
Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives

 ,
, 
 , , , , , , ,
, , , , , , ,  , ,
, ,  and
and
Abstract
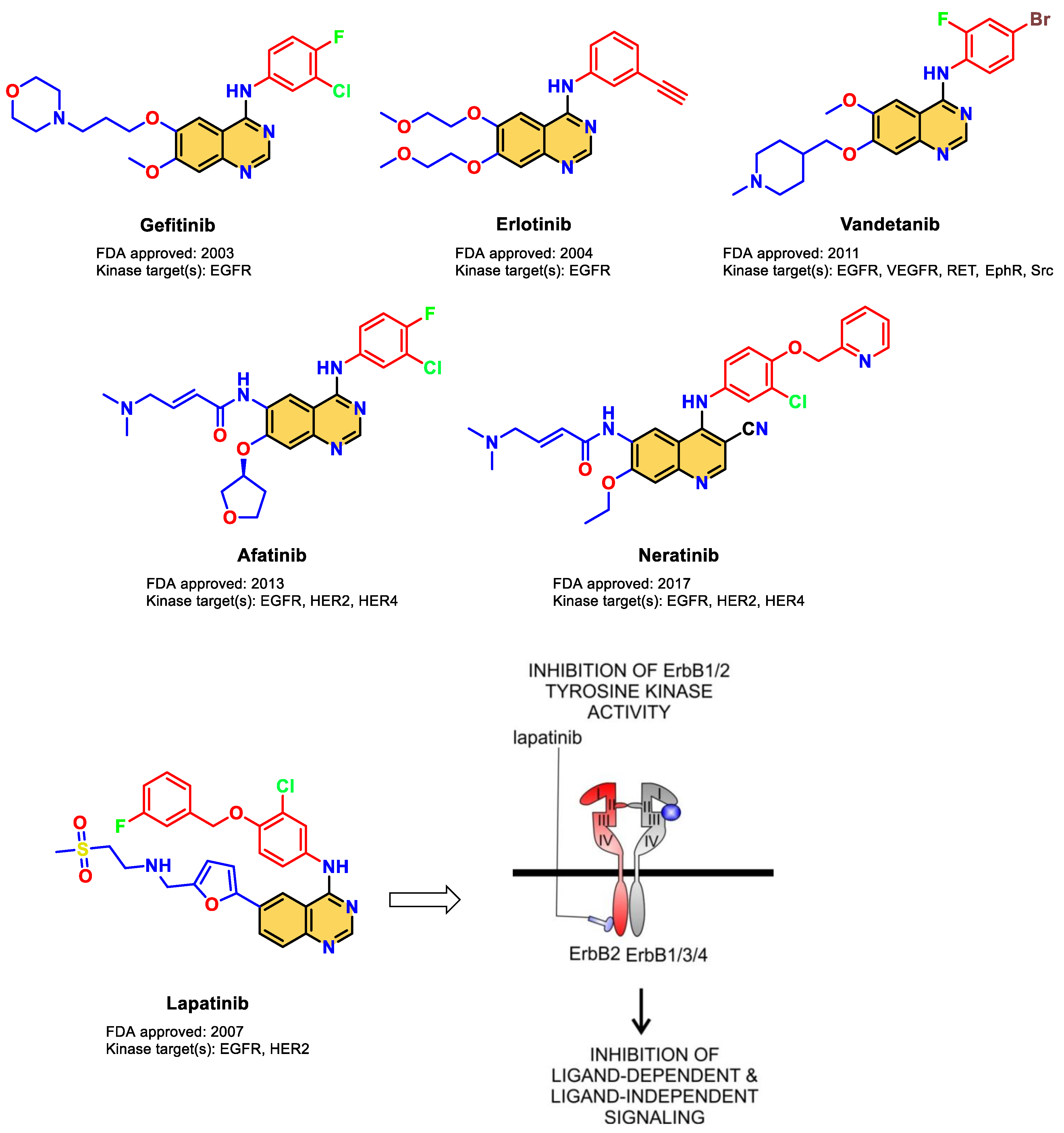
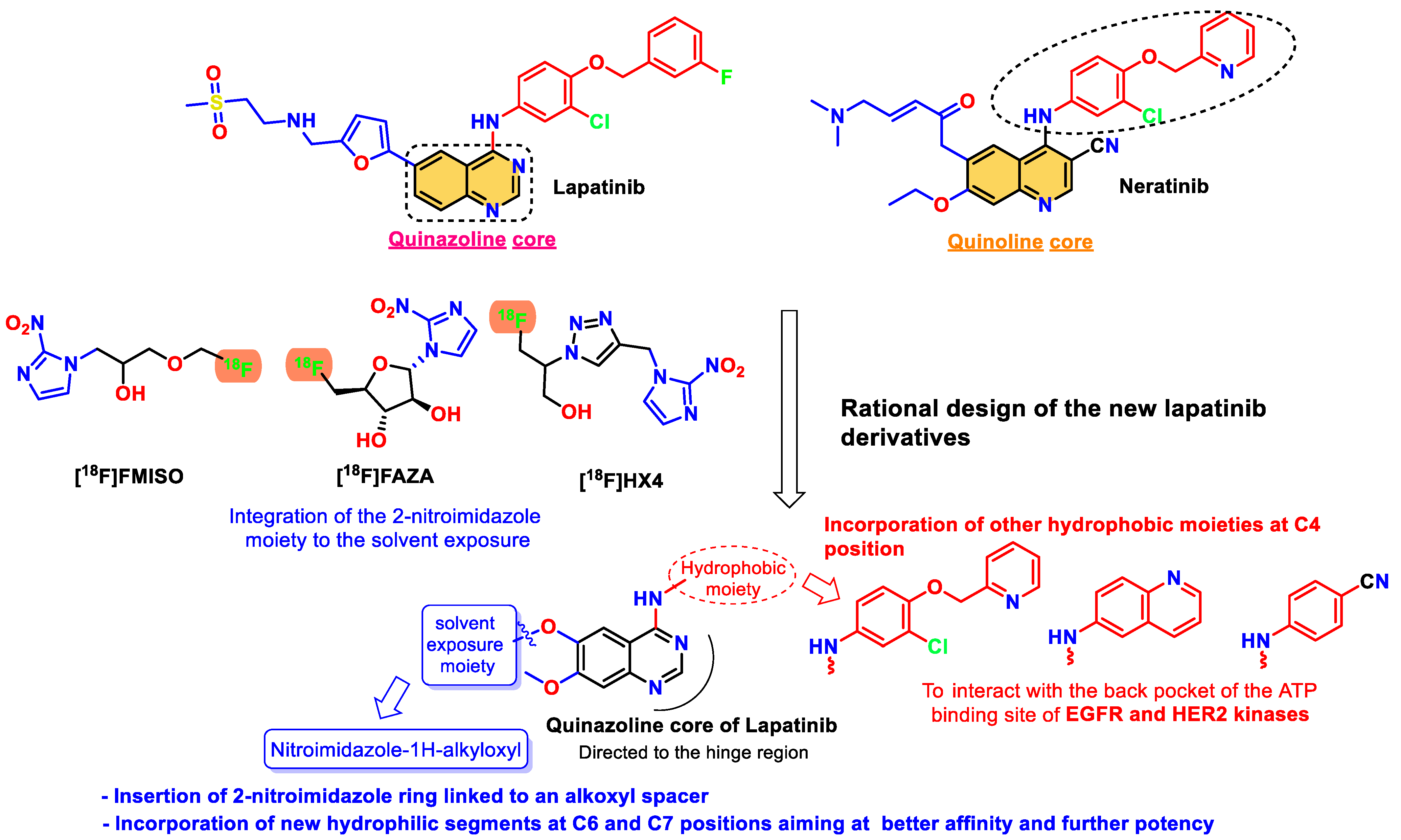
:1. Introduction

2. Materials and Methods
2.1. Chemical Reagents, Purification, and Instrumentation
2.2. Synthesis of 1-(n-Bromoalkyl)-2-nitro-1H-imidazoles (2a–d)
2.2.1. 1-(3-Bromopropyl)-2-nitro-1H-imidazole (2a)
2.2.2. 1-(4-Bromobutyl)-2-nitro-1H-imidazole (2b)
2.2.3. 1-(5-Bromopentyl)-2-nitro-1H-imidazole (2c)
2.2.4. 1-(6-Bromohexyl)-2-nitro-1H-imidazole (2d)
2.3. Synthesis of Intermediate Acetates 4a–c
2.3.1. 7-Methoxy-4-(quinolin-6-ylamino)quinazolin-6-yl acetate (4a)
2.3.2. 4-((4-Cyanophenyl)amino)-7-methoxyquinazolin-6-yl acetate (4b)
2.3.3. 4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino)-7-methoxyquinazolin-6-yl acetate (4c)
2.4. Synthesis of Pre-Final Intermediates 5a–c
2.4.1. 7-Methoxy-4-(quinolin-6-ylamino)quinazolin-6-ol (5a)
2.4.2. 4-((6-Hydroxy-7-methoxyquinazolin-4-yl)amino)benzonitrile (5b)
2.4.3. 4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino)-7-methoxyquinazolin-6-ol (5c)
2.5. Synthesis of the Target Lapatinib Derivatives 6a–l
2.5.1. 7-Methoxy-6-(3-(2-nitro-1H-imidazol-1-yl)propoxy)-N-(quinolin-6-yl)quinazolin-4-amine (6a)
2.5.2. 7-Methoxy-6-(4-(2-nitro-1H-imidazol-1-yl)butoxy)-N-(quinolin-6-yl)quinazolin-4-amine (6b)
2.5.3. 7-Methoxy-6-((5-(2-nitro-1H-imidazol-1-yl)pentyl)oxy)-N-(quinolin-6-yl)quinazolin-4-amine (6c)
2.5.4. 7-Methoxy-6-((6-(2-nitro-1H-imidazol-1-yl)hexyl)oxy)-N-(quinolin-6-yl)quinazolin-4-amine (6d)
2.5.5. 4-((7-Methoxy-6-(3-(2-nitro-1H-imidazol-1-yl)propoxy)quinazolin-4-yl)amino)benzonitrile (6e)
2.5.6. 4-((7-Methoxy-6-(4-(2-nitro-1H-imidazol-1-yl)butoxy)quinazolin-4-yl)amino)benzonitrile (6f)
2.5.7. 4-((7-Methoxy-6-((5-(2-nitro-1H-imidazol-1-yl)pentyl)oxy)quinazolin-4-yl)amino)benzonitrile (6g)
2.5.8. 4-((7-Methoxy-6-((6-(2-nitro-1H-imidazol-1-yl)hexyl)oxy)quinazolin-4-yl)amino)benzonitrile (6h)
2.5.9. N-(3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)-7-methoxy-6-(3-(2-nitro-1H-imidazol-1-yl)propoxy)quinazolin-4-amine (6i)
2.5.10. N-(3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)-7-methoxy-6-(4-(2-nitro-1H-imidazol-1-yl)butoxy)quinazolin-4-amine (6j)
2.5.11. N-(3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)-7-methoxy-6-((5-(2-nitro-1H-imidazol-1-yl)pentyl)oxy)quinazolin-4-amine (6k)
2.5.12. N-(3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)-7-methoxy-6-((6-(2-nitro-1H-imidazol-1-yl)hexyl)oxy)quinazolin-4-amine (6l)
2.6. In Vitro Kinase Assays
2.7. Molecular Docking Studies
2.8. Molecular Dynamics (MD) Simulations and MM-GBSA Study
3. Results and Discussion
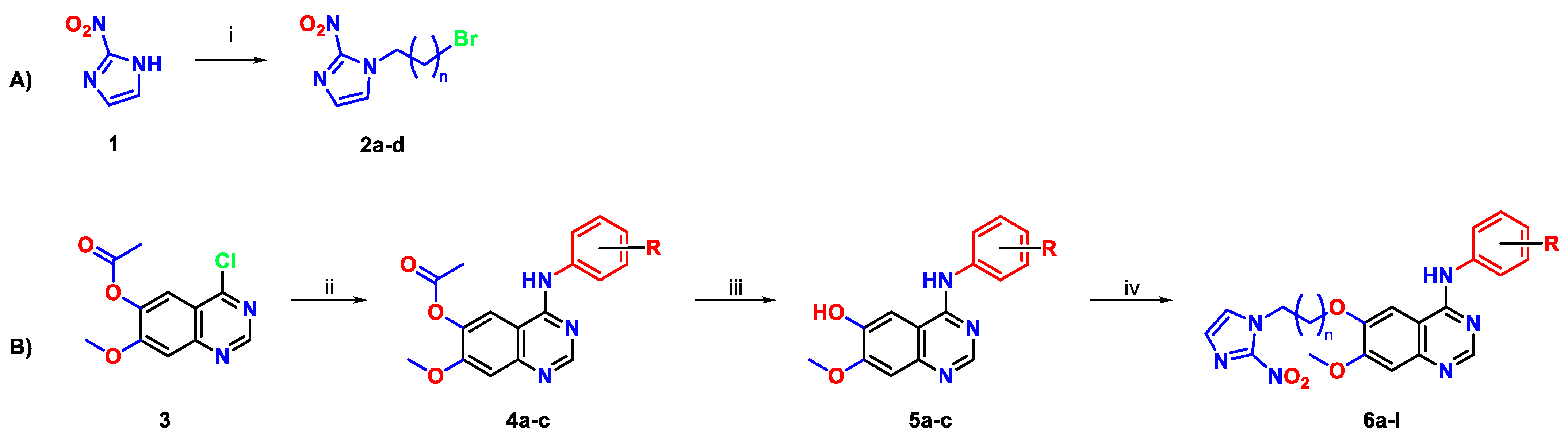
3.1. Chemical Synthesis
3.2. Structure Elucidation of the Newly Synthesized Lapatinib Derivatives 6a–l
3.3. In Silico Druggability Studies of the Newly Synthesized Lapatinib Derivatives 6a–l
3.4. Biological Evaluaiton
3.4.1. EGFR and HER2 Kinase Assay of Compounds 6a–l
3.4.2. Dose-Dependent Evaluation of Compound 6j over EGFR and HER2
3.4.3. Kinase Selectivity Panel of Compound 6j
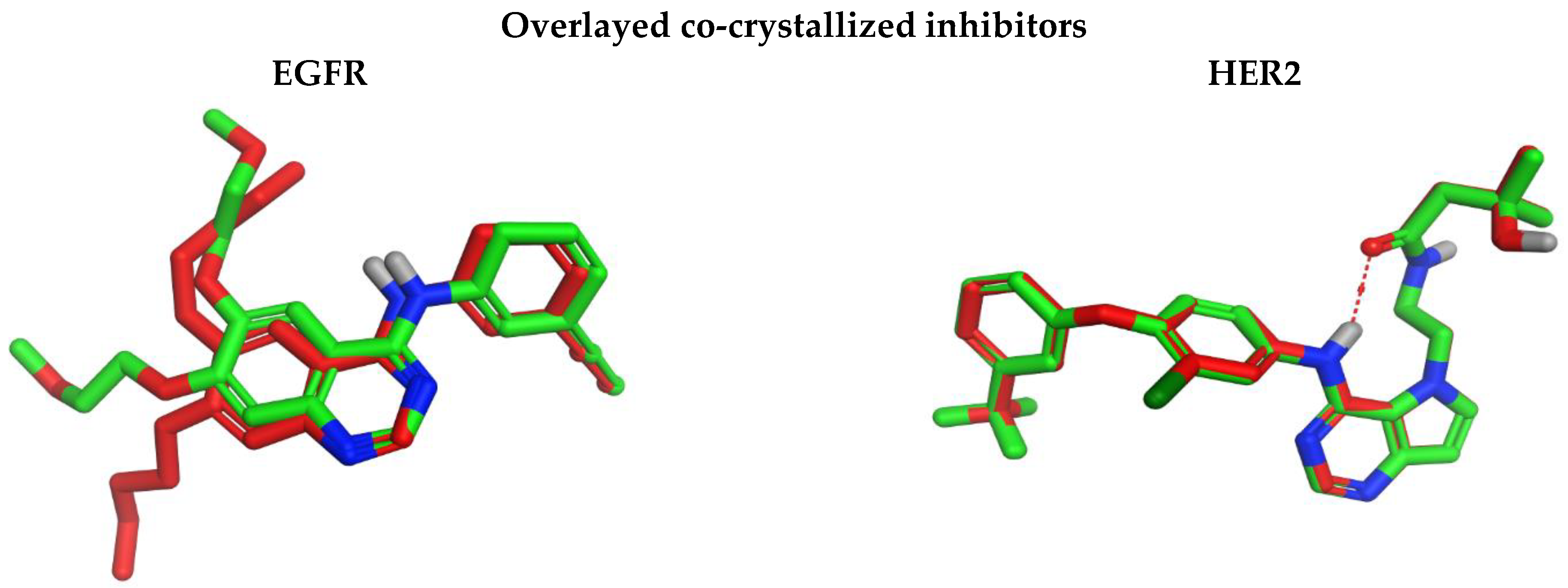
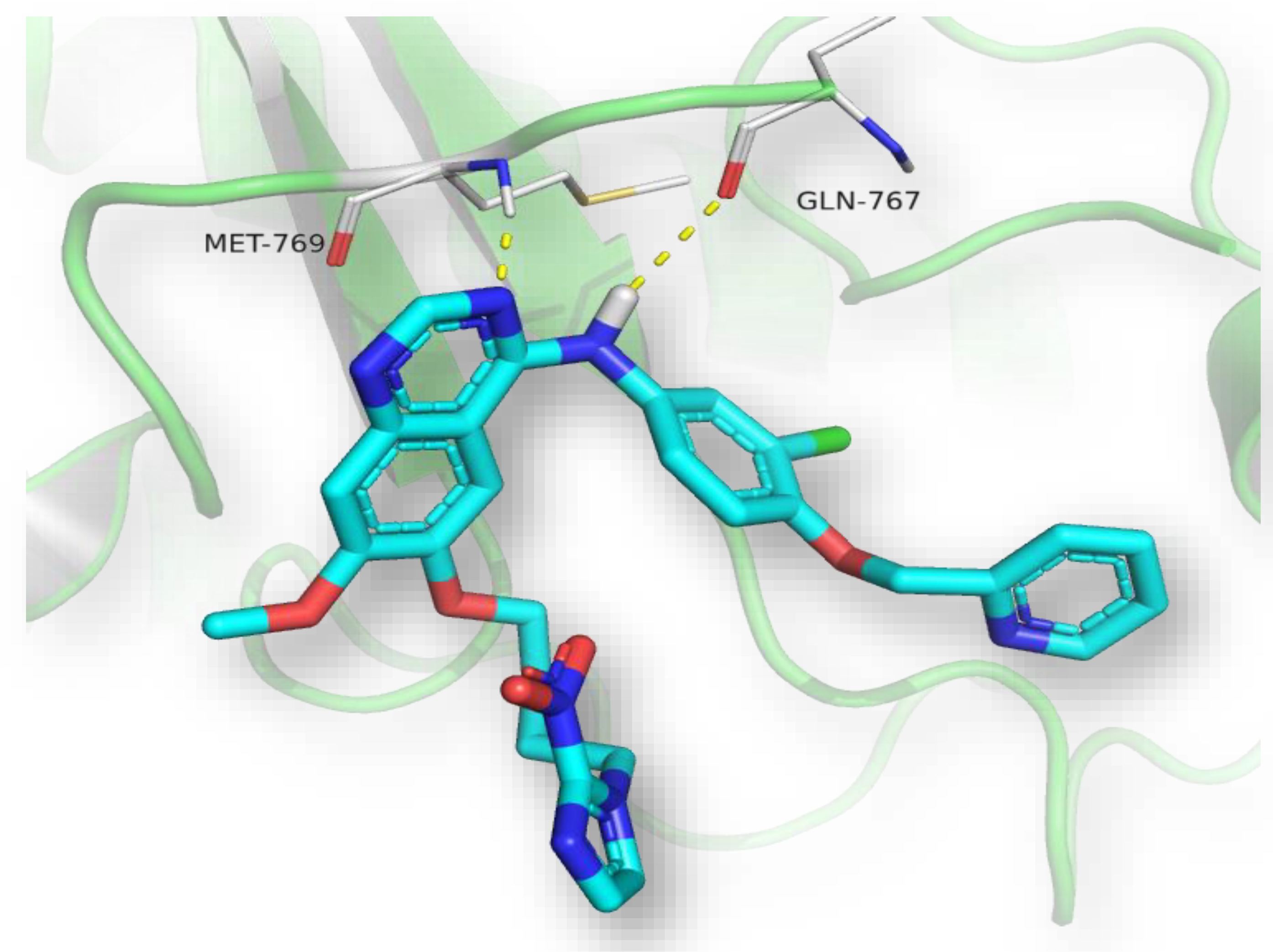
3.4.4. Molecular Docking
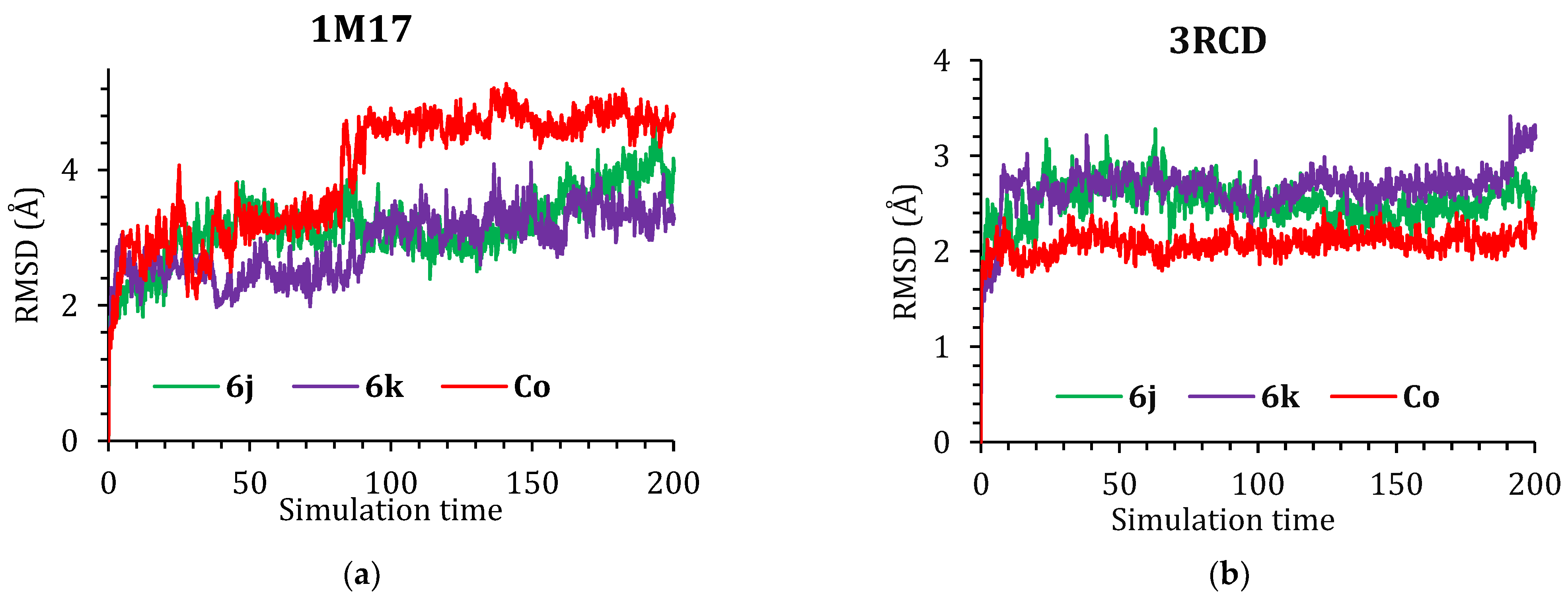
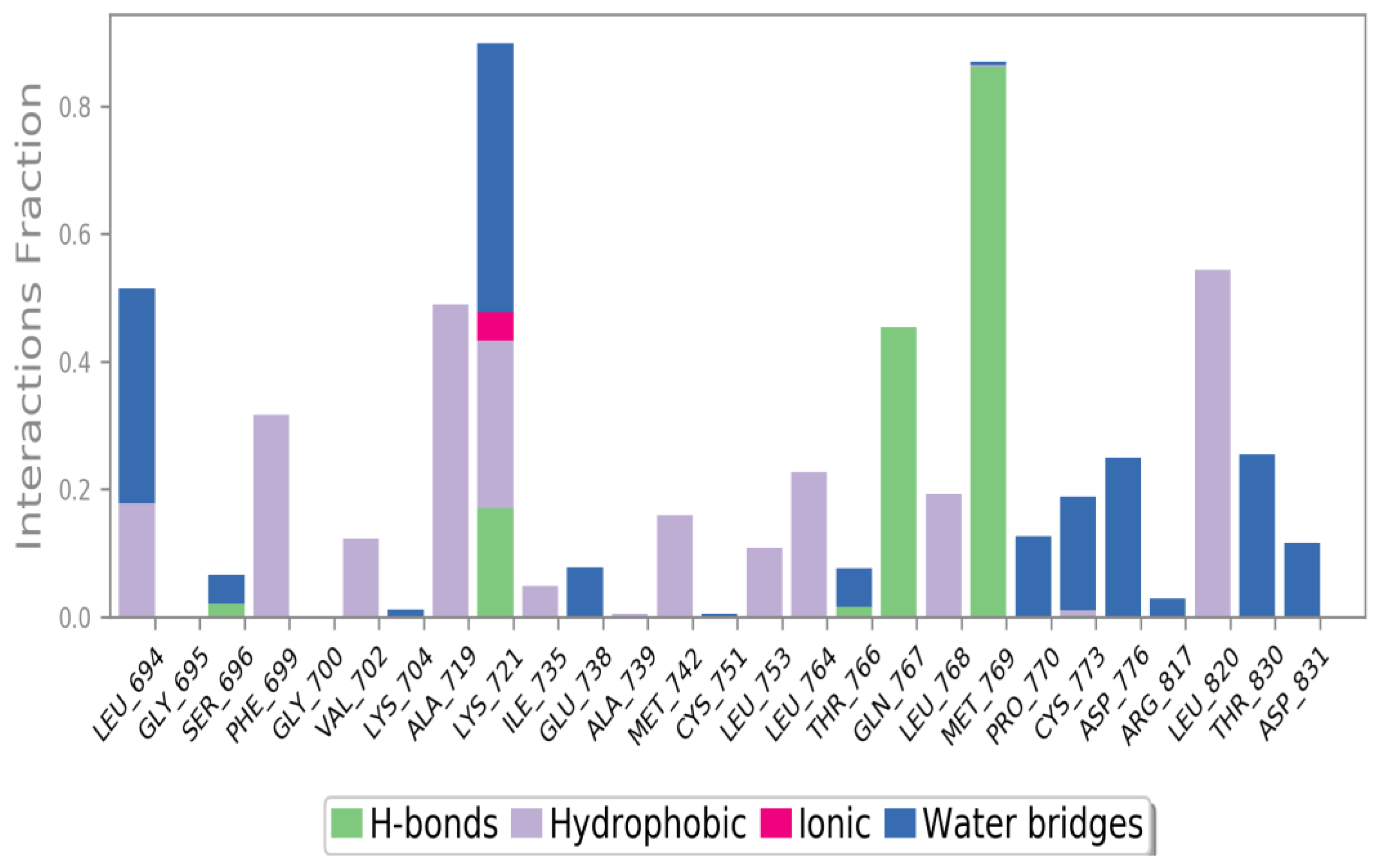
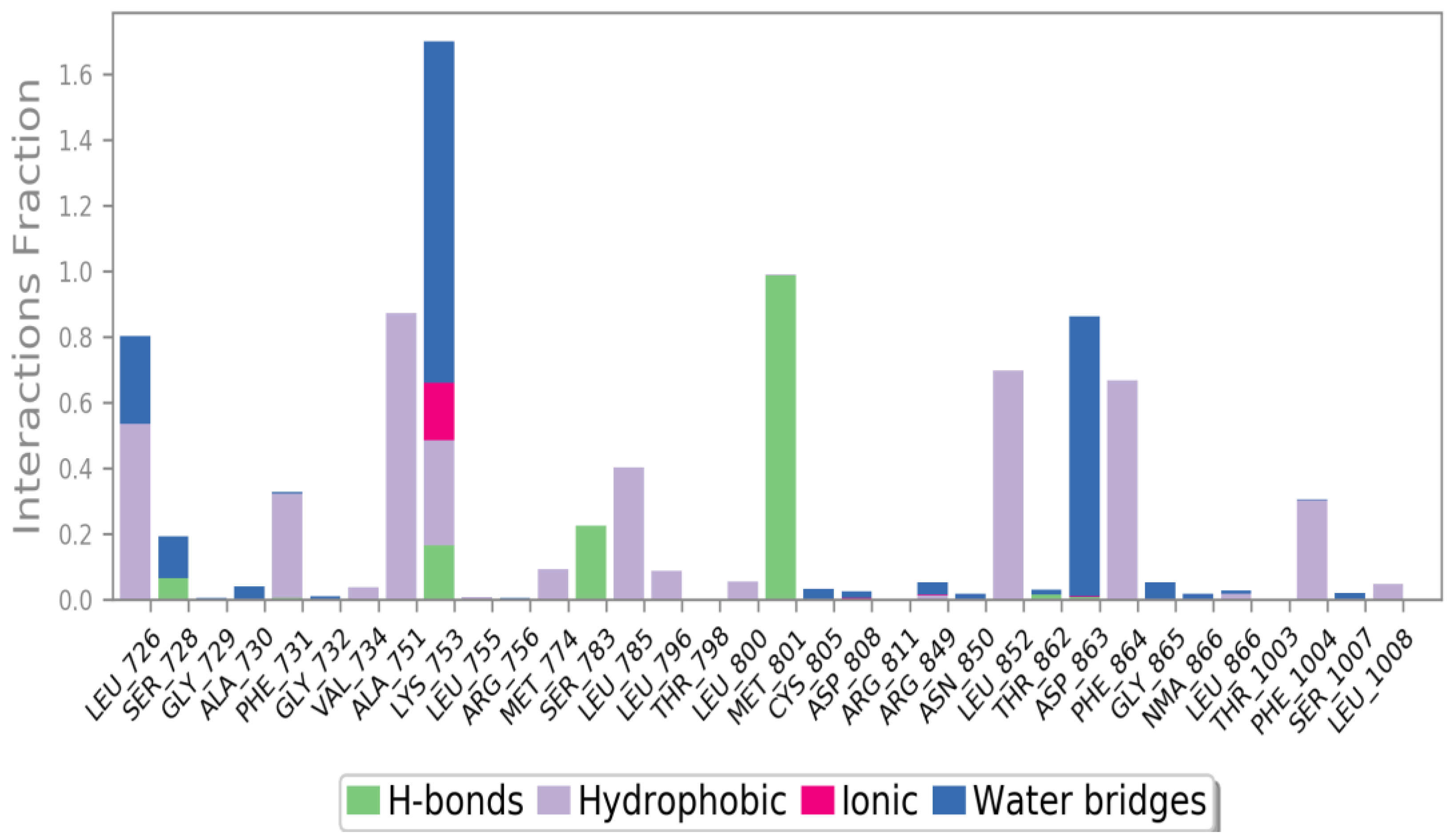
3.4.5. Molecular Dynamics (MD) Simulations
3.4.6. MM-GBSA Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Supuran, C.T.; Scozzafava, A. Protein tyrosine kinase inhibitors as anticancer agents. Expert Opin. Ther. Pat. 2004, 14, 35–53. [Google Scholar] [CrossRef]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Li, F.; Wei, Y.; Zhang, Y.; Cui, J.; Dai, R.; Chen, H.; Xie, J.; Cai, P. EGFR, HER2, and HER3 protein expression in paired primary tumor and lymph node metastasis of colorectal cancer. Sci. Rep. 2022, 12, 12894. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Subbiah, V.; Russo, A.; Banna, G.L.; Malapelle, U.; Rolfo, C.; Addeo, A. EGFR and HER2 exon 20 insertions in solid tumours: From biology to treatment. Nat. Rev. Clin. Oncol. 2022, 19, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Pu, T.; Chen, S.; Qiu, Y.; Zhong, X.; Zheng, H.; Chen, L.; Bu, H.; Ye, F. Breast cancers with EGFR and HER2 co-amplification favor distant metastasis and poor clinical outcome. Oncol. Lett. 2017, 14, 6562–6570. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Varella-Garcia, M.; Cappuzzo, F. Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene 2009, 28, S32–S37. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.; Vidal, L.; Shaw, H.; de Bono, J. Dual inhibition of ErbB1 (EGFR/HER1) and ErbB2 (HER2/neu). Eur. J. Cancer 2007, 43, 481–489. [Google Scholar] [CrossRef]
- Hynes, N.E.; Stern, D.F. The biology of erbB-2/nue/HER-2 and its role in cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 1994, 1198, 165–184. [Google Scholar] [CrossRef]
- Dowsett, M.; Cooke, T.; Ellis, I.; Gullick, W.J.; Gusterson, B.; Mallon, E.; Walker, R. Assessment of HER2 status in breast cancer: Why, when and how? Eur. J. Cancer 2000, 36, 170–176. [Google Scholar] [CrossRef]
- Kim, H.; Muller, W.J. The Role of the Epidermal Growth Factor Receptor Family in Mammary Tumorigenesis and Metastasis. Exp. Cell Res. 1999, 253, 78–87. [Google Scholar] [CrossRef]
- Koeppen, H.K.W.; Wright, B.D.; Burt, A.D.; Quirke, P.; McNicol, A.M.; Dybdal, N.O.; Sliwkowski, M.X.; Hillan, K.J. Overexpression of HER2/neu in solid tumours: An immunohistochemical survey. Histopathology 2001, 38, 96–104. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.R.; Mishra, P.; Abraham, J. Neratinib, A Novel HER2-Targeted Tyrosine Kinase Inhibitor. Clin. Breast Cancer 2016, 16, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.; Leary, A. Lapatinib: A novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs Today 2006, 42, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, P.; Sharma, V.; Alam, O.; Manaithiya, A.; Alam, P.; Kahksha; Alam, M.T.; Imran, M. Novel quinazoline-based EGFR kinase inhibitors: A review focussing on SAR and molecular docking studies (2015–2019). Eur. J. Med. Chem. 2020, 204, 112640. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Moghimi, S.; Toolabi, M.; Foroumadi, A. Pyrimidine-based EGFR TK inhibitors in targeted cancer therapy. Eur. J. Med. Chem. 2021, 221, 113523. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Farag, A.K.; Viswanath, A.N.; Bedair, T.M.; Leem, D.G.; Lee, K.T.; Pae, A.N.; Roh, E.J. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: Design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg. Med. Chem. Lett. 2015, 25, 5147–5154. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; et al. Design and Synthesis of Novel Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitors Bearing a Pyrrolo[3,2-d]pyrimidine Scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Verma, C.; Guzman, M.; Jimenez, J.; Parra, J.L.; Pedersen, K.; Smith, D.J.; Landolfi, S.; Ramon y Cajal, S.; Arribas, J.; et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 2009, 28, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib): Relationships among Protein Conformation, Inhibitor Off-Rate, and Receptor Activity in Tumor Cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.-M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations that Are Resistant to Gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Ji, H.; Yuza, Y.; Meyerson, M.; Wong, K.-K.; Tenen, D.G.; Halmos, B. An Alternative Inhibitor Overcomes Resistance Caused by a Mutation of the Epidermal Growth Factor Receptor. Cancer Res. 2005, 65, 7096–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.-M.; Gilmer, T.M. Novel Mechanism of Lapatinib Resistance in HER2-Positive Breast Tumor Cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Quan, H.; Zhao, J.; Xie, C.; Wang, L.; Lou, L. RON confers lapatinib resistance in HER2-positive breast cancer cells. Cancer Lett. 2013, 340, 43–50. [Google Scholar] [CrossRef]
- Rexer, B.N.; Ham, A.J.L.; Rinehart, C.; Hill, S.; de Matos Granja-Ingram, N.; Gonzalez-Angulo, A.M.; Mills, G.B.; Dave, B.; Chang, J.C.; Liebler, D.C.; et al. Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene 2011, 30, 4163–4174. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, P.J.A.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-Kinase Hyperactivation Results in Lapatinib Resistance that Is Reversed by the mTOR/Phosphatidylinositol 3-Kinase Inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [Green Version]
- Lancini, G.C.; Arioli, V.; Lazzari, E.; Bellani, P. Synthesis and relationship between structure and activity of 2-nitroimidazole derivatives. J. Med. Chem. 1969, 12, 775–780. [Google Scholar] [CrossRef]
- Noss, M.B.; Panicucci, R.; McClelland, R.A.; Rauth, A.M. 1-Methyl-2-nitrosoimidazole: Cytotoxic and glutathione depleting capabilities. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Hoigebazar, L.; Jeong, J.M.; Lee, J.Y.; Shetty, D.; Yang, B.Y.; Lee, Y.S.; Lee, D.S.; Chung, J.K.; Lee, M.C. Syntheses of 2-nitroimidazole derivatives conjugated with 1,4,7-triazacyclononane-N,N’-diacetic acid labeled with F-18 using an aluminum complex method for hypoxia imaging. J. Med. Chem. 2012, 55, 3155–3162. [Google Scholar] [CrossRef] [PubMed]
- Hervent, A.S.; De Keulenaer, G.W. Molecular mechanisms of cardiotoxicity induced by ErbB receptor inhibitor cancer therapeutics. Int. J. Mol. Sci. 2012, 13, 12268–12286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Shustov, G.; Liang, H.; Khlebnikov, V.; Zheng, W.; Yang, X.H.; Cheeseman, C.; Wiebe, L.I. Design, synthesis, and preliminary biological evaluation of 6-O-glucose-azomycin adducts for diagnosis and therapy of hypoxic tumors. J. Med. Chem. 2012, 55, 6033–6046. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, J.; Janssen, M.H.; Ollers, M.; Aerts, H.J.; Dubois, L.; Hochstenbag, M.; Dingemans, A.M.; Lalisang, R.; Brans, B.; Windhorst, B.; et al. PET imaging of hypoxia using [18F]HX4: A phase I trial. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1663–1668. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Zhu, S.; Ma, X.; Qiu, N.; Peng, P.; Sheng, R.; Hu, Y. Design, synthesis and biological evaluation of 6-(nitroimidazole-1H-alkyloxyl)-4-anilinoquinazolines as efficient EGFR inhibitors exerting cytotoxic effects both under normoxia and hypoxia. Eur. J. Med. Chem. 2015, 89, 826–834. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Kim, H.J.; Elsherbeny, M.H.; Paik, S.; Park, J.H.; Gotina, L.; Abdellattif, M.H.; Gouda, N.A.; Cho, J.; Lee, K.; et al. Discovery of 3,4-dichloro-N-(1H-indol-5-yl)benzamide: A highly potent, selective, and competitive hMAO-B inhibitor with high BBB permeability profile and neuroprotective action. Bioorg. Chem. 2021, 116, 105352. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Paik, S.; Kim, H.J.; Park, J.H.; Londhe, A.M.; Lee, K.; Pae, A.N.; Park, K.D.; Roh, E.J. Discovery of N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide: A novel, selective, and competitive indole-based lead inhibitor for human monoamine oxidase B. J. Enzym. Inhib. Med. Chem. 2020, 35, 1568–1580. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Woo, J.; Gouda, N.A.; Kim, J.; Nada, H.; Roh, E.J.; Park, K.D.; Cho, J.; Lee, K. Melatonin Analogues Potently Inhibit MAO-B and Protect PC12 Cells against Oxidative Stress. Antioxidants 2021, 10, 1604. [Google Scholar] [CrossRef]
- Elsherbeny, M.H.; Kim, J.; Gouda, N.A.; Gotina, L.; Cho, J.; Pae, A.N.; Lee, K.; Park, K.D.; Elkamhawy, A.; Roh, E.J. Highly Potent, Selective, and Competitive Indole-Based MAO-B Inhibitors Protect PC12 Cells against 6-Hydroxydopamine- and Rotenone-Induced Oxidative Stress. Antioxidants 2021, 10, 1641. [Google Scholar] [CrossRef]
- Cheng, W.; Yuan, Y.; Qiu, N.; Peng, P.; Sheng, R.; Hu, Y. Identification of novel 4-anilinoquinazoline derivatives as potent EGFR inhibitors both under normoxia and hypoxia. Bioorganic Med. Chem. 2014, 22, 6796–6805. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Zhang, Q.; Zhang, Y.; Shao, J.; Ding, L.; Tang, C.; Feng, B. Synthesis and biological evaluation of new series of quinazoline derivatives as EGFR/HER2 dual-target inhibitors. Bioorganic Med. Chem. Lett. 2022, 67, 128703. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Nada, H.; Byun, H.J.; Lee, C.H.; Elkamhawy, A. Hit Identification of a Novel Quinazoline Sulfonamide as a Promising EphB3 Inhibitor: Design, Virtual Combinatorial Library, Synthesis, Biological Evaluation, and Docking Simulation Studies. Pharmaceuticals 2021, 14, 1247. [Google Scholar] [CrossRef] [PubMed]
- Elsherbeny, M.H.; Ammar, U.M.; Abdellattif, M.H.; Abourehab, M.A.S.; Abdeen, A.; Ibrahim, S.F.; Abdelrahaman, D.; Mady, W.; Roh, E.J.; Elkamhawy, A. 2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic Acid as a Novel and Selective Aurora A Kinase Inhibitory Lead with Apoptosis Properties: Design, Synthesis, In Vitro and In Silico Biological Evaluation. Life 2022, 12, 876. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2016; p. 1010. [Google Scholar]
- Madbouly, E.; Lashine, E.-S.; Al-Karmalawy, A.A.; Sebaiy, M.; Pratsinis, H.; Kletsas, D.; Metwally, K. Design and Synthesis of Novel Quinazolinone-Chalcone Hybrids as Potential Apoptotic Candidates Targeting Caspase-3 and PARP-1: In Vitro, Molecular docking, and SAR Studies. New J. Chem. 2022, 46. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [Green Version]
- Mansour, K.A.; Elbermawi, A.; Al-Karmalawy, A.A.; Lahloub, M.-F.; El-Neketi, M. Cytotoxic effects of extracts obtained from plants of the Oleaceae family: Bio-guided isolation and molecular docking of new secoiridoids from Jasminum humile. Pharm. Biol. 2022, 60, 1374–1383. [Google Scholar] [CrossRef]
- Salem, M.A.; El-Shiekh, R.A.; Aborehab, N.M.; Al-Karmalawy, A.A.; Ezzat, S.M.; Alseekh, S.; Fernie, A.R. Metabolomics driven analysis of Nigella sativa seeds identifies the impact of roasting on the chemical composition and immunomodulatory activity. Food Chem. 2022, 398, 133906. [Google Scholar] [CrossRef]
- Kutkat, O.; Moatasim, Y.; Al-Karmalawy, A.A.; Abulkhair, H.S.; Gomaa, M.R.; El-Taweel, A.N.; Abo Shama, N.M.; GabAllah, M.; Mahmoud, D.B.; Kayali, G.; et al. Robust antiviral activity of commonly prescribed antidepressants against emerging coronaviruses: In vitro and in silico drug repurposing studies. Sci. Rep. 2022, 12, 12920. [Google Scholar] [CrossRef]
- Mahmoud, A.; Mostafa, A.; Al-Karmalawy, A.A.; Zidan, A.; Abulkhair, H.S.; Mahmoud, S.H.; Shehata, M.; Elhefnawi, M.M.; Ali, M.A. Telaprevir is a potential drug for repurposing against SARS-CoV-2: Computational and in vitro studies. Heliyon 2021, 7, e07962. [Google Scholar] [CrossRef]
- Hammoud, M.M.; Khattab, M.; Abdel-Motaal, M.; Van der Eycken, J.; Alnajjar, R.; Abulkhair, H.; Al-Karmalawy, A.A. Synthesis, structural characterization, DFT calculations, molecular docking, and molecular dynamics simulations of a novel ferrocene derivative to unravel its potential antitumor activity. J. Biomol. Struct. Dyn. 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ezz Eldin, R.R.; Saleh, M.A.; Alotaibi, M.H.; Alsuair, R.K.; Alzahrani, Y.A.; Alshehri, F.A.; Mohamed, A.F.; Hafez, S.M.; Althoqapy, A.A.; Khirala, S.K.; et al. Ligand-based design and synthesis of N’-Benzylidene-3,4-dimethoxybenzohydrazide derivatives as potential antimicrobial agents; evaluation by in vitro, in vivo, and in silico approaches with SAR studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1098–1119. [Google Scholar] [CrossRef] [PubMed]
- El-Masry, R.M.; Al-Karmalawy, A.A.; Alnajjar, R.; Mahmoud, S.H.; Mostafa, A.; Kadry, H.H.; Abou-Seri, S.M.; Taher, A.T. Newly synthesized series of oxoindole–oxadiazole conjugates as potential anti-SARS-CoV-2 agents: In silico and in vitro studies. New J. Chem. 2022, 46, 5078–5090. [Google Scholar] [CrossRef]
- Release, S. 3: Desmond Molecular Dynamics System, DE Shaw Research, Schrödinger; Maestro-Desmond Interoperability Tools: New York, NY, USA, 2017. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.; Knight, J.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Manual, D.U. Desmond2. 2. 2009.
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Chemical Structure | Isolated Yield (%) |
|---|---|---|
| 6a |  | 50.3 |
| 6b |  | 83.6 |
| 6c |  | 69.1 |
| 6d |  | 70.0 |
| 6e |  | 43.7 |
| 6f |  | 61.4 |
| 6g |  | 81.7 |
| 6h |  | 69.9 |
| 6i |  | 55.9 |
| 6j |  | 83.9 |
| 6k |  | 76.5 |
| 6l |  | 79.8 |
| 6a | 6b | 6c | 6d | 6e | 6f | 6g | 6h | 6i | 6j | 6k | 6l | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Physicochemical properties | Molar Refractivity | 132.37 | 137.18 | 141.89 | 146.79 | 121.78 | 126.59 | 131.40 | 136.21 | 150.85 | 155.66 | 160.47 | 165.27 |
| TPSA (Å2) | 132.80 | 143.70 | 142.03 | ||||||||||
| Log P o/w (WLOGP) | 4.50 | 4.89 | 5.28 | 5.67 | 3.83 | 4.22 | 4.61 | 5.00 | 5.43 | 5.82 | 6.21 | 6.60 | |
| Consensus Log P o/w | 2.82 | 3.16 | 3.47 | 3.81 | 2.36 | 2.74 | 3.00 | 3.23 | 3.55 | 3.86 | 4.24 | 4.60 | |
| Water solubility | Moderately Soluble | Poorly Soluble | Insoluble | ||||||||||
| Pharmacokinetics | GI absorption | Low | |||||||||||
| BBB permeant | No | ||||||||||||
| P-gp substrate | Yes | No | |||||||||||
| CYP1A2 inhibitor | Yes | No | |||||||||||
| CYP3A4 inhibitor | Yes | ||||||||||||
| CYP2C9 inhibitor | Yes | ||||||||||||
| CYP2C19 inhibitor | Yes | ||||||||||||
| CYP2D6 inhibitor | Yes | No | |||||||||||
| Lead likeness | Drug likeness (Lipinski) | Yes | No | ||||||||||
| Cpd | % Kinase Inhibition at 10 µM | |
|---|---|---|
| EGFR | HER2 | |
| 6a | 98.79 | 74.72 |
| 6b | 99.34 | 78.04 |
| 6c | 99.32 | 89.99 |
| 6d | 99.09 | 79.68 |
| 6e | 67.27 | 8.78 |
| 6f | 76.55 | 11.69 |
| 6g | 91.80 | 43.06 |
| 6h | 94.67 | 47.34 |
| 6i | 97.65 | 87.16 |
| 6j | 99.03 | 96.73 |
| 6k | 98.64 | 91.16 |
| 6l | 98.58 | 89.05 |
| Cpd | Kinase Assay | |
|---|---|---|
| EGFR IC50 (nM) | HER2 IC50 (nM) | |
| 6j | 1.8 | 87.8 |
| Staurosporine | 88.1 | 35.5 |
| Lapatinib | 10.0 | 9.0 |
| Cpd | % Kinase Inhibition at 10 µM | ||||
|---|---|---|---|---|---|
| CDK2/cyclin A | c-MET | FGFR1 | KDR/VEGFR2 | P38a/MAPK14 | |
| 6j | –0.83 | 19.27 | 13.87 | 10.32 | –33.31 |
| 3D Interactions | 3D Pocket Positioning |
|---|---|
| Co-Crystallized AQ4 Inhibitor | |
 |  |
| 6j | |
 |  |
| 6k | |
 |  |
| 3D Interactions | 3D Pocket Positioning |
|---|---|
| Co-Crystallized 03P Inhibitor | |
 |  |
| 6j | |
 |  |
| 6k | |
 |  |
| ΔG Binding | Coulomb | Covalent | H-Bond | Lipo | Solv_GB | vdW | |
|---|---|---|---|---|---|---|---|
| 6j-1m17 | −67.76 | −3.38 | 0.40 | −1.09 | −18.50 | 14.09 | −58.88 |
| 6k-1m17 | −57.51 | 2.03 | 2.48 | −0.09 | −18.07 | 11.43 | −54.62 |
| AQ4-1M17 | −63.27 | −15.75 | 1.91 | −0.57 | −18.90 | 25.98 | −55.93 |
| 6j-3RCD | −102.57 | −6.71 | 5.25 | −0.97 | −34.49 | 15.59 | −79.99 |
| 6k-3RCD | −93.50 | −4.02 | 4.26 | −0.63 | −31.98 | 18.14 | −77.68 |
| 03P-3RCD | −75.81 | −10.36 | 0.63 | −0.84 | −21.37 | 24.94 | −68.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkamhawy, A.; Son, S.; Lee, H.Y.; El-Maghrabey, M.H.; Hamd, M.A.E.; Alshammari, S.O.; Abdelhameed, A.A.; Alshammari, Q.A.; Abdeen, A.; Ibrahim, S.F.; et al. Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives. Pharmaceuticals 2023, 16, 43. https://doi.org/10.3390/ph16010043
Elkamhawy A, Son S, Lee HY, El-Maghrabey MH, Hamd MAE, Alshammari SO, Abdelhameed AA, Alshammari QA, Abdeen A, Ibrahim SF, et al. Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives. Pharmaceuticals. 2023; 16(1):43. https://doi.org/10.3390/ph16010043
Chicago/Turabian StyleElkamhawy, Ahmed, Seohyun Son, Hwa Young Lee, Mahmoud H. El-Maghrabey, Mohamed A. El Hamd, Saud O. Alshammari, Abeer A. Abdelhameed, Qamar A. Alshammari, Ahmed Abdeen, Samah F. Ibrahim, and et al. 2023. "Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives" Pharmaceuticals 16, no. 1: 43. https://doi.org/10.3390/ph16010043
APA StyleElkamhawy, A., Son, S., Lee, H. Y., El-Maghrabey, M. H., Hamd, M. A. E., Alshammari, S. O., Abdelhameed, A. A., Alshammari, Q. A., Abdeen, A., Ibrahim, S. F., Mahdi, W. A., Alshehri, S., Alnajjar, R., Choi, W. J., Al-Karmalawy, A. A., & Lee, K. (2023). Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives. Pharmaceuticals, 16(1), 43. https://doi.org/10.3390/ph16010043