
Exploring Probenecid Derived 1,3,4-Oxadiazole-Phthalimide Hybrid as α-Amylase Inhibitor: Synthesis, Structural Investigation, and Molecular Modeling

 ,
,  , , , , , , , ,
, , , , , , , ,  and
and
Abstract
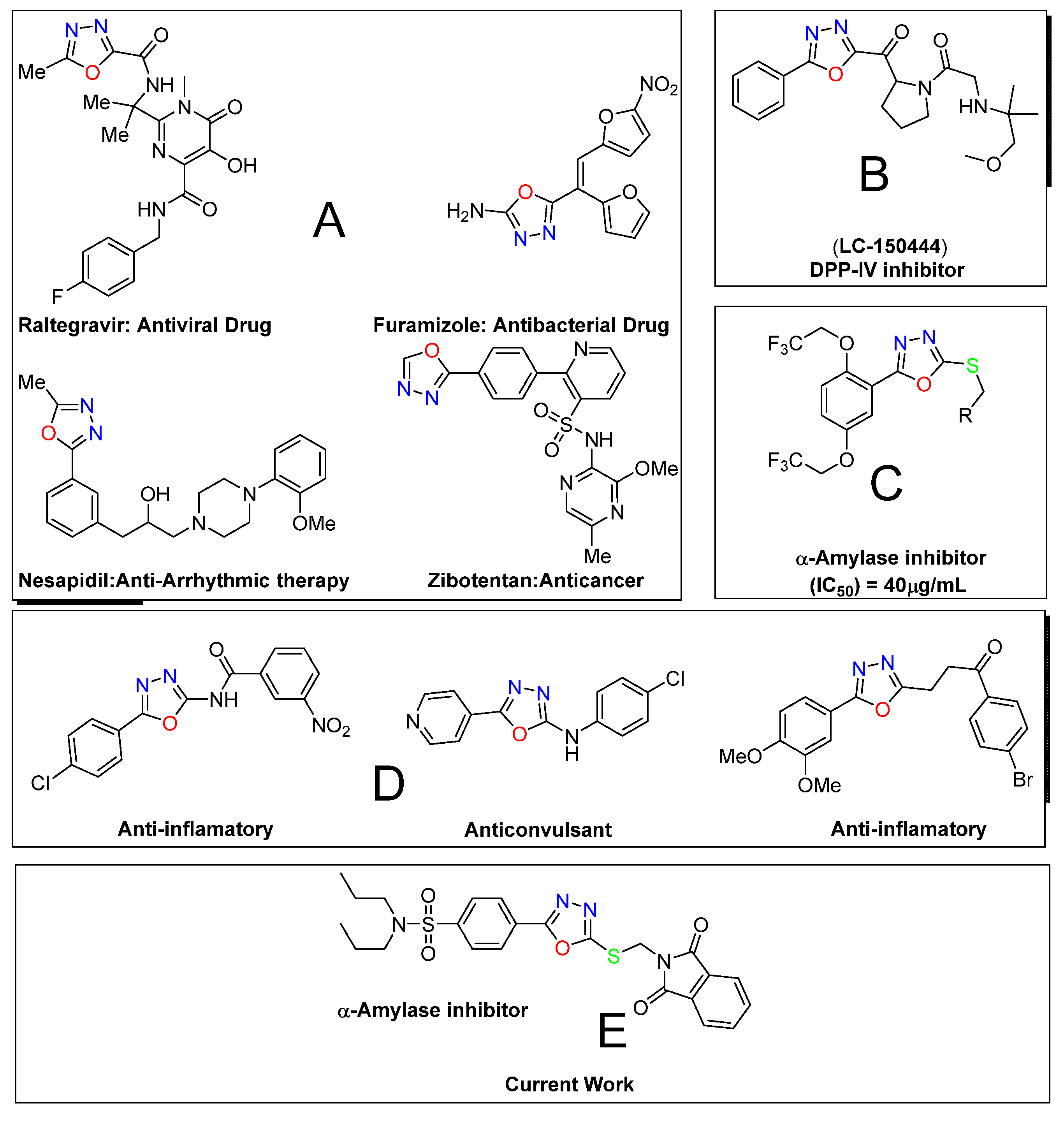
:1. Introduction
2. Results and Discussion
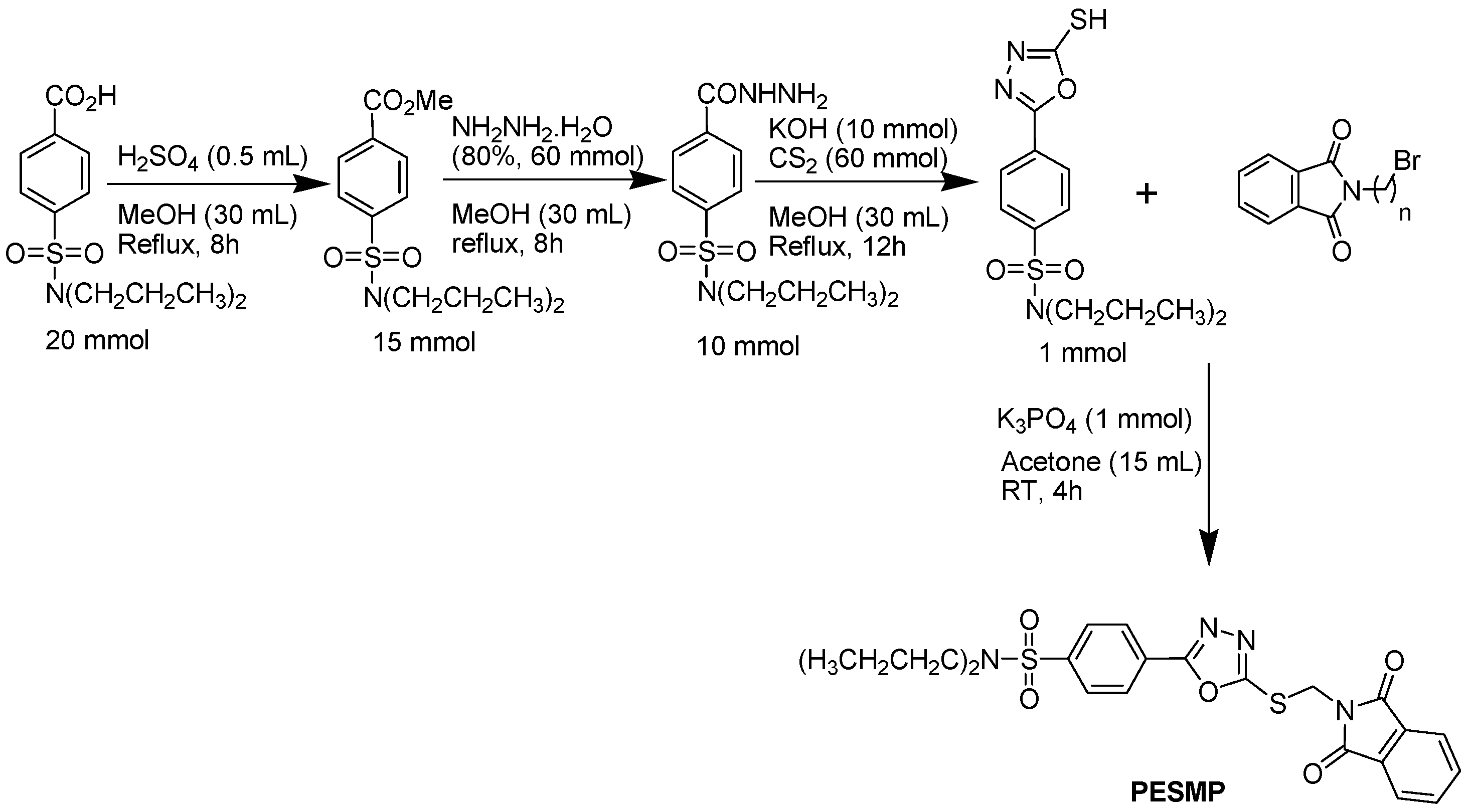
2.1. Chemistry
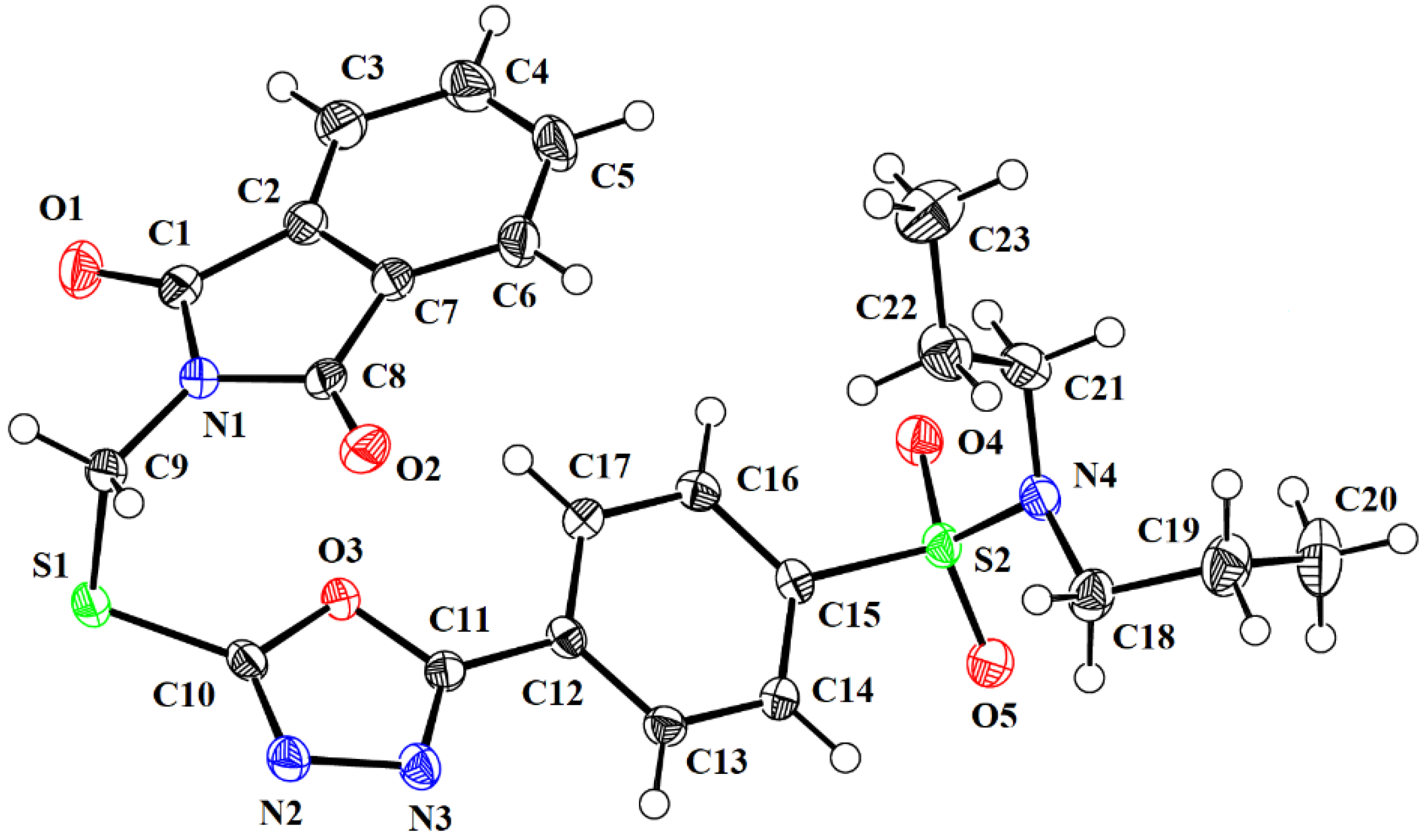
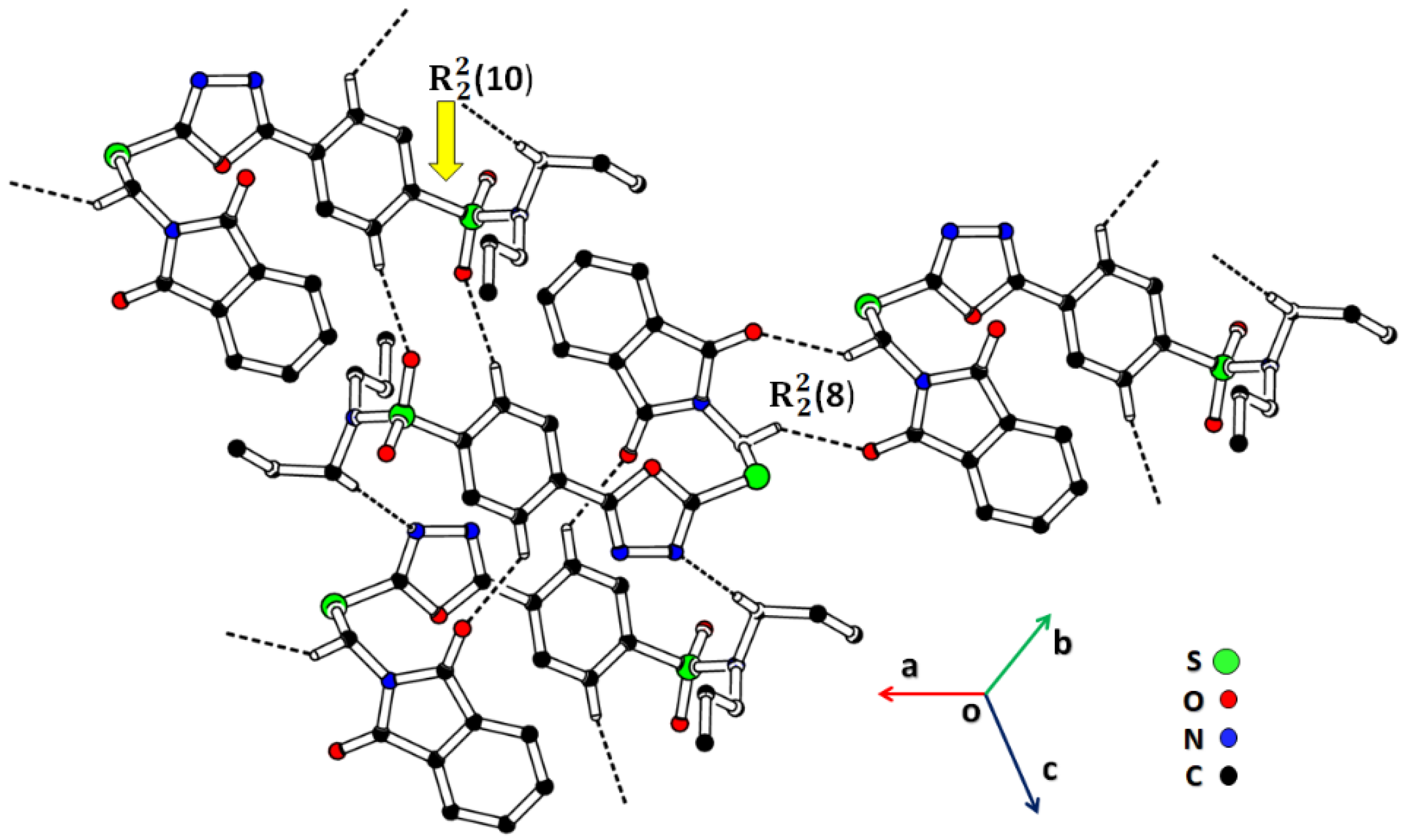
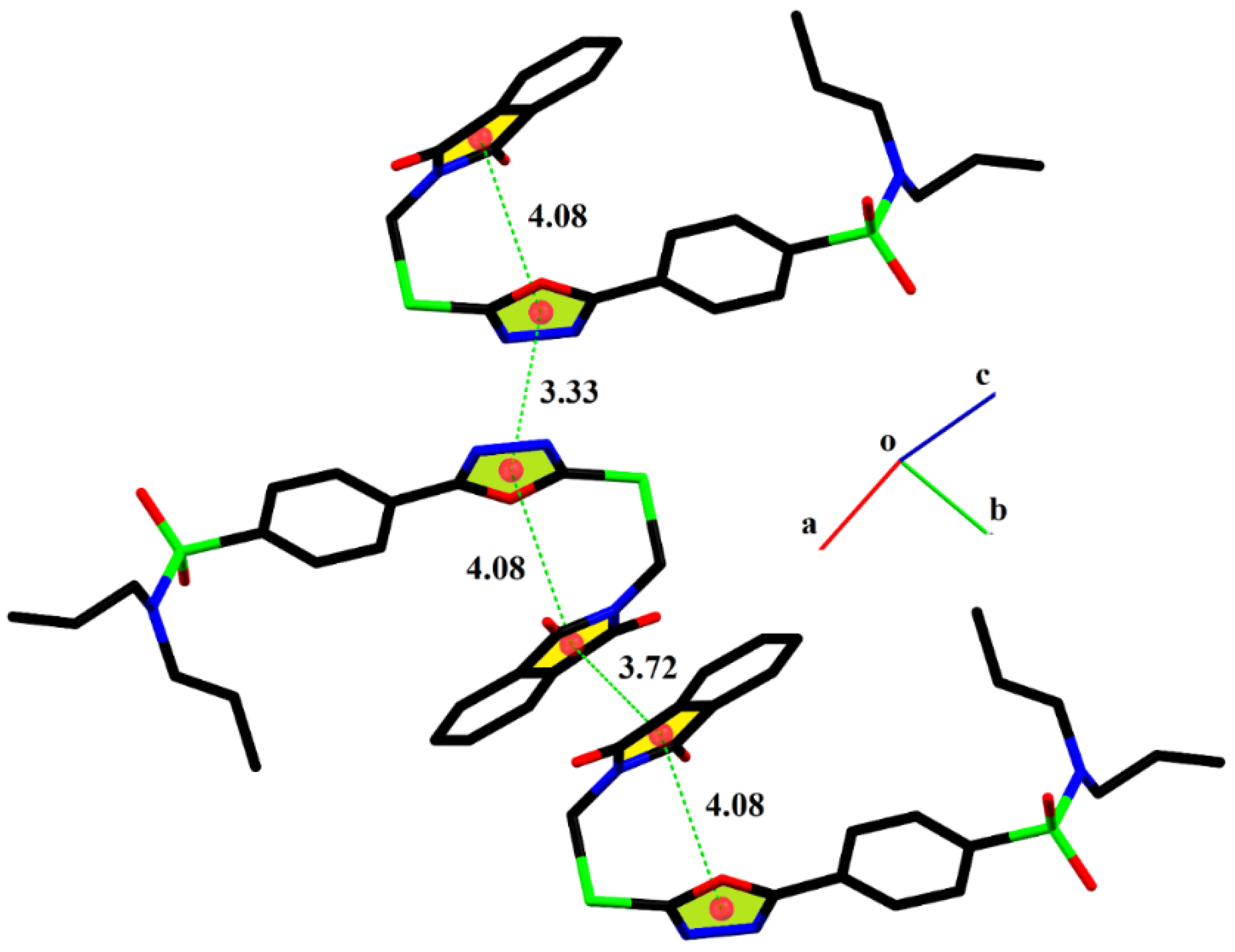
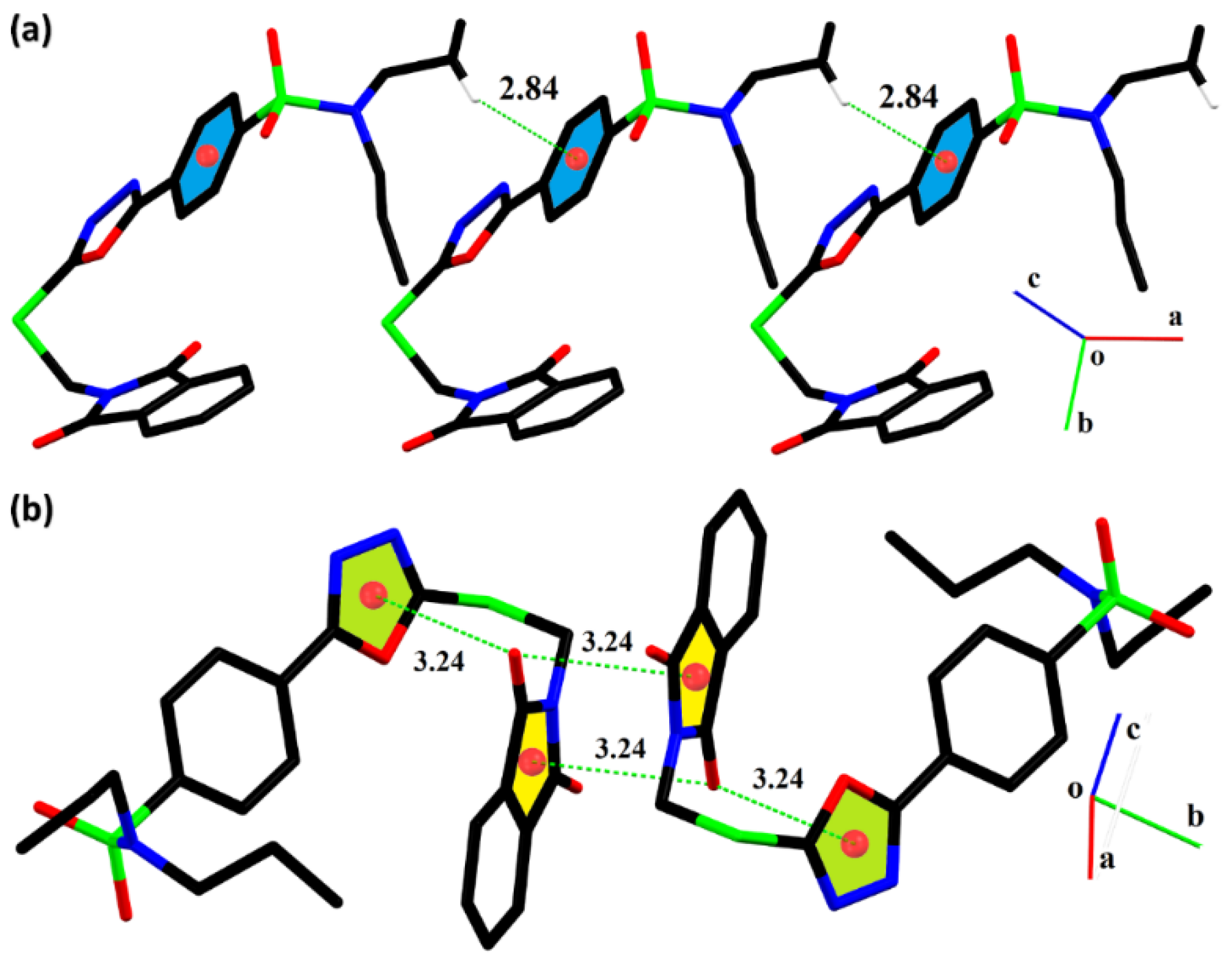
2.2. Single-Crystal Analysis
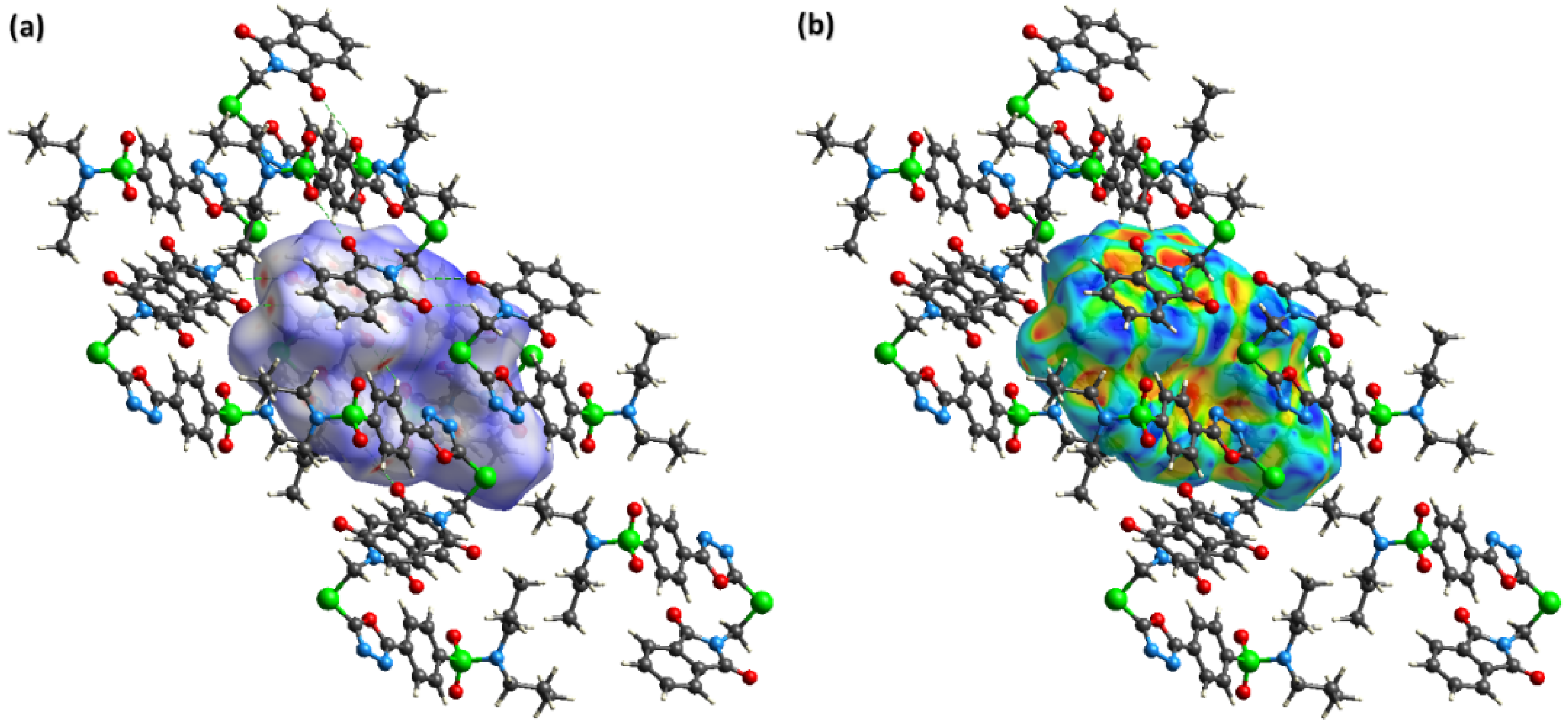
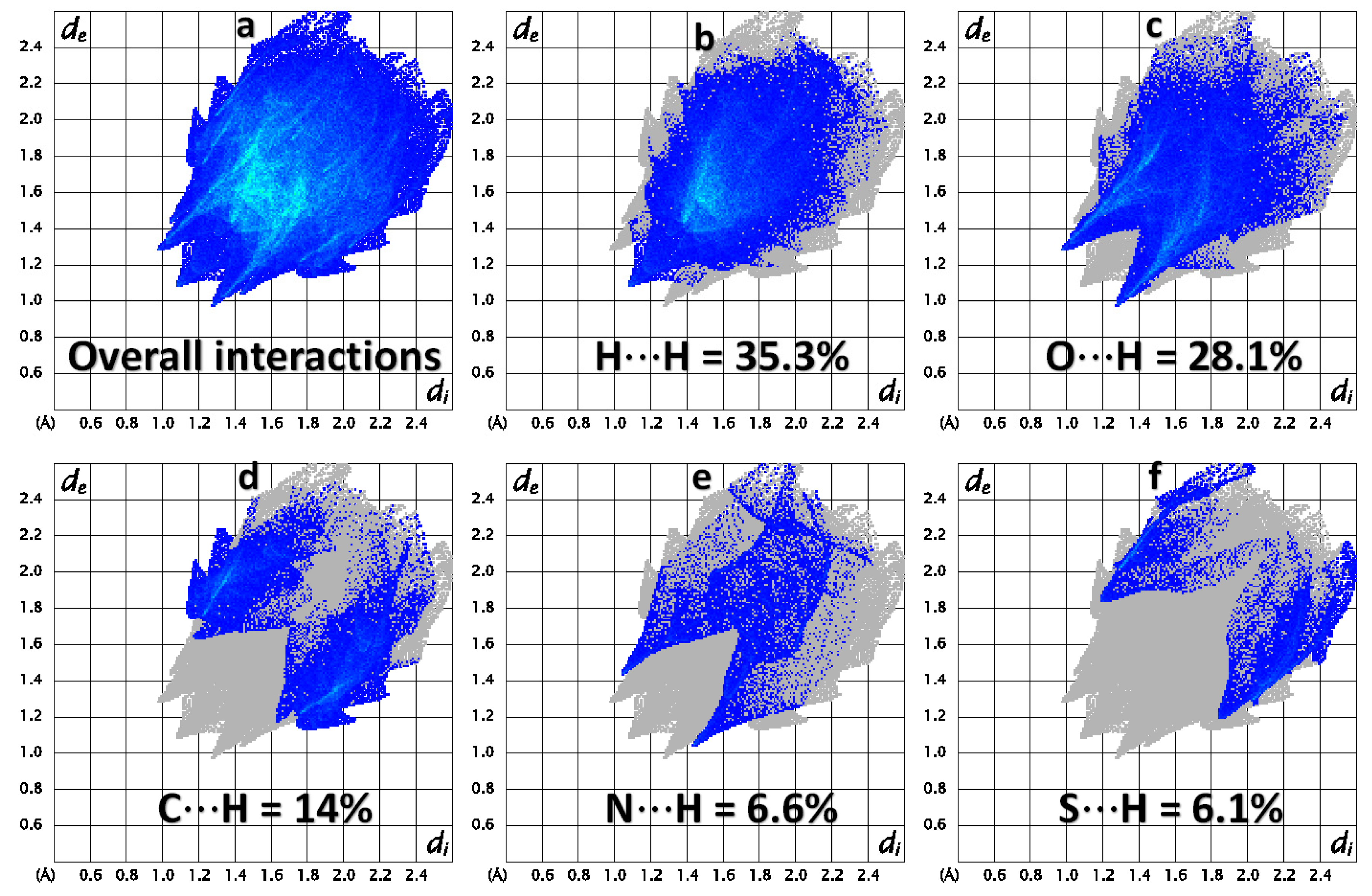
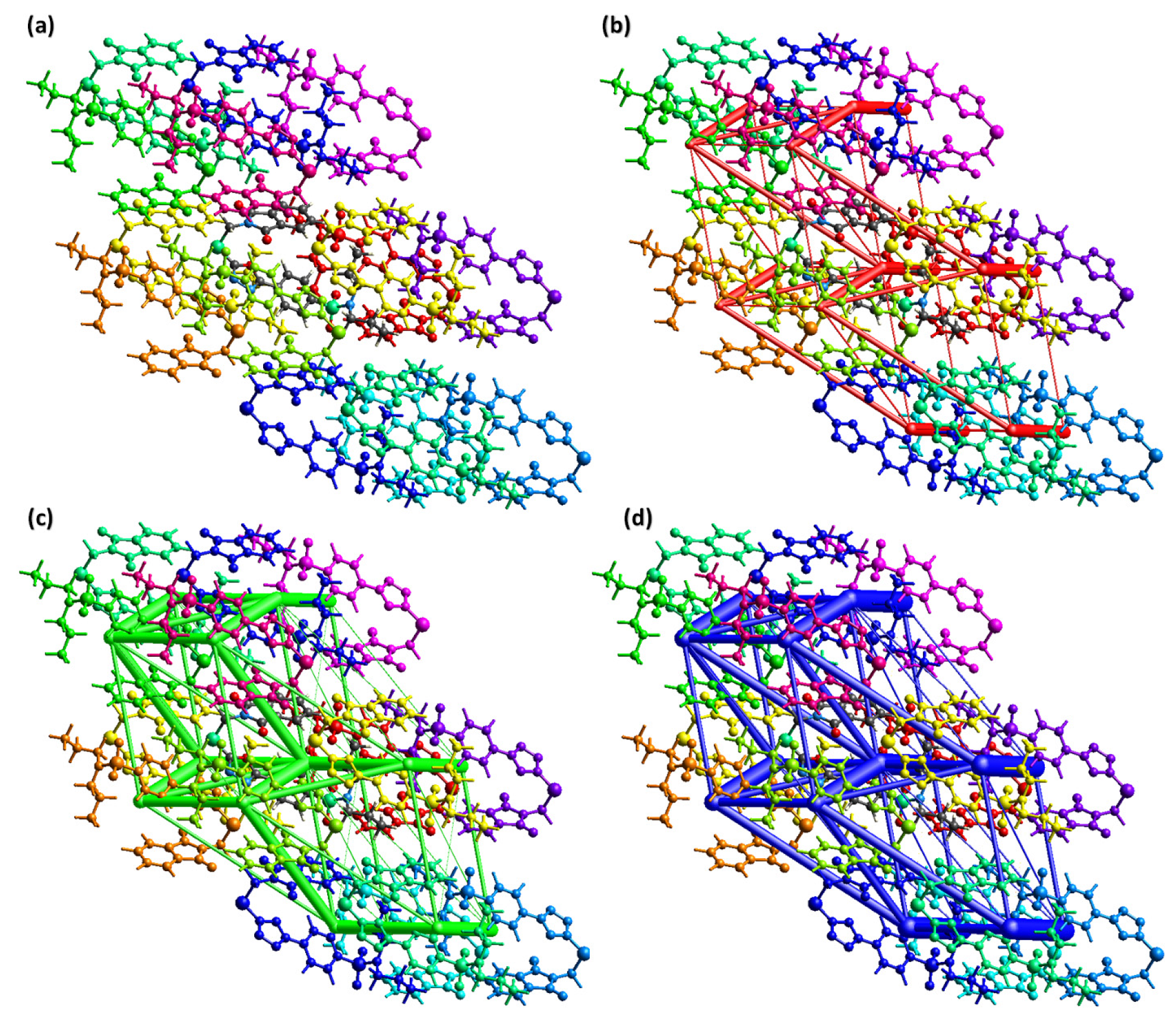
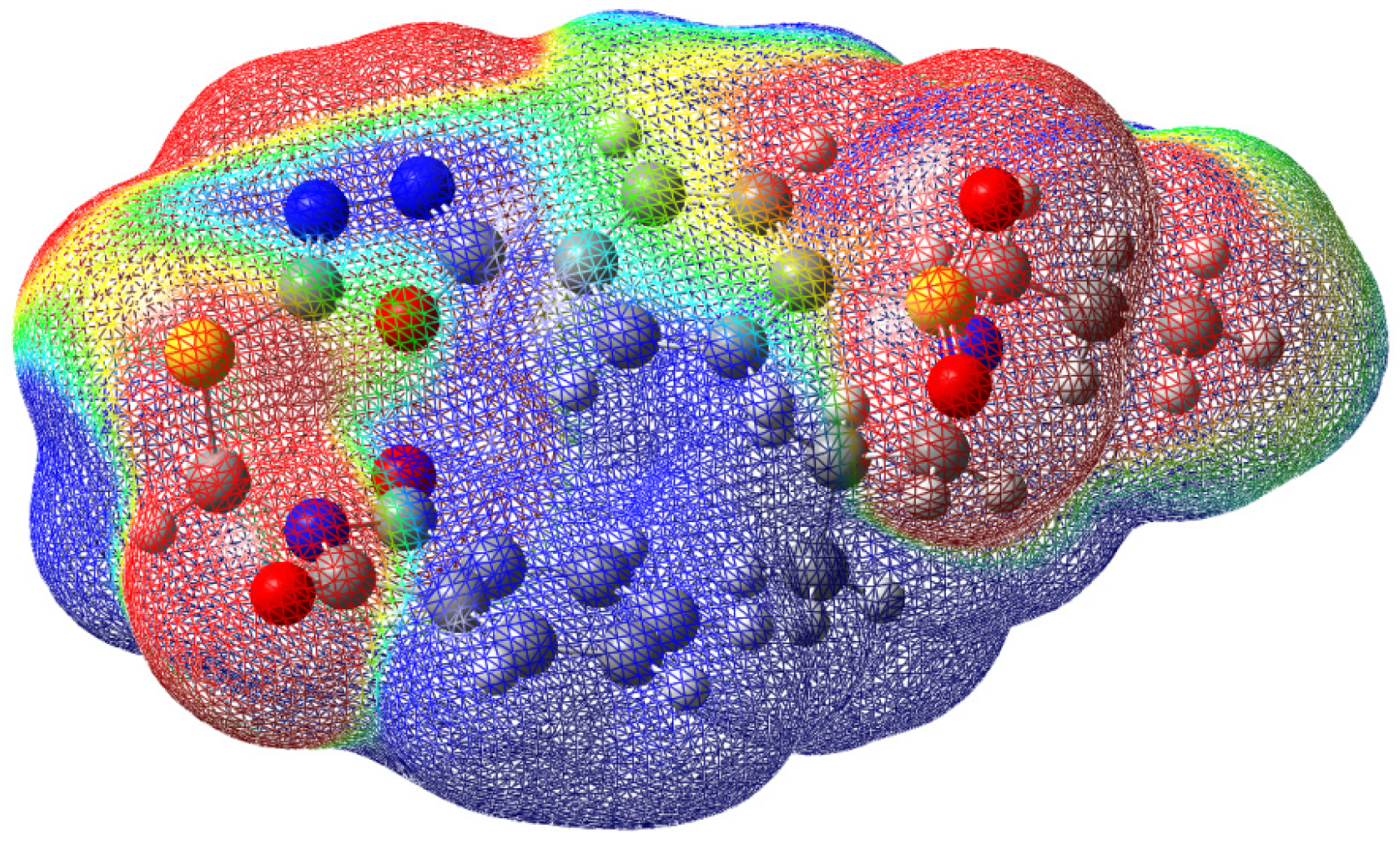
2.3. Hirshfeld Surface Analysis and Interaction Energy Calculation
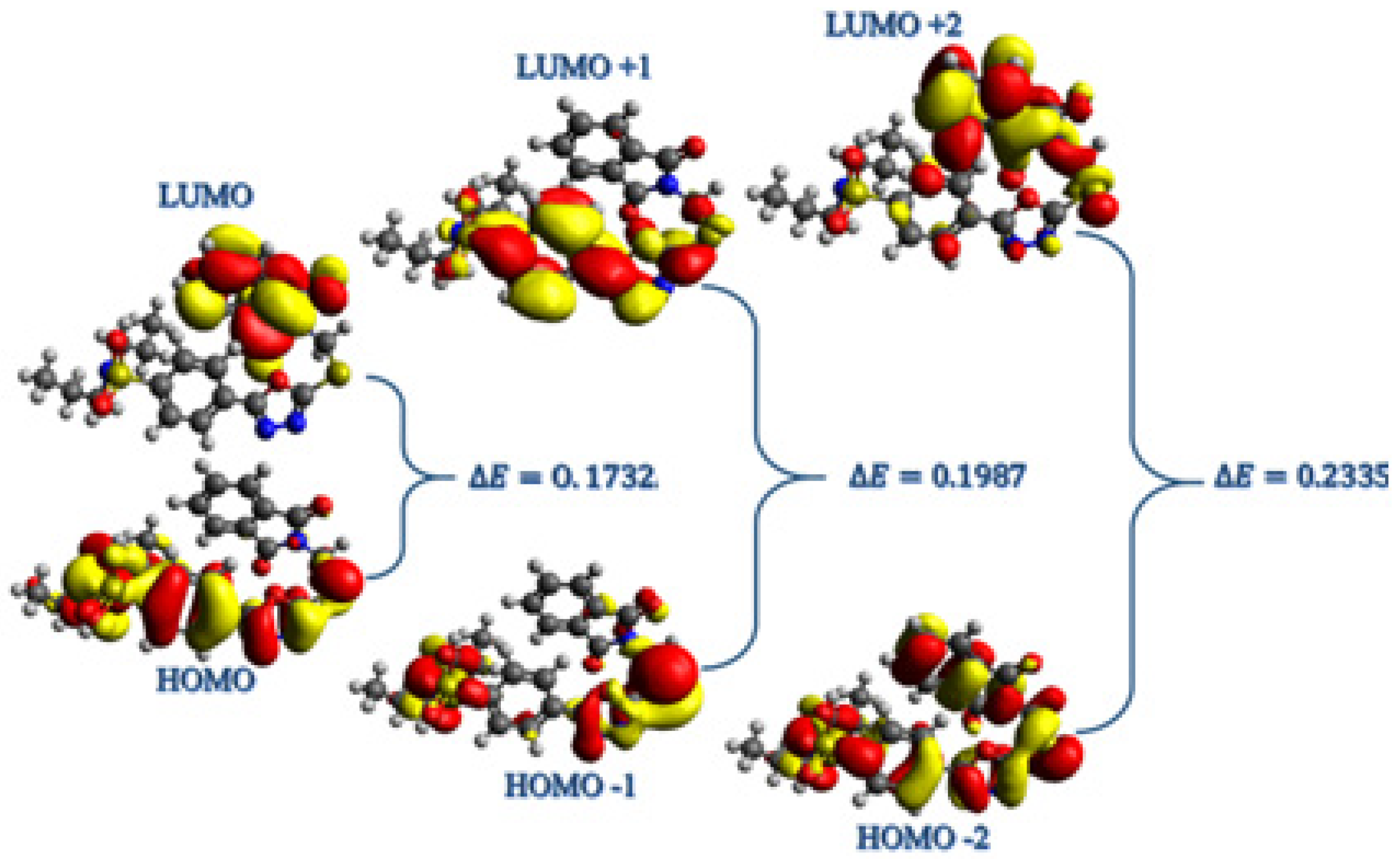
2.4. DFT Calculations
2.4.1. Frontier Molecular Orbitals Analysis
2.4.2. Global Reactivity Parameters (GRPs)
2.4.3. Molecular Electrostatic Potential (MEP)
2.5. α-Amylase Inhibition Activity
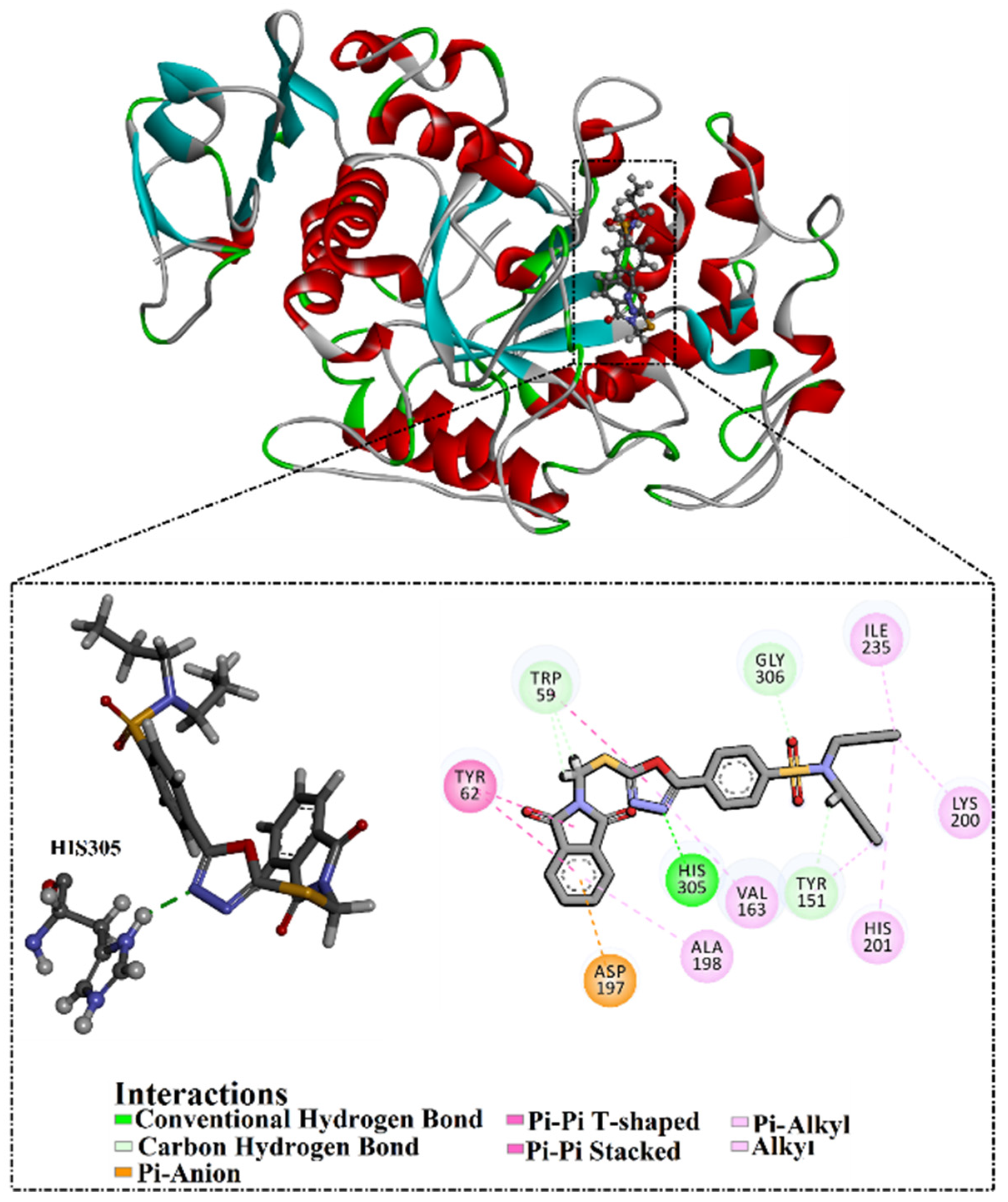
2.6. Molecular Docking
3. Experimental Section
3.1. Experimental Details
3.1.1. Synthetic Procedure for the Preparation of 4-(5-((1,3-Dioxoisoindolin-2-yl)methylthio)-1,3,4-oxadiazol-2-yl)-N,N-dipropylbenzene-sulfonamide (PESMP)
3.1.2. Methyl 4-(dipropylsulfamoyl)benzoate (MPE)
3.1.3. 4-(Dipropylsulfamoyl)benzoic acid hydrazide (PEH)
3.1.4. N,N-dipropyl-4-(5-thioxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)benzene sulfonamide (PEO)
3.1.5. 2-((5-Phenyl-2-thioxo-1,3,4-oxadiazol-3(2H)-yl)methyl)isoindoline-1,3-dione (PESMP)
3.2. Computational Details
3.2.1. Hirshfeld Surface (HS) Analysis
3.2.2. Density Functional Theory (DFT) Analysis
3.3. Enzyme Inhibition Activity
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Diffusion of eHealth: Making Universal Health Coverage Achievable: Report of the Third Global Survey on eHealth; World Health Organization: Geneva, Switzerland, 2017.
- Hunt, D.; Hemmingsen, B.; Matzke, A.; Varghese, C.; Hammerich, A.; Luciani, S.; Hennis, A.; Branca, F.; Bull, F.; Berdzuli, N.; et al. The WHO Global Diabetes Compact: A new initiative to support people living with diabetes. Lancet Diabetes Endocrinol. 2021, 9, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Kumar, V.; Nayak, S.K.; Wadhwa, P.; Kaur, P.; Sahu, S.K. Alpha-amylase as molecular target for treatment of diabetes mellitus: A comprehensive review. Chem. Biol. Drug Des. 2021, 98, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Rana, K.; Salahuddin; Sahu, J.K. Significance of 1,3,4-Oxadiazole Containing Compounds in New Drug Development. Curr. Drug Res. Rev. 2021, 13, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Hkiri, S.; Hafidh, A.; Cavalier, J.F.; Touil, S.; Samarat, A. Design, synthesis, antimicrobial evaluation, and molecular docking studies of novel symmetrical 2,5-difunctionalized 1,3,4-oxadiazoles. J. Heterocycl. Chem. 2020, 57, 1044–1054. [Google Scholar] [CrossRef]
- Wang, S.; Liu, H.; Wang, X.; Lei, K.; Li, G.; Li, J.; Liu, R.; Quan, Z. Synthesis of 1,3,4-oxadiazole derivatives with anticonvulsant activity and their binding to the GABAA receptor. Eur. J. Med. Chem. 2020, 206, 112672. [Google Scholar] [CrossRef]
- Glomb, T.; Szymankiewicz, K.; Swiatek, P. Anti-Cancer Activity of Derivatives of 1,3,4-Oxadiazole. Molecules 2018, 23, 3361. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, A.; Pathak, D.; Shah, K. 1,3,4-oxadiazole and its derivatives: A review on recent progress in anticancer activities. Chem. Biol. Drug Des. 2021, 97, 572–591. [Google Scholar] [CrossRef]
- Gani, R.S.; Kudva, A.K.; Timanagouda, K.; Raghuveer; Mujawar, S.B.H.; Joshi, S.D.; Raghu, S.V. Synthesis of novel 5-(2,5-bis(2,2,2-trifluoroethoxy)phenyl)-1,3,4-oxadiazole-2-thiol derivatives as potential glucosidase inhibitors. Bioorg. Chem. 2021, 114, 105046. [Google Scholar] [CrossRef]
- Hamdani, S.S.; Khan, B.A.; Hameed, S.; Batool, F.; Saleem, H.N.; Mughal, E.U.; Saeed, M. Synthesis and evaluation of novel S-benzyl- and S-alkylphthalimide- oxadiazole -benzenesulfonamide hybrids as inhibitors of dengue virus protease. Bioorg. Chem. 2020, 96, 103567. [Google Scholar] [CrossRef]
- Peng, F.; Liu, T.; Wang, Q.; Liu, F.; Cao, X.; Yang, J.; Liu, L.; Xie, C.; Xue, W. Antibacterial and Antiviral Activities of 1,3,4-Oxadiazole Thioether 4H-Chromen-4-one Derivatives. J. Agric. Food Chem. 2021, 69, 11085–11094. [Google Scholar] [CrossRef]
- Chawla, G.; Naaz, B.; Siddiqui, A.A. Exploring 1,3,4-Oxadiazole Scaffold for Anti-inflammatory and Analgesic Activities: A Review of Literature From 2005–2016. Mini-Rev. Med. Chem. 2018, 18, 216–233. [Google Scholar] [CrossRef]
- Sun, J.; Makawana, J.A.; Zhu, H.L. 1,3,4-oxadiazole derivatives as potential biological agents. Mini-Rev. Med. Chem. 2013, 13, 1725–1743. [Google Scholar] [CrossRef]
- Croxtall, J.D.; Keam, S.J. Raltegravir: A review of its use in the management of HIV infection in treatment-experienced patients. Drugs 2009, 69, 1059–1075. [Google Scholar] [CrossRef]
- Seo, I. Antibacterial Activity of Furamizole on Mycoplasma gallisepticum. Korean J. Vet. Res. 1973, 13, 35–38. [Google Scholar]
- Bora, R.O.; Dar, B.; Pradhan, V.; Farooqui, M. [1, 2, 4]-oxadiazoles: Synthesis and biological applications. Mini-Rev. Med. Chem. 2014, 14, 355–369. [Google Scholar] [CrossRef]
- Fizazi, K.; Higano, C.S.; Nelson, J.B.; Gleave, M.; Miller, K.; Morris, T.; Nathan, F.E.; McIntosh, S.; Pemberton, K.; Moul, J.W. Phase III, randomized, placebo-controlled study of docetaxel in combination with zibotentan in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2013, 31, 1740–1747. [Google Scholar] [CrossRef]
- Verma, G.; Khan, M.F.; Akhtar, W.; Alam, M.M.; Akhter, M.; Shaquiquzzaman, M. A Review Exploring Therapeutic Worth of 1,3,4-Oxadiazole Tailored Compounds. Mini-Rev. Med. Chem. 2019, 19, 477–509. [Google Scholar] [CrossRef]
- Maryan, L.; Marta, M.; Myroslava, K.; Iryna, D.; Stefan, H.; Taras, C.; Ihor, C.; Vasyl, M. Approaches for synthesis and chemical modification of non-condensed heterocyclic systems based on 1,3,4-oxadiazole ring and their biological activity: A review. J. Appl. Pharm. Sci. 2020, 10, 151–165. [Google Scholar] [CrossRef]
- Robbins, N.; Koch, S.E.; Tranter, M.; Rubinstein, J. The history and future of probenecid. Cardiovasc. Toxicol 2012, 12, 1–9. [Google Scholar] [CrossRef]
- D’Ascenzio, M.; Carradori, S.; Secci, D.; Vullo, D.; Ceruso, M.; Akdemir, A.; Supuran, C.T. Selective inhibition of human carbonic anhydrases by novel amide derivatives of probenecid: Synthesis, biological evaluation and molecular modelling studies. Bioorg. Med. Chem. 2014, 22, 3982–3988. [Google Scholar] [CrossRef]
- Chawla, G. 1,2,4-Oxadiazole as a Privileged Scaffold for Anti-inflammatory and Analgesic Activities: A Review. Mini-Rev. Med. Chem. 2018, 18, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, F.; Jafari, E.; Shojaei, F.; Sadeghi-Aliabadi, H. Synthesis and cytotoxic activity evaluation of some new 1, 3, 4-oxadiazole, 1, 3, 4-thiadiazole and 1, 2, 4- triazole derivatives attached to phthalimide. Res. Pharm. Sci. 2021, 16, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Antunes, R.; Batista, H.; Srivastava, R.A.; Thomas, G.; Araujo, C.C.; Longo, R.L.; Magalhaes, W.; Leao, M.B.C.; Pavao, A.C. Synthesis, characterization and interaction mechanism of new oxadiazolo-phthalimides as peripheral analgesics. IV. J. Mol. Struct. 2003, 660, 1–13. [Google Scholar] [CrossRef]
- Khan, B.A.; Hamdani, S.S.; Ahmed, M.N.; Hameed, S.; Ashfaq, M.; Shawky, A.M.; Ibrahim, M.A.A.; Sidhom, P.A. Synthesis, X-ray diffraction analysis, quantum chemical studies and alpha-amylase inhibition of probenecid derived S-alkylphthalimide-oxadiazole-benzenesulfonamide hybrids. J. Enzym. Inhib. Med. Chem. 2022, 37, 1464–1478. [Google Scholar] [CrossRef]
- Li, P.; Vik, E.C.; Maier, J.M.; Karki, I.; Strickland, S.M.S.; Umana, J.M.; Smith, M.D.; Pellechia, P.J.; Shimizu, K.D. Electrostatically Driven CO-pi Aromatic Interactions. J. Am. Chem. Soc. 2019, 141, 12513–12517. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Madni, M.; Ahmed, M.N.; Abbasi, G.; Hameed, S.; Ibrahim, M.A.A.; Tahir, M.N.; Ashfaq, M.; Gil, D.M.; Gomila, R.M.; Frontera, A. Synthesis and X-ray Characterization of 4,5-Dihydropyrazolyl-Thiazoles Bearing a Coumarin Moiety: On the Importance of Antiparallel π-Stacking. ChemistrySelect 2022, 7, e202202287. [Google Scholar] [CrossRef]
- Malik, H.; Akhter, Z.; Shahbaz, M.; Yousuf, S.; Munawar, K.S.; Muhammad, S.; Qamar, S.; Abbas, A.; Ashfaq, M.; Ahmad, T. Synthesis, spectroscopic characterization, single crystal, theoretical investigation, and biological screenings of azo-based moieties. J. Mol. Struct. 2022, 1270, 133867. [Google Scholar] [CrossRef]
- Faihan, A.S.; Aziz, N.M.; Ashfaq, M.; Hassan, W.M.I.; Al-Jibori, S.A.; Al-Janabi, A.S.; Tahir, M.N.; Al-barwari, A.S.M.O. Synthesis, characterization, and X-ray crystallography of unexpected chloro-substitution on 1-(4-chlorophenyl)-3-phenylthiourea platinum(II) complex with tertiary phosphine ligand. J. Mol. Struct. 2022, 1270, 133985. [Google Scholar] [CrossRef]
- Ashfaq, M.; Ali, A.; Tahir, M.N.; Khalid, M.; Assiri, M.A.; Imran, M.; Munawar, K.S.; Habiba, U. Synthetic approach to achieve halo imine units: Solid-state assembly, DFT based electronic and non linear optical behavior. Chem. Phys. Lett. 2022, 803, 139843. [Google Scholar] [CrossRef]
- Taia, A.; El Ibrahimi, B.; Benhiba, F.; Ashfaq, M.; Tahir, M.N.; Essaber, M.; Aatif, A.; Hokelek, T.; Mague, J.T.; Sebbar, N.K.; et al. Syntheses, single crystal X-ray structure, Hirshfeld surface analyses, DFT computations and Monte Carlo simulations of New Eugenol derivatives bearing 1,2,3-triazole moiety. J. Mol. Struct. 2021, 1234, 130189. [Google Scholar] [CrossRef]
- Ahmed, M.N.; Madni, M.; Anjum, S.; Andleeb, S.; Hameed, S.; Khan, A.M.; Ashfaq, M.; Tahir, M.N.; Gil, D.M.; Frontera, A. Crystal engineering with pyrazolyl-thiazole derivatives: Structure-directing role of π-stacking and σ-hole interactions. CrystEngComm 2021, 23, 3276–3287. [Google Scholar] [CrossRef]
- Malik, A.N.; Kuznetsov, A.; Ali, A.; Ashfaq, M.; Tahir, M.N.; Siddique, A. Imine-based Zwitterion: Synthesis, single-crystal characterization, and computational investigation. J. Mol. Struct. 2022, 1253, 132237. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. ChemComm 2007, 37, 3814–3816. [Google Scholar] [CrossRef]
- Kargar, H.; Ashfaq, M.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Munawar, K.S.; Tahir, M.N. Unsymmetrical Ni(II) Schiff base complex: Synthesis, spectral characterization, crystal structure analysis, Hirshfeld surface investigation, theoretical studies, and antibacterial activity. J. Mol. Struct. 2022, 1265, 133381. [Google Scholar] [CrossRef]
- Kargar, H.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Bahadori, M.; Moghadam, M.; Ashfaq, M.; Munawar, K.S.; Tahir, M.N. Spectroscopic investigation, molecular structure, catalytic activity with computational studies of a novel Pd(II) complex incorporating unsymmetrical tetradentate Schiff base ligand. Inorg. Chem. Commun. 2022, 142, 109697. [Google Scholar] [CrossRef]
- Iqbal, Y.; Haroon, M.; Akhtar, T.; Ashfaq, M.; Tahir, M.N.; Rasheed, L.; Yousuf, M.; Zia, M.A. Synthesis, spectroscopic characterization, single crystal XRD, Hirshfeld Surface analysis and theoretical studies (DFT) of 4-adamantyl-(2-(substitutedbenzylidene)hydrazinyl)thiazoles. J. Mol. Struct. 2022, 1267, 133620. [Google Scholar] [CrossRef]
- Ahmed, M.N.; Ghias, M.; Shah, S.W.A.; Shoaib, M.; Tahir, M.N.; Ashfaq, M.; Ibrahim, M.A.A.; Andleeb, H.; Gil, D.M.; Frontera, A. X-ray characterization, Hirshfeld surface analysis, DFT calculations, in vitroandin silicolipoxygenase inhibition (LOX) studies of dichlorophenyl substituted 3-hydroxy-chromenones. New J. Chem. 2021, 45, 19928–19940. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Ali, A.; Khalid, M.; Ashfaq, M.; Malik, A.N.; Tahir, M.N.; Assiri, M.A.; Imran, M.; AlcantaraMorais, S.F.; Braga, A.A.C. Preparation, QTAIM and Single-Crystal Exploration of the Pyrimethamine-Based Co-Crystal Salts with Substituted Benzoic Acids. Chemistryselect 2022, 7, e202200349. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Visualisation and characterisation of voids in crystalline materials. Crystengcomm 2011, 13, 1804–1813. [Google Scholar] [CrossRef]
- Ashfaq, M.; Tahir, M.N.; Muhammad, S.; Munawar, K.S.; Ali, A.; Bogdanov, G.; Alarfaji, S.S. Single-Crystal Investigation, Hirshfeld Surface Analysis, and DFT Study of Third-Order NLO Properties of Unsymmetrical Acyl Thiourea Derivatives. ACS Omega 2021, 6, 31211–31225. [Google Scholar] [CrossRef] [PubMed]
- Kargar, H.; Ashfaq, M.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Munawar, K.S.; Tahir, M.N. Synthesis, characterization, SC-XRD, HSA and DFT study of a novel copper(I) iodide complex with 2-(thiophen-2-yl)-4,5-dihydro-1H-imidazole ligand: An experimental and theoretical approach. J. Mol. Struct. 2022, 1253, 132264. [Google Scholar] [CrossRef]
- Ashfaq, M.; Khalid, M.; Tahir, M.N.; Ali, A.; Arshad, M.N.; Asiri, A.M. Synthesis of Crystalline Fluoro-Functionalized Imines, Single Crystal Investigation, Hirshfeld Surface Analysis, and Theoretical Exploration. ACS Omega 2022, 7, 9867–9878. [Google Scholar] [CrossRef] [PubMed]
- Khattak, Z.A.K.; Ahmad, N.; Younus, H.A.; Ullah, H.; Yu, B.Y.; Munawar, K.S.; Ashfaq, M.; Ali, S.; Shahadat, H.M.; Verpoort, F. Synthesis of 3D Cadmium(II)-Carboxylate Framework Having Potential for Co-Catalyst Free CO2 Fixation to Cyclic Carbonates. Inorganics 2022, 10, 162. [Google Scholar] [CrossRef]
- Ashfaq, M.; Ali, A.; Tahir, M.N.; Kuznetsov, A.; Munawar, K.S.; Muhammad, S. Synthesis, single-crystal exploration, hirshfeld surface analysis, and DFT investigation of the thiosemicarbazones. J. Mol. Struct. 2022, 1262, 133088. [Google Scholar] [CrossRef]
- Khan, M.U.; Iqbal, J.; Khalid, M.; Hussain, R.; Braga, A.A.C.; Hussain, M.; Muhammad, S. Designing triazatruxene-based donor materials with promising photovoltaic parameters for organic solar cells. RSC Adv. 2019, 9, 26402–26418. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, R.D.; Vivas, M.G.; Silva, D.L.; Eucat, G.; Bretonniere, Y.; Andraud, C.; De Boni, L.; Mendonca, C.R. First-Order Hyperpolarizability of Triphenylamine Derivatives Containing Cyanopyridine: Molecular Branching Effect. J. Phys. Chem. C 2018, 122, 1770–1778. [Google Scholar] [CrossRef]
- Wielopolski, M.; Kim, J.-H.; Jung, Y.-S.; Yu, Y.-J.; Kay, K.-Y.; Holcombe, T.W.; Zakeeruddin, S.M.; Grätzel, M.; Moser, J.-E. Position-Dependent Extension of π-Conjugation in D-π-A Dye Sensitizers and the Impact on the Charge-Transfer Properties. J. Phys. Chem. C 2013, 117, 13805–13815. [Google Scholar] [CrossRef] [Green Version]
- Garza, A.J.; Osman, O.I.; Wazzan, N.A.; Khan, S.B.; Asiri, A.M.; Scuseria, G.E. A computational study of the nonlinear optical properties of carbazole derivatives: Theory refines experiment. Theor. Chem. Acc. 2014, 133, 1458. [Google Scholar] [CrossRef]
- Appleton, A.L.; Brombosz, S.M.; Barlow, S.; Sears, J.S.; Bredas, J.L.; Marder, S.R.; Bunz, U.H. Effects of electronegative substitution on the optical and electronic properties of acenes and diazaacenes. Nat Commun 2010, 1, 91. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. USA 1986, 83, 8440–8441. [Google Scholar] [CrossRef] [Green Version]
- Luque, F.J.; Lopez, J.M.; Orozco, M. Perspective on "Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects"-Miertus S, Scrocco E, Tomasi J (1981) Chem Phys 55: 117. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar] [CrossRef]
- Kanis, D.R.; Ratner, M.A.; Marks, T.J. Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects. Chem. Rev. 2002, 94, 195–242. [Google Scholar] [CrossRef]
- Gilles, C.; Astier, J.P.; Marchis-Mouren, G.; Cambillau, C.; Payan, F. Crystal structure of pig pancreatic alpha-amylase isoenzyme II, in complex with the carbohydrate inhibitor acarbose. Eur. J. Biochem. 1996, 238, 561–569. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Frisch, A. Gaussian 09W Reference; Gaussian: Wallingford, CT, USA, 2009; Volume 470, 25p. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0. 16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Jaragh-Alhadad, L.A.; Atia, M.A.M.; Alzahrani, O.R.; Ahmed, M.N.; Moustafa, M.S.; Soliman, M.E.S.; Shawky, A.M.; Pare, P.W.; et al. Exploring Toxins for Hunting SARS-CoV-2 Main Protease Inhibitors: Molecular Docking, Molecular Dynamics, Pharmacokinetic Properties, and Reactome Study. Pharmaceuticals 2022, 15, 153. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Mekhemer, G.A.H.; Shawky, A.M.; Moustafa, M.F.; Atia, M.A.M. In Silico targeting human multidrug transporter ABCG2 in breast cancer: Database screening, molecular docking, and molecular dynamics study. Mol. Inform. 2022, 41, e2060039. [Google Scholar] [CrossRef]
- Almansour, N.M.; Abdelrahman, A.H.M.; Ismail Fagiree, E.; Ibrahim, M.A.A. In silico drug repurposing and lipid bilayer molecular dynamics puzzled out potential breast cancer resistance protein (BCRP/ABCG2) inhibitors. J. Biomol. Struct. Dyn. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- OpenEye, SZYBKI 1.9.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Gasteiger, J.; Marsili, M. Iterative Partial Equalization of Orbital Electronegativity-a Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Dassault Systèmes BIOVIA, Discovery Studio Visualize, Version 2019; Dassault Systèmes: San Diego, CA, USA, 2019.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | PESMP |
|---|---|
| CCDC | 2184321 |
| Chemical formula | C23H24N4O5S2 |
| Mr | 500.58 |
| Crystal system, space group | Triclinic |
| Temperature (K) | 293 |
| a, b, c (Å) | 7.9647 (1), 11.4141 (1), 13.5283 (2) |
| α, β, γ° | 96.606 (1), 98.617 (1), 100.899 (1) |
| V (Å3) | 1180.95 (3) |
| Z | 2 |
| Density (calculated)g/cm−3 | 1.408 |
| F(000) | 524 |
| Radiation type | Cu Kα |
| Wavelength (λ) | 1.54184 |
| µ (mm−1) | 2.411 |
| Crystal size (mm) | 0.08 × 0.06 × 0.05 |
| Data Collection | |
| Diffractometer | Bruker Kappa APEXII CCD |
| Absorption correction | Multi-scan (SADABS; Bruker, 2007) |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 10075, 4146, 3939 |
| Rint | 0.021 |
| Theta range for data collection (°) | 3.341 to 67.404 |
| Index ranges | −4 ≤ h ≤ 8, −13 ≤ k ≤ 11, −16 ≤ l ≤ 16 |
| (sin θ/λ)max (Å−1) | 0.599 |
| Data Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.030, 0.079, 1.06 |
| No. of reflections | 4146 |
| No. of parameters | 309 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.25, −0.39 |
| D—H···A | D—H | H···A | D···A | <(D—H···A)° |
|---|---|---|---|---|
| C9—H9A···O1i | 0.97 | 2.44 | 3.1659 (18) | 132 |
| C16—H16···O4ii | 0.93 | 2.53 | 3.4137 (17) | 160 |
| C18—H18A···O5 | 0.97 | 2.41 | 2.9028 (19) | 111 |
| C18—H18B···N2iii | 0.97 | 2.62 | 3.5809 (19) | 173 |
| C21—H21A···O4 | 0.97 | 2.39 | 2.8934 (18) | 112 |
| C—H···π | C—H | H···π | C···π | <(C—H···π)° |
| C19—H19A···Cg1iv | 0.97 | 2.84 | 3.6326 (18) | 140 |
| C—O···π | C—O | O···π | C···π | <(C—O···π)° |
| C8—O2···Cg2 | 1.2093 (17) | 3.2430 (12) | 3.4758 (15) | 90.74 (9) |
| C8—O2···Cg3v | 1.2093 (17) | 3.2417 (12) | 3.2765 (15) | 80.93 (8) |
| Contact % | Atom | H | C | N | O | S |
| H | 35.3 | 14 | 6.6 | 28.1 | 6.1 | |
| C | 14 | 0.2 | 4.7 | 1.7 | 0.1 | |
| N | 6.6 | 4.7 | 0.3 | 0.7 | 1 | |
| O | 28.1 | 1.7 | 0.7 | 0.4 | 0.9 | |
| S | 6.1 | 0.1 | 1 | 0.9 | 0 | |
| Surface% | 62.7 | 10.45 | 6.8 | 16.1 | 4.05 | |
| Random Contacts % | Atom | H | C | N | O | S |
| H | 39.31 | |||||
| C | 13.10 | 1.09 | ||||
| N | 8.53 | 1.42 | 0.46 | |||
| O | 20.19 | 3.36 | 2.19 | 2.59 | ||
| S | 5.08 | 0.85 | 0.55 | 1.30 | 0.16 | |
| Enrichment ratio | Atom | H | C | N | O | S |
| H | 0.90 | |||||
| C | 1.07 | 0.18 | ||||
| N | 0.77 | 3.31 | ||||
| O | 1.39 | 0.51 | 0.32 | 0.15 | ||
| S | 1.20 | 0.69 |
| MOs | E | ΔE |
|---|---|---|
| LUMO | −0.0946 | 0.1732 |
| HOMO | −0.2678 | |
| LUMO + 1 | −0.0822 | 0.1987 |
| HOMO − 1 | −0.2808 | |
| LUMO + 2 | −0.0562 | 0.2335 |
| HOMO − 2 | −0.2898 |
| Compound | Concentration (μg/mL) | % of Inhibition | IC50 Value (μg/mL) |
|---|---|---|---|
| PESMP | 10 50 100 150 200 | 48.00 57.00 68.68 75.49 81.06 | 10.60 ± 0.16 |
| Acarbose | 10 100 200 | 55.21 73.83 82.55 | 8.80 ± 0.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, B.A.; Hamdani, S.S.; Khalid, M.; Ashfaq, M.; Munawar, K.S.; Tahir, M.N.; Braga, A.A.C.; Shawky, A.M.; Alqahtani, A.M.; Abourehab, M.A.S.; et al. Exploring Probenecid Derived 1,3,4-Oxadiazole-Phthalimide Hybrid as α-Amylase Inhibitor: Synthesis, Structural Investigation, and Molecular Modeling. Pharmaceuticals 2023, 16, 424. https://doi.org/10.3390/ph16030424
Khan BA, Hamdani SS, Khalid M, Ashfaq M, Munawar KS, Tahir MN, Braga AAC, Shawky AM, Alqahtani AM, Abourehab MAS, et al. Exploring Probenecid Derived 1,3,4-Oxadiazole-Phthalimide Hybrid as α-Amylase Inhibitor: Synthesis, Structural Investigation, and Molecular Modeling. Pharmaceuticals. 2023; 16(3):424. https://doi.org/10.3390/ph16030424
Chicago/Turabian StyleKhan, Bilal Ahmad, Syeda Shamila Hamdani, Muhammad Khalid, Muhammad Ashfaq, Khurram Shahzad Munawar, Muhammad Nawaz Tahir, Ataualpa A. C. Braga, Ahmed M. Shawky, Alaa M. Alqahtani, Mohammed A. S. Abourehab, and et al. 2023. "Exploring Probenecid Derived 1,3,4-Oxadiazole-Phthalimide Hybrid as α-Amylase Inhibitor: Synthesis, Structural Investigation, and Molecular Modeling" Pharmaceuticals 16, no. 3: 424. https://doi.org/10.3390/ph16030424
APA StyleKhan, B. A., Hamdani, S. S., Khalid, M., Ashfaq, M., Munawar, K. S., Tahir, M. N., Braga, A. A. C., Shawky, A. M., Alqahtani, A. M., Abourehab, M. A. S., Gabr, G. A., Ibrahim, M. A. A., & Sidhom, P. A. (2023). Exploring Probenecid Derived 1,3,4-Oxadiazole-Phthalimide Hybrid as α-Amylase Inhibitor: Synthesis, Structural Investigation, and Molecular Modeling. Pharmaceuticals, 16(3), 424. https://doi.org/10.3390/ph16030424