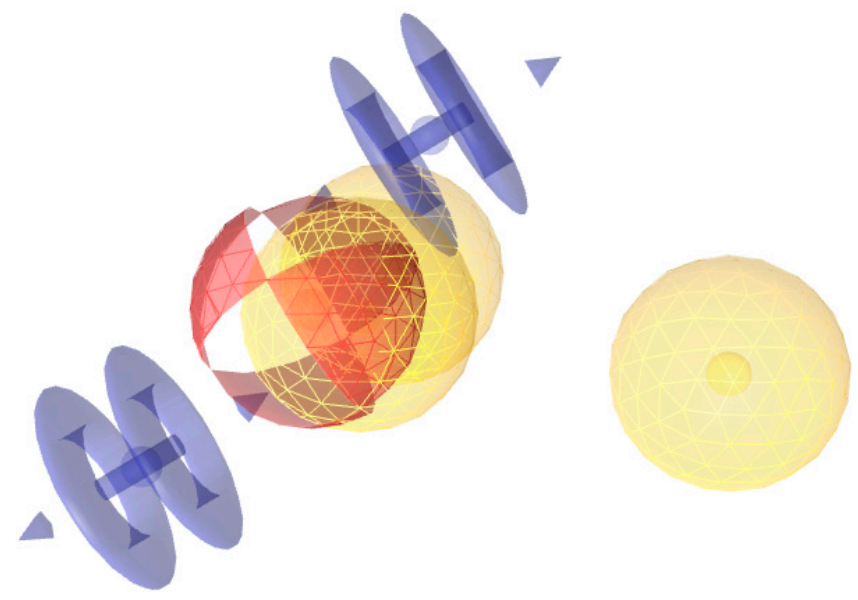
2.1. Generation of a Refined Pharmacophore for Virtual Screening
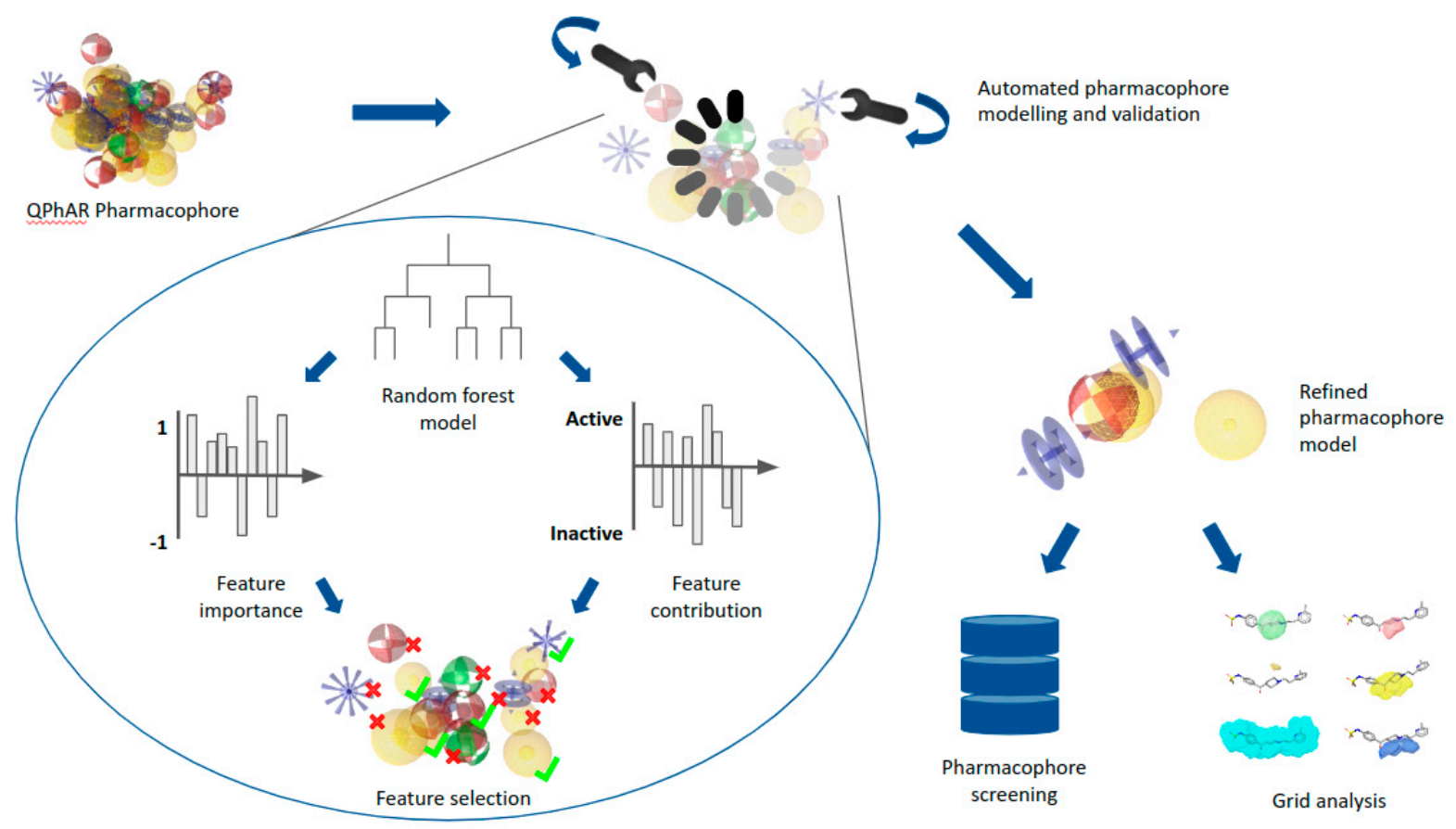
2.2. End-to-End Pharmacophore Modelling
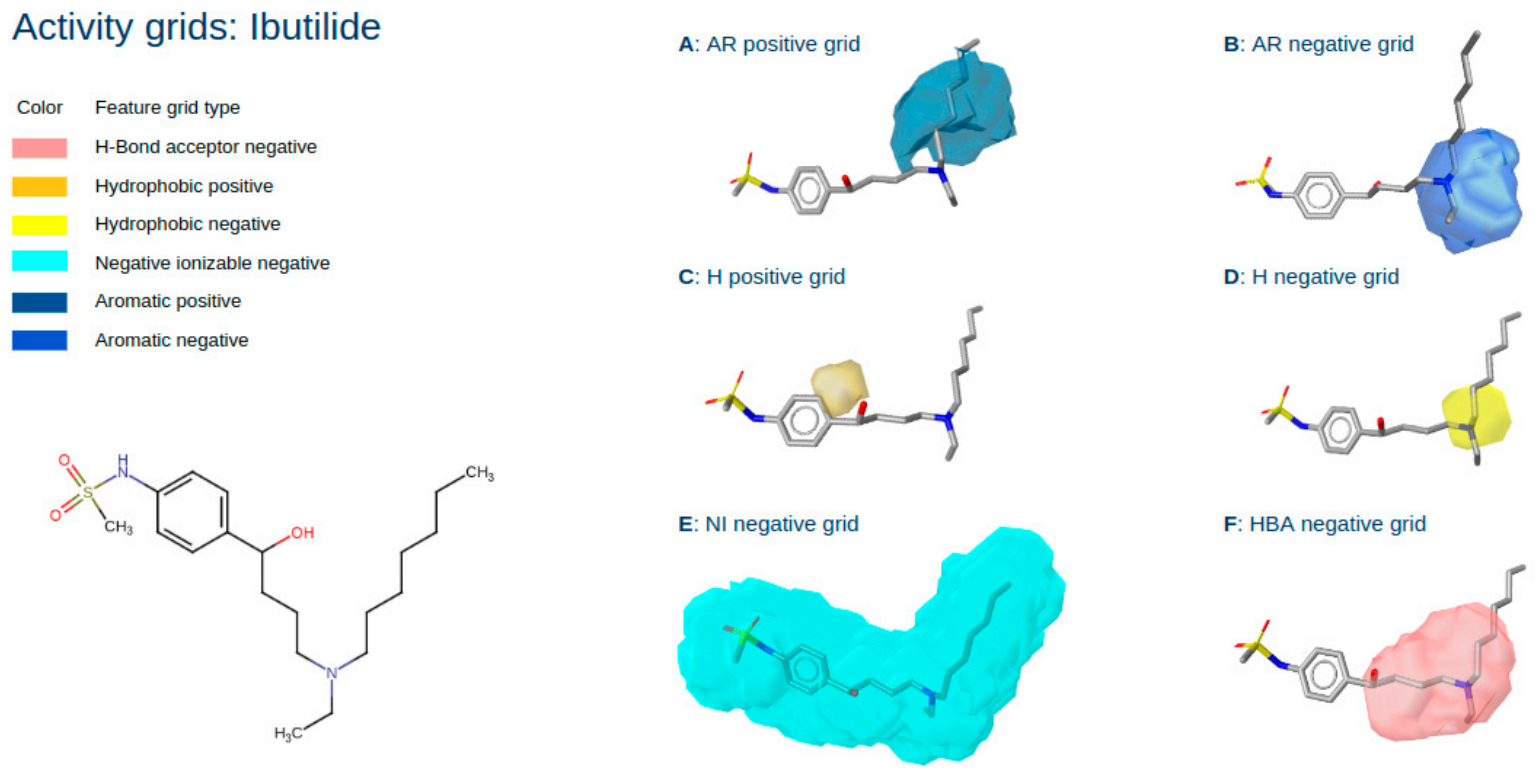
2.3. Three-Dimensional Pharmacophore Activity Profiling
3. Materials and Methods
3.1. Datasets and Training of QPhAR Models
- A separate training and test set has been defined previously.
- The training set contains between 15 and 30 molecules.
- Activity values for each compound in the dataset were measured in Ki or IC50 values.
- To avoid modelling experimental noise, the associated activity values range by at least three orders of magnitude.
3.2. Screening Baselines
3.3. Hyperparameter Optimisation
- Weight features by importance: True, False.
- Set exclusion volumes: True, False.
- Calculate feature contribution from ML (alternatively from QPhAR model): True, False.
- Number of resulting features: [4, 8].
3.4. Refined Pharmacophore Generation Algorithms
- Determination of feature importance.
- Determination of feature contribution.
- Processing negatively contributing features.
- Selection of features for refined pharmacophore.
3.5. Determination of Feature Importance
3.6. Determination of Feature Contribution
- Feature contribution information derived from the QPhAR pharmacophore model: As explained in the QPhAR publication [13], the QPhAR algorithm associates each newly generated pharmacophore feature with a list of activities. These activities will not only be used to determine the relevance of the feature—whether it is actual information or just adds noise to the model—but also to determine the contribution of a pharmacophore feature to the models’ predictions. The mean activity based on the list of associated features is calculated for each feature, resulting in one feature-activity for each pharmacophore feature. Finally, the feature-activities are compared against each other and scaled by their variance. Features with a positive sign of its scaled activity are considered to contribute positively to the prediction of the QPhAR model. Features with a negative sign contribute negatively to the prediction.
- Feature contribution information derived from the RF model: To extract feature contributions from an RF model in a deterministic way, two assumptions are made. First, the data provided to the machine learning model in the QPhAR algorithm represent the pairwise distances between features of the QPhAR model and the pharmacophore to predict. Second, applying the splitting criterion of each node in a tree of the random forest model will yield the left-child node for input values below or equal to the splitting threshold and the right-child node for input values above the splitting threshold. Both these assumptions are ensured by the implementation of the QPhAR algorithm as well as scikit-learn’s RF implementation.
- Following this logic, a simple algorithm can be devised to determine whether a feature contributes positively or negatively to the prediction of a sample. For each node in each tree, the node’s value is obtained and compared against its neighbouring node. Suppose the left child node has the higher predicted activity. In that case, we can assume that this feature contributes positively to activity since the left child node represents a smaller distance of pairwise pharmacophore features. At the same time, the right child node yields the lower activity prediction, which is associated with a larger distance of pharmacophore feature pairs. On the other hand, if the left child node yields the lower predicted activity, which is associated with a smaller feature pair distance, then the feature can be considered to contribute negatively to activity.
- During this process, the feature-id of each node is obtained, which corresponds to the pharmacophore feature it represents. The value of the feature with the corresponding feature-id is aggregated as the mean value of all nodes that either obtain their value from this feature-id or have a child node that obtains the prediction processing this feature-id. Once all trees and nodes are processed, a value representing the activity is obtained for each pharmacophore feature. These values are scaled as above by their variance. Once again, features with a positive sign are considered to contribute positively to the activity, whereas features with a negative sign are considered to contribute negatively to the activity.
3.7. Processing Negatively Contributing Features
3.8. Selection of Features for the Refined Output Pharmacophore
3.9. 3D Activity Profiling
3.10. Metrics
3.10.1. Fβ-Score
3.10.2. FSpecificity-Score
3.10.3. FComposite-Score
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caporuscio, F.; Tafi, A. Pharmacophore Modelling: A Forty Year Old Approach and Its Modern Synergies. Curr. Med. Chem. 2011, 18, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.-J.; Klebe, G.; Kubinyi, H. Wirkstoffdesign; Spektrum Akademischer Verlag: Heidelberg, Germany, 1996. [Google Scholar]
- Leach, A.R.; Gillet, V.J.; Lewis, R.A.; Taylor, R. Three-Dimensional Pharmacophore Methods in Drug Discovery. J. Med. Chem. 2010, 53, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Güner, O.F. Pharmacophore Perception, Development, and Use in Drug Design; Internat’l University Line: San Diego, CA, USA, 2000; pp. 6–8. [Google Scholar]
- Yang, S.-Y. Pharmacophore Modeling and Applications in Drug Discovery: Challenges and Recent Advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.O.; Dahabiyeh, L.A.; Bustanji, Y.; Zalloum, H.; Saleh, S. Combining Ligand-Based Pharmacophore Modeling, Quantitative Structure−Activity Relationship Analysis and in Silico Screening for the Discovery of New Potent Hormone Sensitive Lipase Inhibitors. J. Med. Chem. 2008, 51, 6478–6494. [Google Scholar] [CrossRef] [PubMed]
- Kurogi, Y.; Güner, O.F. Pharmacophore Modeling and Three-Dimensional Database Searching for Drug Design Using Catalyst. Curr. Med. Chem. 2001, 8, 1035–1055. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Neidhart, W.; Giller, T.; Schmid, G. “Scaffold-Hopping” by Topological Pharmacophore Search: A Contribution to Virtual Screening. Angew. Chem. Int. Ed. 1999, 38, 2894–2896. [Google Scholar] [CrossRef]
- Vuorinen, A.; Schuster, D. Methods for Generating and Applying Pharmacophore Models as Virtual Screening Filters and for Bioactivity Profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef]
- Mason, J.S.; Good, A.C.; Martin, E.J. 3-D Pharmacophores in Drug Discovery. Curr. Pharm. Des. 2001, 7, 567–597. [Google Scholar] [CrossRef]
- Chen, X.; Rusinko, A., 3rd; Tropsha, A.; Young, S.S. Automated pharmacophore identification for large chemical data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 887–896. [Google Scholar] [CrossRef]
- Baber, J.C.; Shirley, W.A.; Gao, Y.; Feher, M. The Use of Consensus Scoring in Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2006, 46, 277–288. [Google Scholar] [CrossRef]
- Kohlbacher, S.M.; Langer, T.; Seidel, T. QPHAR: Quantitative Pharmacophore Activity Relationship: Method and Validation. J. Cheminform. 2021, 13, 57. [Google Scholar] [CrossRef]
- Garg, D.; Gandhi, T.; Gopi Mohan, C. Exploring QSTR and Toxicophore of HERG K+ Channel Blockers Using GFA and HypoGen Techniques. J. Mol. Graph. Model. 2008, 26, 966–976. [Google Scholar] [CrossRef]
- Ece, A.; Sevin, F. The Discovery of Potential Cyclin A/CDK2 Inhibitors: A Combination of 3D QSAR Pharmacophore Modeling, Virtual Screening, and Molecular Docking Studies. Med. Chem. Res. 2013, 22, 5832–5843. [Google Scholar] [CrossRef]
- Ma, Y.; Li, H.-L.; Chen, X.-B.; Jin, W.-Y.; Zhou, H.; Ma, Y.; Wang, R.-L. 3D QSAR Pharmacophore Based Virtual Screening for Identification of Potential Inhibitors for CDC25B. Comput. Biol. Chem. 2018, 73, 1–12. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Li, L.-L.; Cao, Z.-X.; Luo, S.-D.; Wei, Y.-Q.; Yang, S.-Y. A Specific Pharmacophore Model of Aurora B Kinase Inhibitors and Virtual Screening Studies Based on It. Chem. Biol. Drug Des. 2009, 73, 115–126. [Google Scholar] [CrossRef]
- Krovat, E.M.; Langer, T. Non-Peptide Angiotensin II Receptor Antagonists: Chemical Feature Based Pharmacophore Identification. J. Med. Chem. 2003, 46, 716–726. [Google Scholar] [CrossRef]
- Schmid, M. Validation of the Novel Quantitative Pharmacophore Modeling Algorithm QPhAR. Master’s Thesis, University of Vienna, Vienna, Austria, 2022. [Google Scholar]
- Perry, M.; Sanguinetti, M.; Mitcheson, J. Revealing the Structural Basis of Action of HERG Potassium Channel Activators and Blockers. J. Physiol. 2010, 588, 3157–3167. [Google Scholar] [CrossRef]
- Garrido, A.; Lepailleur, A.; Mignani, S.M.; Dallemagne, P.; Rochais, C. HERG Toxicity Assessment: Useful Guidelines for Drug Design. Eur. J. Med. Chem. 2020, 195, 112290. [Google Scholar] [CrossRef]
- Zhang, J.; Gan, Y.; Li, H.; Yin, J.; He, X.; Lin, L.; Xu, S.; Fang, Z.; Kim, B.; Gao, L.; et al. Inhibition of the CDK2 and Cyclin A Complex Leads to Autophagic Degradation of CDK2 in Cancer Cells. Nat. Commun. 2022, 13, 2835. [Google Scholar] [CrossRef]
- Ece, A.; Sevin, F. Exploring QSAR on 4-Cyclohexylmethoxypyrimidines as Antitumor Agents for Their Inhibitory Activity of CDK2. Lett. Drug Des. Discov. 2010, 7, 625–631. [Google Scholar] [CrossRef]
- Seidel, T. Chemical Data Processing Toolkit, GitHub Repository. Available online: https://github.com/aglanger/CDPKit (accessed on 19 March 2021).
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kutlushina, A.; Khakimova, A.; Madzhidov, T.; Polishchuk, P. Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules 2018, 23, 3094. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Taha, A.A.; Hanbury, A. Metrics for Evaluating 3D Medical Image Segmentation: Analysis, Selection, and Tool. BMC Med. Imaging 2015, 15, 29. [Google Scholar] [CrossRef] [Green Version]
- Chinchor, N. MUC-4 Evaluation Metrics. In Proceedings of the 4th Conference on Message Understanding, McLean, VA, USA, 16 June 1992; pp. 22–29. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Source | FComposite-Score | QphAR Model Performance | ||
|---|---|---|---|---|
| Baseline | QphAR | R2 | RMSE | |
| Ece et al. [15] | 0.38 | 0.58 | 0.88 | 0.41 |
| Garg et al. [14] | 0.00 | 0.40 | 0.67 | 0.56 |
| Ma et al. [16] | 0.57 | 0.73 | 0.58 | 0.44 |
| Wang et al. [17] | 0.69 | 0.58 | 0.56 | 0.46 |
| Krovat et al. [18] | 0.94 | 0.56 | 0.50 | 0.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohlbacher, S.M.; Schmid, M.; Seidel, T.; Langer, T. Applications of the Novel Quantitative Pharmacophore Activity Relationship Method QPhAR in Virtual Screening and Lead-Optimisation. Pharmaceuticals 2022, 15, 1122. https://doi.org/10.3390/ph15091122
Kohlbacher SM, Schmid M, Seidel T, Langer T. Applications of the Novel Quantitative Pharmacophore Activity Relationship Method QPhAR in Virtual Screening and Lead-Optimisation. Pharmaceuticals. 2022; 15(9):1122. https://doi.org/10.3390/ph15091122
Chicago/Turabian StyleKohlbacher, Stefan Michael, Matthias Schmid, Thomas Seidel, and Thierry Langer. 2022. "Applications of the Novel Quantitative Pharmacophore Activity Relationship Method QPhAR in Virtual Screening and Lead-Optimisation" Pharmaceuticals 15, no. 9: 1122. https://doi.org/10.3390/ph15091122
APA StyleKohlbacher, S. M., Schmid, M., Seidel, T., & Langer, T. (2022). Applications of the Novel Quantitative Pharmacophore Activity Relationship Method QPhAR in Virtual Screening and Lead-Optimisation. Pharmaceuticals, 15(9), 1122. https://doi.org/10.3390/ph15091122