
Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis
 , , and
, , and 
Abstract
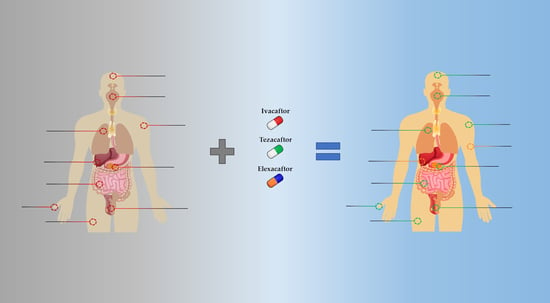
:
1. Introduction
2. Clinical Trials: From Monotherapy to Triple Combination Therapy
2.1. Ivacaftor Monotherapy
2.2. Lumacaftor-Ivacaftor Therapy
2.3. Tezacaftor-Ivacaftor Therapy
2.4. Triple Combination Therapy
3. Clinical Outcomes of ETI Therapy in Case Reports, Observational, and Real-Life Studies
3.1. Clinical Studies in PwCF Carrying at Least One F508del-CFTR Allele and Advanced Lung Disease
3.2. Case Reports in Rare (Non-F508del) CF Genotypes
3.3. Additional Respiratory Implications
3.4. Pre- and Post-Transplant Implications
3.5. Gastrointestinal Implications
3.6. Fertility and Pregnancy Implications
3.7. Skin Implications
3.8. Other Implications
4. Outlook and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviation
References
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Garratt, A.; Hill, A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J. Cyst. Fibros. 2022, 21, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.C.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Shteinberg, M.; Taylor-Cousar, J.L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 2020, 29, 190112. [Google Scholar] [CrossRef] [Green Version]
- Cystic Fibrosis Foundation Patient Registry–2021 Annual Data Report. Available online: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf (accessed on 31 January 2023).
- Lopez, A.; Daly, C.; Vega-Hernandez, G.; MacGregor, G.; Rubin, J.L. Elexacaftor/tezacaftor/ivacaftor projected survival and long-term health outcomes in people with cystic fibrosis homozygous for F508del. J. Cyst. Fibros. 2023, in press. [Google Scholar] [CrossRef]
- Rachel, M.; Topolewicz, S.; Śliwczyński, A.; Galiniak, S. Managing Cystic Fibrosis in Polish Healthcare. Int. J. Environ. Res. Public Health 2020, 17, 7630. [Google Scholar] [CrossRef]
- da Silva Filho, L.V.R.F.; Zampoli, M.; Cohen-Cymberknoh, M.; Kabra, S.K. Cystic fibrosis in low and middle-income countries (LMIC): A view from four different regions of the world. Paediatr. Respir. Rev. 2020, 38, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The impact of FDA and EMA regulatory decision–making process on the access to CFTR modulators for the treatment of cystic fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef]
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfsen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J. Cyst. Fibros. 2019, 18, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, D.; Carnovale, V.; Iacotucci, P.; Braggion, C.; Castellani, C.; Cimino, G.; Colangelo, C.; Francalanci, M.; Leonetti, G.; Lucidi, V.; et al. Effectivenesss of ivacaftor in severe cystic fibrosis patients and non-G551D gating mutations. Pediatr. Pulmonol. 2019, 54, 1398–1403. [Google Scholar] [CrossRef]
- Munck, A.; Kerem, E.; Ellemunter, H.; Campbell, D.; Wang, L.T.; Ahluwalia, N.; Owen, C.A.; Wainwright, C. Tezacaftor/ivacaftor in people with cystic fibrosis heterozygous for minimal function CFTR mutations. J. Cyst. Fibros. 2020, 19, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.T.; Taylor-Cousar, J.L.; Davies, J.; Gibson, R.L.; Mall, M.A.; McKone, E.F.; McNally, P.; Ramsey, B.W.; Rayment, J.H.; Rowe, S.M.; et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2021, 203, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.J.; Mall, M.A.; Álvarez, A.; Colombo, C.; de Winter-de Groot, K.M.; Fajac, I.; McBennett, K.A.; McKone, E.F.; Ramsey, B.W.; Sutharsan, S.; et al. Triple Therapy for Cystic Fibrosis Phe508del-Gating and -Residual Function Genotypes. N. Engl. J. Med. 2021, 385, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef]
- Sutharsan, S.; McKone, E.F.; Downey, D.G.; Duckers, J.; MacGregor, G.; Tullis, E.; Van Braeckel, E.; Wainwright, C.E.; Watson, D.; Ahluwalia, N.; et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: A 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir. Med. 2022, 10, 267–277. [Google Scholar] [CrossRef]
- Mall, M.A.; Brugha, R.; Gartner, S.; Legg, J.; Moeller, A.; Mondejar-Lopez, P.; Prais, D.; Pressler, T.; Ratjen, F.; Reix, P.; et al. Efficacy and Safety of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 Through 11 Years of Age with Cystic Fibrosis Heterozygous for F508del and a Minimal Function Mutation: A Phase 3b, Randomized, Placebo-controlled Study. Am. J. Respir. Crit. Care Med. 2022, 206, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.P.; Paynter, A.C.; Heltshe, S.L.; Donaldson, S.H.; Frederick, C.A.; Freedman, S.D.; Gelfond, D.; Hoffman, L.R.; Kelly, A.; Narkewicz, M.R.; et al. Clinical Effectiveness of Elexacaftor/Tezacaftor/Ivacaftor in People with Cystic Fibrosis: A Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 529–539. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Ratjen, F.; Russell, R.; Donaldson, S.H.; Riekert, K.A.; Sawicki, G.S.; Odem-Davis, K.; Young, J.K.; Rosenbluth, D.; Taylor-Cousar, J.L.; et al. Discontinuation versus continuation of hypertonic saline or dornase alfa in modulator treated people with cystic fibrosis (SIMPLIFY): Results from two parallel, multicentre, open-label, randomised, controlled, non-inferiority trials. Lancet Respir. Med. 2022. [Google Scholar] [CrossRef]
- Uluer, A.Z.; MacGregor, G.; Azevedo, P.; Indihar, V.; Keating, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Rubenstein, R.C.; et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: Randomised, double-blind, controlled, phase 2 trials. Lancet Respir. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.-R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 64–73. [Google Scholar] [CrossRef]
- Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Ferrillo, L.; Pepe, A.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; Celardo, A.; et al. Elexacaftor/Tezacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation and Advanced Lung Disease: A 48-Week Observational Study. J. Clin. Med. 2022, 11, 1021. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, K.M.; O’Carroll, O.M.; Carroll, C.; Grogan, B.; Connolly, A.; O’Shaughnessy, L.; Nicholson, T.T.; Gallagher, C.G.; McKone, E.F. Efficacy of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease. Eur. Respir. J. 2021, 57, 2003079. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Colangelo, C.; Marsicovetere, G.; D’Andria, M.; Francalanci, M.; Innocenti, D.; Masi, E.; Avarello, A.; Taccetti, G.; Amato, F.; et al. Effectiveness of Elexacaftor/Tezacaftor/Ivacaftor Therapy in Three Subjects with the Cystic Fibrosis Genotype Phe508del/Unknown and Advanced Lung Disease. Genes 2021, 12, 1178. [Google Scholar] [CrossRef]
- Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Medio, P.; Ferrillo, L.; De Gregorio, F.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; et al. Effectiveness and safety of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease with the Phe508del/minimal function genotype. Respir. Med. 2021, 189, 106646. [Google Scholar] [CrossRef]
- Salvatore, D.; Colangelo, C.; D’Andria, M.; Marsicovetere, G.; Passarella, D. Elexacaftor/tezacaftor/ivacaftor as rescue therapy in a patient with the cystic fibrosis genotype F508DEL/G1244E. Clin. Case Rep. 2021, 9, e04713. [Google Scholar] [CrossRef]
- Gur, M.; Bar-Yoseph, R.; Toukan, Y.; Hanna, M.; Masarweh, K.; Bentur, L. Twelve years of progressive Mycobacterium abscessus lung disease in CF-Response to Trikafta. Pediatr. Pulmonol. 2021, 56, 4048–4050. [Google Scholar] [CrossRef]
- Migliorisi, G.; Collura, M.; Ficili, F.; Pensabene, T.; Bongiorno, D.; Collura, A.; Di Bernardo, F.; Stefani, S. Elexacaftor-Tezacaftor-Ivacaftor as a Final Frontier in the Treatment of Cystic Fibrosis: Definition of the Clinical and Microbiological Implications in a Case-Control Study. Pharmaceuticals 2022, 15, 606. [Google Scholar] [CrossRef]
- Macconi, L.; Galici, V.; Di Maurizio, M.; Rossi, E.; Taccetti, G.; Terlizzi, V. Early Effects of Elexacaftor-Tezacaftor-Ivacaftor Therapy on Magnetic Resonance Imaging in Patients with Cystic Fibrosis and Advanced Lung Disease. J. Clin. Med. 2022, 11, 4277. [Google Scholar] [CrossRef]
- Stylemans, D.; Darquenne, C.; Schuermans, D.; Verbanck, S.; Vanderhelst, E. Peripheral lung effect of elexacaftor/tezacaftor/ivacaftor in adult cystic fibrosis. J. Cyst. Fibros. 2022, 21, 160–163. [Google Scholar] [CrossRef]
- Kos, R.; Neerincx, A.H.; Fenn, D.W.; Brinkman, P.; Lub, R.; Vonk, S.E.M.; Roukema, J.; Reijers, M.H.; Terheggen-Lagro, S.W.J.; Altenburg, J.; et al. Real-life efficacy and safety of elexacaftor/tezacaftor/ivacaftor on severe cystic fibrosis lung disease patients. Pharmacol. Res. Perspect. 2022, 10, e01015. [Google Scholar] [CrossRef]
- Salvatore, D.; Cimino, G.; Troiani, P.; Bignamini, E.; Esposito, I.; Leonetti, G.; Zanda, M.; Manunza, D.; Pepe, A. Elexacaftor/tezacaftor/ivacaftor in children aged 6–11 years with cystic fibrosis, at least one F508DEL allele, and advanced lung disease: A 24-week observational study. Pediatr. Pulmonol. 2022, 57, 2253–2256. [Google Scholar] [CrossRef] [PubMed]
- Dhote, T.; Martin, C.; Regard, L.; Pesenti, L.; Kanaan, R.; Carlier, N.; Honoré, I.; Da Silva, J.; Witko-Sarsat, V.; Burgel, P.-R. Normalisation of circulating neutrophil counts after 12 months of elexacaftor-tezacaftor-ivacaftor in patients with advanced cystic fibrosis. Eur. Respir. J. 2023, 61, 2202096. [Google Scholar] [CrossRef]
- McCoy, K.S.; Blind, J.; Johnson, T.; Olson, P.; Raterman, L.; Bai, S.; Eisner, M.; Sheikh, S.I.; Druhan, S.; Young, C.; et al. Clinical change 2 years from start of elexacaftor-tezacaftor-ivacaftor in severe cystic fibrosis. Pediatr. Pulmonol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Comegna, M.; Terlizzi, V.; Salvatore, D.; Colangelo, C.; Di Lullo, A.M.; Zollo, I.; Taccetti, G.; Castaldo, G.; Amato, F. Elexacaftor-Tezacaftor-Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype. Antibiotics 2021, 10, 828. [Google Scholar] [CrossRef]
- Huang, Y.; Paul, G.; Lee, J.; Yarlagadda, S.; McCoy, K.; Naren, A.P. Elexacaftor/Tezacaftor/Ivacaftor Improved Clinical Outcomes in a Patient with N1303K-CFTR Based on In Vitro Experimental Evidence. Am. J. Respir. Crit. Care Med. 2021, 204, 1231–1235. [Google Scholar] [CrossRef]
- Elson, E.C.; Capel, P.; Haynes, J.; Duehlmeyer, S.; Fischer, M.; Escobar, H. CFTR Modulator Therapy in an Individual With Cystic Fibrosis Caused by a N1303K CFTR Variant and Infected With Mycobacterium abscessus. J. Pediatr. Pharmacol. Ther. 2022, 27, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.-R.; Sermet-Gaudelus, I.; Durieu, I.; Kanaan, R.; Macey, J.; Grenet, D.; Porzio, M.; Coolen-Allou, N.; Chiron, R.; Marguet, C.; et al. The French Compassionate Program of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis with advanced lung disease and no F508del CFTR variant. Eur. Respir. J. 2023. [Google Scholar] [CrossRef]
- Stekolchik, E.; Saul, D.; Chidekel, A. Clinical efficacy of elexacaftor-tezacaftor-ivacaftor in an adolescent with homozygous G85E cystic fibrosis. Respir. Med. Case Rep. 2022, 40, 101775. [Google Scholar] [CrossRef]
- Livnat, G.; Dagan, A.; Heching, M.; Shmueli, E.; Prais, D.; Yaacoby-Bianu, K.; Stein, N.; Mei-Zahav, M.; Gur, M.; Cohen-Cymberknoh, M.; et al. Treatment effects of Elexacaftor/Tezacaftor/Ivacaftor in people with CF carrying non-F508del mutations. J. Cyst. Fibros. 2022. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulou, G.; Brandenberg, N.; Hoehnel, S.; Ceroni, C.; Balmpouzis, Z.; Blanchon, S.; Dorta, G.; Sauty, A.; Koutsokera, A. Rectal organoid-guided CFTR modulator therapy restores lung function in a CF patient with the rare 1677delTA/R334W genotype. Eur. Respir. J. 2022, 60, 2201341. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, L.; Triphan, S.M.F.; Wege, S.; Kauczor, H.-U.; Heussel, C.P.; Schmitt, N.; Wuennemann, F.; Mayer, V.L.; Sommerburg, O.; Mall, M.A.; et al. Magnetic resonance imaging detects improvements of pulmonary and paranasal sinus abnormalities in response to elexacaftor/tezacaftor/ivacaftor therapy in adults with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 1053–1060. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Renz, D.M.; Stahl, M.; Pallenberg, S.T.; Sommerburg, O.; Naehrlich, L.; Berges, J.; Dohna, M.; Ringshausen, F.C.; Doellinger, F.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on Lung Clearance Index and Magnetic Resonance Imaging in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2022, 206, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Streibel, C.; Willers, C.C.; Pusterla, O.; Bauman, G.; Stranzinger, E.; Brabandt, B.; Bieri, O.; Curdy, M.; Bullo, M.; Frauchiger, B.S.; et al. Effects of elexacaftor/tezacaftor/ivacaftor therapy in children with cystic fibrosis–A comprehensive assessment using lung clearance index, spirometry, and functional and structural lung MRI. J. Cyst. Fibros. 2023. [Google Scholar] [CrossRef]
- Stapleton, A.L.; Kimple, A.J.; Goralski, J.L.; Nouraie, S.M.; Branstetter, B.F.; Shaffer, A.D.; Pilewski, J.M.; Senior, B.A.; Lee, S.E.; Zemke, A.C. Elexacaftor-Tezacaftor-Ivacaftor improves sinonasal outcomes in cystic fibrosis. J. Cyst. Fibros. 2022, 21, 792–799. [Google Scholar] [CrossRef]
- Lee, T.; Sawicki, G.S.; Altenburg, J.; Millar, S.J.; Geiger, J.M.; Jennings, M.T.; Lou, Y.; McGarry, L.J.; Van Brunt, K.; Linnemann, R.W. Effect of Elexacaftor/Tezacaftor/Ivacaftor on Annual Rate of Lung Function Decline in People with Cystic Fibrosis. J. Cyst. Fibros. 2022. [Google Scholar] [CrossRef]
- Bec, R.; Reynaud-Gaubert, M.; Arnaud, F.; Naud, R.; Dufeu, N.; Di Bisceglie, M.; Coiffard, B.; Gaubert, J.-Y.; Bermudez, J.; Habert, P. Chest computed tomography improvement in patients with cystic fibrosis treated with elexacaftor-tezacaftor-ivacaftor: Early report. Eur. J. Radiol. 2022, 154, 110421. [Google Scholar] [CrossRef]
- DiMango, E.; Overdevest, J.; Keating, C.; Francis, S.F.; Dansky, D.; Gudis, D. Effect of highly effective modulator treatment on sinonasal symptoms in cystic fibrosis. J. Cyst. Fibros. 2021, 20, 460–463. [Google Scholar] [CrossRef]
- Causer, A.J.; Shute, J.K.; Cummings, M.H.; Shepherd, A.I.; Wallbanks, S.R.; Pulsford, R.M.; Bright, V.; Connett, G.; Saynor, Z.L. Elexacaftor–Tezacaftor–Ivacaftor improves exercise capacity in adolescents with cystic fibrosis. Pediatr. Pulmonol. 2022, 57, 2652–2658. [Google Scholar] [CrossRef]
- Martin, C.; Burnet, E.; Ronayette-Preira, A.; de Carli, P.; Martin, J.; Delmas, L.; Prieur, B.; Burgel, P.-R. Patient perspectives following initiation of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis and advanced lung disease. Respir. Med. Res. 2021, 80, 100829. [Google Scholar] [CrossRef]
- Bacon, D.R.; Stapleton, A.; Goralski, J.L.; Ebert, C.S.; Thorp, B.D.; Nouraie, M.; Shaffer, A.D.; Senior, B.A.; Lee, S.E.; Zemke, A.C.; et al. Olfaction before and after initiation of elexacaftor-tezacaftor-ivacaftor in a cystic fibrosis cohort. Int. Forum Allergy Rhinol. 2022, 12, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Beswick, D.M.; Humphries, S.M.; Balkissoon, C.D.; Strand, M.; Vladar, E.K.; Lynch, D.A.; Taylor-Cousar, J.L. Impact of Cystic Fibrosis Transmembrane Conductance Regulator Therapy on Chronic Rhinosinusitis and Health Status: Deep Learning CT Analysis and Patient-reported Outcomes. Ann. Am. Thorac. Soc. 2022, 19, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, C.X.; Osterbauer, B.; Hasday, S.; Keens, T.G.; Koempel, J.; Ference, E.H. Improvement in sinonasal quality-of-life indicators for pediatric patients with cystic fibrosis treated with elexacaftor-tezacaftor-ivacaftor. Int. Forum Allergy Rhinol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.; Britt, R.D.; Ryan-Wenger, N.A.; Khan, A.Q.; Lewis, B.W.; Gushue, C.; Ozuna, H.; Jaganathan, D.; McCoy, K.; Kopp, B.T. Impact of elexacaftor–tezacaftor–ivacaftor on bacterial colonization and inflammatory responses in cystic fibrosis. Pediatr. Pulmonol. 2022. [Google Scholar] [CrossRef]
- Bode, S.F.N.; Rapp, H.; Lienert, N.; Appel, H.; Fabricius, D. Effects of CFTR-modulator triple therapy on sinunasal symptoms in children and adults with cystic fibrosis. Eur. Arch. Otorhino-Laryngol. 2023. [Google Scholar] [CrossRef]
- Giallongo, A.; Parisi, G.F.; Papale, M.; Manti, S.; Mulé, E.; Aloisio, D.; Terlizzi, V.; Rotolo, N.; Leonardi, S. Effects of Elexacaftor/Tezacaftor/Ivacaftor on Cardiorespiratory Polygraphy Parameters and Respiratory Muscle Strength in Cystic Fibrosis Patients with Severe Lung Disease. Genes 2023, 14, 449. [Google Scholar] [CrossRef]
- Beck, M.R.; Hornick, D.B.; Pena, T.A.; Singh, S.B.; Wright, B.A. Impact of elexacaftor/tezacaftor/ivacaftor on bacterial cultures from people with cystic fibrosis. Pediatr. Pulmonol. 2023. [Google Scholar] [CrossRef]
- Wise, S.K.; Lin, S.Y.; Toskala, E.; Orlandi, R.R.; Akdis, C.A.; Alt, J.A.; Azar, A.; Baroody, F.M.; Bachert, C.; Canonica, G.W.; et al. International Consensus Statement on Allergy and Rhinology: Allergic Rhinitis. Int. Forum Allergy Rhinol. 2018, 8, 108–352. [Google Scholar] [CrossRef]
- Birket, S.E.; Chu, K.K.; Houser, G.H.; Liu, L.; Fernandez, C.M.; Solomon, G.M.; Lin, V.; Shastry, S.; Mazur, M.; Sloane, P.A.; et al. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. Am. J. Physiol.–Lung Cell. Mol. Physiol. 2016, 310, L928–L939. [Google Scholar] [CrossRef] [Green Version]
- Lindig, J.; Steger, C.; Beiersdorf, N.; Michl, R.; Beck, J.F.; Hummel, T.; Mainz, J.G. Smell in cystic fibrosis. Eur. Arch. Otorhinolaryngol. 2013, 270, 915–921. [Google Scholar] [CrossRef]
- Saynor, Z.L.; Barker, A.R.; Oades, P.J.; Williams, C.A. The Effect of Ivacaftor in Adolescents With Cystic Fibrosis (G551D Mutation). Pediatr. Phys. Ther. 2014, 26, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Edgeworth, D.; Keating, D.; Ellis, M.; Button, B.; Williams, E.; Clark, D.; Tierney, A.; Heritier, S.; Kotsimbos, T.; Wilson, J. Improvement in exercise duration, lung function and well-being in G551D-cystic fibrosis patients: A double-blind, placebo-controlled, randomized, cross-over study with ivacaftor treatment. Clin. Sci. 2017, 131, 2037–2045. [Google Scholar] [CrossRef] [PubMed]
- Savi, D.; Schiavetto, S.; Simmonds, N.J.; Righelli, D.; Palange, P. Effects of Lumacaftor/Ivacaftor on physical activity and exercise tolerance in three adults with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Almulhem, M.; Harnett, N.; Graham, S.; Haq, I.; Visram, S.; Ward, C.; Brodlie, M. Exploring the impact of elexacaftor-tezacaftor-ivacaftor treatment on opinions regarding airway clearance techniques and nebulisers: TEMPO a qualitative study in children with cystic fibrosis, their families and healthcare professionals. BMJ Open Respir. Res. 2022, 9, e001420. [Google Scholar] [CrossRef]
- Bermingham, B.; Rueschhoff, A.; Ratti, G.; Nesmith, A.; Goodwin, D.; Gray, S.; Garcia, B. Short-term effect of elexacaftor-tezacaftor-ivacaftor on lung function and transplant planning in cystic fibrosis patients with advanced lung disease. J. Cyst. Fibros. 2021, 20, 768–771. [Google Scholar] [CrossRef]
- Martin, C.; Reynaud-Gaubert, M.; Hamidfar, R.; Durieu, I.; Murris-Espin, M.; Danner-Boucher, I.; Chiron, R.; Leroy, S.; Douvry, B.; Grenet, D.; et al. Sustained effectiveness of elexacaftor-tezacaftor-ivacaftor in lung transplant candidates with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 489–496. [Google Scholar] [CrossRef]
- Benninger, L.A.; Trillo, C.; Lascano, J. CFTR modulator use in post lung transplant recipients. J. Hear. Lung Transpl. 2021, 40, 1498–1501. [Google Scholar] [CrossRef]
- Doligalski, C.T.; McKinzie, C.J.; Yang, A.; Lobo, L.J.; Coakley, R. Poor tolerability of cystic fibrosis transmembrane conductance regulator modulator therapy in lung transplant recipients. Pharmacotherapy 2022, 42, 580–584. [Google Scholar] [CrossRef]
- Ramos, K.J.; Guimbellot, J.S.; Valapour, M.; Bartlett, L.E.; Wai, T.H.; Goss, C.H.; Pilewski, J.M.; Faro, A.; Diamond, J.M. CFLTC Study Group Use of elexacaftor/tezacaftor/ivacaftor among cystic fibrosis lung transplant recipients. J. Cyst. Fibros. 2022, 21, 745–752. [Google Scholar] [CrossRef]
- McKinzie, C.J.; Doligalski, C.T.; Lobritto, S.J.; Coakley, R.D.; Gower, W.A. Use of elexacaftor/tezacaftor/ivacaftor in liver transplant patients with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 227–229. [Google Scholar] [CrossRef]
- Ragan, H.; Autry, E.; Bomersback, T.; Hewlett, J.; Kormelink, L.; Safirstein, J.; Shanley, L.; Lubsch, L. The use of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis postliver transplant: A case series. Pediatr. Pulmonol. 2022, 57, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Ørum, M.B.; Rönsholt, F.F.; Jeppesen, M.; Bendstrup, E.; Katzenstein, T.L.; Ott, P.; Perch, M.; Pressler, T.; Qvist, T.; Jensen-Fangel, S. Outcome of elexacaftor/tezacaftor/ivacaftor therapy in patients with cystic fibrosis and solid organ transplantation. Pediatr. Pulmonol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenberg, S.J.; Vu, P.T.; Skalland, M.; Hoffman, L.R.; Pope, C.; Gelfond, D.; Narkewicz, M.R.; Nichols, D.P.; Heltshe, S.L.; Donaldson, S.H.; et al. Elexacaftor/tezacaftor/ivacaftor and gastrointestinal outcomes in cystic fibrosis: Report of promise-GI. J. Cyst. Fibros. 2022. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Begnel, L.; Wallendorf, M.; Litvin, M. Effect of elexacaftor-tezacaftor-ivacaftor on body weight and metabolic parameters in adults with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 265–271. [Google Scholar] [CrossRef]
- Scully, K.J.; Marchetti, P.; Sawicki, G.S.; Uluer, A.; Cernadas, M.; Cagnina, R.E.; Kennedy, J.C.; Putman, M.S. The effect of elexacaftor/tezacaftor/ivacaftor (ETI) on glycemia in adults with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 258–263. [Google Scholar] [CrossRef]
- Wright, B.A.; Ketchen, N.K.; Rasmussen, L.N.; Bartels, A.R.; Singh, S.B. Impact of elexacaftor/tezacaftor/ivacaftor on vitamin D absorption in cystic fibrosis patients. Pediatr. Pulmonol. 2022, 57, 655–657. [Google Scholar] [CrossRef]
- Shakir, S.; Echevarria, C.; Doe, S.; Brodlie, M.; Ward, C.; Bourke, S.J. Elexacaftor-Tezacaftor-Ivacaftor improve Gastro-Oesophageal reflux and Sinonasal symptoms in advanced cystic fibrosis. J. Cyst. Fibros. 2022, 21, 807–810. [Google Scholar] [CrossRef]
- Safirstein, J.; Grant, J.J.; Clausen, E.; Savant, D.; Dezube, R.; Hong, G. Biliary disease and cholecystectomy after initiation of elexacaftor/ivacaftor/tezacaftor in adults with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 506–510. [Google Scholar] [CrossRef]
- Lowry, S.; Mogayzel, P.J.; Oshima, K.; Karnsakul, W. Drug-induced liver injury from elexacaftor/ivacaftor/tezacaftor. J. Cyst. Fibros. 2022, 21, e99–e101. [Google Scholar] [CrossRef]
- Korten, I.; Kieninger, E.; Krueger, L.; Bullo, M.; Flück, C.E.; Latzin, P.; Casaulta, C.; Boettcher, C. Short-Term Effects of Elexacaftor/Tezacaftor/Ivacaftor Combination on Glucose Tolerance in Young People With Cystic Fibrosis-An Observational Pilot Study. Front. Pediatr. 2022, 10, 852551. [Google Scholar] [CrossRef]
- Chan, C.L.; Granados, A.; Moheet, A.; Singh, S.; Vigers, T.; Arbeláez, A.M.; Yi, Y.; Hu, S.; Norris, A.W.; Ode, K.L. Glycemia and β-cell function before and after elexacaftor/tezacaftor/ivacaftor in youth and adults with cystic fibrosis. J. Clin. Transl. Endocrinol. 2022, 30, 100311. [Google Scholar] [CrossRef] [PubMed]
- Mainz, J.G.; Zagoya, C.; Polte, L.; Naehrlich, L.; Sasse, L.; Eickmeier, O.; Smaczny, C.; Barucha, A.; Bechinger, L.; Duckstein, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor Treatment Reduces Abdominal Symptoms in Cystic Fibrosis-Early results Obtained With the CF-Specific CFAbd-Score. Front. Pharmacol. 2022, 13, 877118. [Google Scholar] [CrossRef]
- Steinack, C.; Ernst, M.; Beuschlein, F.; Hage, R.; Roeder, M.; Schuurmans, M.M.; Schmid, C.; Gaisl, T. Improved glucose tolerance after initiation of Elexacaftor/Tezacaftor/Ivacaftor in adults with cystic fibrosis. J. Cyst. Fibros. 2023. [Google Scholar] [CrossRef]
- Tewkesbury, D.H.; Athwal, V.; Bright-Thomas, R.J.; Jones, A.M.; Barry, P.J. Longitudinal effects of elexacaftor/tezacaftor/ivacaftor on liver tests at a large single adult cystic fibrosis centre. J. Cyst. Fibros. 2023. [Google Scholar] [CrossRef]
- Francalanci, M.; Terlizzi, V.; Fevola, C.; Di Rosa, G.; Pierattini, V.; Roselli, E.; Bonomi, P.; Cavicchi, M.C.; Galici, V.; Neri, A.S.; et al. Nutritional Status and Circulating Levels of Fat-Soluble Vitamins in Cystic Fibrosis Patients: A Cohort Study and Evaluation of the Effect of CFTR Modulators. Children 2023, 10, 252. [Google Scholar] [CrossRef]
- Schnell, A.; Jüngert, J.; Klett, D.; Hober, H.; Kaiser, N.; Ruppel, R.; Geppert, A.; Tremel, C.; Sobel, J.; Plattner, E.; et al. Increase of liver stiffness and altered bile acid metabolism after triple CFTR modulator initiation in children and young adults with cystic fibrosis. Liver Int. 2023. [Google Scholar] [CrossRef]
- Wood, M.; Babowicz, F.; Kennedy, A.G.; Antell, M.; Gilhooly, E.; Tompkins, B.J.; Reddy, S.S. Incidence of transaminitis in adults with cystic fibrosis taking elexacaftor/tezacaftor/ivacaftor. J. Am. Pharm. Assoc. 2023. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Magaret, A.; Vu, P.T.; VanDalfsen, J.M.; Keller, A.; Wilson, A.; Putman, M.S.; Mayer-Hamblett, N.; Esther, C.R.; Taylor-Cousar, J.L. Prospectively evaluating maternal and fetal outcomes in the era of CFTR modulators: The MAYFLOWERS observational clinical trial study design. BMJ Open Respir. Res. 2022, 9, e001289. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.E.; Goodwin, D.L.; NeSmith, A.; Garcia, B.; Mingora, C.; Ladores, S.L.; Rowe, S.M.; Krick, S.; Solomon, G.M. Elexacafator/tezacaftor/ivacaftor resolves subfertility in females with CF: A two center case series. J. Cyst. Fibros. 2021, 20, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Chamagne, M.; Farabet, C.; Grenet, D.; Ayoubi, J.M. Comparison of two pregnancies with and without elexacaftor-tezacaftor-ivacaftor in a woman with cystic fibrosis. Respir. Med. Res. 2023, 83, 100953. [Google Scholar] [CrossRef]
- Collins, B.; Fortner, C.; Cotey, A.; Esther, C.R.J.; Trimble, A. Drug exposure to infants born to mothers taking Elexacaftor, Tezacaftor, and Ivacaftor. J. Cyst. Fibros. 2022, 21, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Fortner, C.N.; Seguin, J.M.; Kay, D.M. Normal pancreatic function and false-negative CF newborn screen in a child born to a mother taking CFTR modulator therapy during pregnancy. J. Cyst. Fibros. 2021, 20, 835–836. [Google Scholar] [CrossRef] [PubMed]
- Balmpouzis, Z.; Faure van Rossum, A.; Baud, D.; Panchaud, A.; Mitropoulou, G.; Mazza Stalder, J.; Koutsokera, A. Successful pregnancy in a cystic fibrosis patient with a severe impairment of lung function receiving Elexacaftor-Tezacaftor-Ivacaftor. Respir. Med. Case Rep. 2022, 40, 101776. [Google Scholar] [CrossRef] [PubMed]
- Szentpetery, S.; Foil, K.; Hendrix, S.; Gray, S.; Mingora, C.; Head, B.; Johnson, D.; Flume, P.A. A case report of CFTR modulator administration via carrier mother to treat meconium ileus in a F508del homozygous fetus. J. Cyst. Fibros. 2022, 21, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, S.M.; Duehlmeyer, S.; Slack, S.M.; Jacobs, H.R.; Heckman, B. Testicular pain following initiation of elexacaftor/tezacaftor/ivacaftor in males with cystic fibrosis. J. Cyst. Fibros. 2020, 19, e39–e41. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Godfrey, E.M.; Josephy, T.; Aitken, M.L.; Taylor-Cousar, J.L. Pregnancy among cystic fibrosis women in the era of CFTR modulators. J. Cyst. Fibros. 2017, 16, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Yeley, J.; Brown, C.; Wesson, M.; Lesko, B.G.; Slaven, J.E.; Chmiel, J.F.; Jain, R.; Sanders, D.B. Immunoreactive Trypsinogen in Infants Born to Women with Cystic Fibrosis Taking Elexacaftor–Tezacaftor–Ivacaftor. Int. J. Neonatal Screen. 2023, 9, 10. [Google Scholar] [CrossRef]
- Kaplan, E.; Shwachman, H.; Perlmutter, A.D.; Rule, A.; Khaw, K.T.; Holsclaw, D.S. Reproductive failure in males with cystic fibrosis. N. Engl. J. Med. 1968, 279, 65–69. [Google Scholar] [CrossRef]
- Hu, M.K.; Wood, G.; Dempsey, O. “Triple therapy” (elexacaftor, tezacaftor, ivacaftor) skin rash in patients with cystic fibrosis. Postgrad. Med. J. 2022, 98, 86. [Google Scholar] [CrossRef]
- Leonhardt, K.; Autry, E.B.; Kuhn, R.J.; Wurth, M.A. CFTR modulator drug desensitization: Preserving the hope of long term improvement. Pediatr. Pulmonol. 2021, 56, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Stashower, J.; Carr, P.; Miller, V.; Zlotoff, B. Novel reaction to new cystic fibrosis medication Trikafta. Clin. Case Rep. 2021, 9, e04116. [Google Scholar] [CrossRef]
- Goldberg, R.H.; Matthews, N.H.; Hristov, A.C.; Wang, F. Urticaria multiforme-like eruption due to a novel agent elexacaftor/tezacaftor/ivacaftor in a pediatric patient with cystic fibrosis. JAAD case reports 2021, 18, 71–73. [Google Scholar] [CrossRef]
- Cheng, A.; Baker, O.; Hill, U. Elexacaftor, tezacaftor and ivacaftor: A case of severe rash and approach to desensitisation. BMJ Case Rep. 2022, 15, e247042. [Google Scholar] [CrossRef]
- Muirhead, C.; Verzasconi, D.; Joshi, S. At-home compounding preparation of slow desensitization of elexacaftor/tezacaftor/ivacaftor for delayed hypersensitivity rash. Pediatr. Pulmonol. 2022, 57, 1779–1781. [Google Scholar] [CrossRef]
- Bhaskaran, D.; Bateman, K. A case of Elexacaftor-Tezacaftor-Ivacaftor induced rash resolving without interruption of treatment. J. Cyst. Fibros. 2022, 21, 1077–1079. [Google Scholar] [CrossRef]
- Diseroad, E.R.; Mogayzel, P.J.; Pan, A. Rechallenge of Elexacaftor/Tezacaftor/Ivacaftor After Skin Rash in Two Pediatric Patients. J. Pediatr. Pharmacol. Ther. 2022, 27, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.N.; Jacobs, H.R.; Philbrick, A.; Zhou, X.A.; Simonsen, M.M.; Safirstein, J.A.; Rotolo, S.M. Drug-induced acne with elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Li Pomi, F.; Di Bartolomeo, L.; Vaccaro, M.; Lentini, M.; Cristadoro, S.; Lucanto, M.C.; Lombardo, M.; Costa, S.; Borgia, F. Malassezia Folliculitis following Triple Therapy for Cystic Fibrosis. Medicina 2022, 58, 1204. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.; Marmor, I.; Schafer, C.; Ko, J.; Torres Garcia, J.A.; Rosman, I.S.; Coughlin, C.; Coverstone, A.; White, A.J. Serum sickness-like reaction following initiation of elexacaftor/tezacaftor/ivacaftor therapy. Pediatr. Pulmonol. 2020, 55, 2846–2847. [Google Scholar] [CrossRef]
- Sosinski, L.M.; Martin, C.; Neugebauer, K.A.; Ghuneim, L.-A.J.; Guzior, D.V.; Castillo-Bahena, A.; Mielke, J.; Thomas, R.; McClelland, M.; Conrad, D.; et al. A restructuring of microbiome niche space is associated with Elexacaftor-Tezacaftor-Ivacaftor therapy in the cystic fibrosis lung. J. Cyst. Fibros. 2022, 21, 996–1005. [Google Scholar] [CrossRef]
- Miller, A.C.; Harris, L.M.; Cavanaugh, J.E.; Abou Alaiwa, M.; Stoltz, D.A.; Hornick, D.B.; Polgreen, P.M. The Rapid Reduction of Infection-Related Visits and Antibiotic Use Among People With Cystic Fibrosis After Starting Elexacaftor-Tezacaftor-Ivacaftor. Clin. Infect. Dis. 2022, 75, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.; Bass, J.L. The Effect of Elexacaftor/Tezacaftor/Ivacaftor on Hospitalizations and Intravenous Antibiotic Use. Perm. J. 2022, 26, 73–79. [Google Scholar] [CrossRef]
- Shneerson, J.M. Digital clubbing and hyperthrophic osteorthropathy: The underlying mechanisms. Br. J. Dis. Chest 1981, 75, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Augarten, A.; Goldman, R.; Laufer, J.; Szeinberg, A.; Efrati, O.; Barak, A.; Miller, M.S.; Yahav, Y. Reversal of digital clubbing after lung transplantation in cystic fibrosis patients: A clue to the pathogenesis of clubbing. Pediatr. Pulmonol. 2002, 34, 378–380. [Google Scholar] [CrossRef]
- Mahlen, T.; Barton, L.; Roberts, D. Impact of highly effective CFTR modulator therapy on digital clubbing in patients with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Scholte, B.J.; Horati, H.; Veltman, M.; Vreeken, R.J.; Garratt, L.W.; Tiddens, H.A.W.M.; Janssens, H.M.; Stick, S.M. Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) Oxidative stress and abnormal bioactive lipids in early cystic fibrosis lung disease. J. Cyst. Fibros. 2019, 18, 781–789. [Google Scholar] [CrossRef]
- Westhölter, D.; Schumacher, F.; Wülfinghoff, N.; Sutharsan, S.; Strassburg, S.; Kleuser, B.; Horn, P.A.; Reuter, S.; Gulbins, E.; Taube, C.; et al. CFTR modulator therapy alters plasma sphingolipid profiles in people with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 713–720. [Google Scholar] [CrossRef]
- Liessi, N.; Pesce, E.; Braccia, C.; Bertozzi, S.M.; Giraudo, A.; Bandiera, T.; Pedemonte, N.; Armirotti, A. Distinctive lipid signatures of bronchial epithelial cells associated with cystic fibrosis drugs, including Trikafta. JCI Insight 2020, 5, e138722. [Google Scholar] [CrossRef]
- Veltman, M.; De Sanctis, J.B.; Stolarczyk, M.; Klymiuk, N.; Bähr, A.; Brouwer, R.W.; Oole, E.; Shah, J.; Ozdian, T.; Liao, J.; et al. CFTR Correctors and Antioxidants Partially Normalize Lipid Imbalance but not Abnormal Basal Inflammatory Cytokine Profile in CF Bronchial Epithelial Cells. Front. Physiol. 2021, 12, 619442. [Google Scholar] [CrossRef]
- Aspinall, S.A.; Mackintosh, K.A.; Hill, D.M.; Cope, B.; McNarry, M.A. Evaluating the Effect of Kaftrio on Perspectives of Health and Wellbeing in Individuals with Cystic Fibrosis. Int. J. Environ. Res. Public Health 2022, 19, 6114. [Google Scholar] [CrossRef]
- Welsner, M.; Schulte, T.; Dietz-Terjung, S.; Weinreich, G.; Stehling, F.; Taube, C.; Strassburg, S.; Schoebel, C.; Sutharsan, S. Effect of Triple Combination CFTR Modulator Therapy on Sleep in Adult Patients with Cystic Fibrosis. Respiration 2022, 101, 766–774. [Google Scholar] [CrossRef]
- Sergeev, V.; Chou, F.Y.; Lam, G.Y.; Hamilton, C.M.; Wilcox, P.G.; Quon, B.S. The Extrapulmonary Effects of Cystic Fibrosis Transmembrane Conductance Regulator Modulators in Cystic Fibrosis. Ann. Am. Thorac. Soc. 2020, 17, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Tindell, W.; Su, A.; Oros, S.M.; Rayapati, A.O.; Rakesh, G. Trikafta and Psychopathology in Cystic Fibrosis: A Case Report. Psychosomatics 2020, 61, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.; Young, D.C.; Safirstein, J.; Bourque, B.; Antell, M.H.; Diloreto, S.; Rotolo, S.M. Mental status changes during elexacaftor/tezacaftor/ivacaftor therapy. J. Cyst. Fibros. 2022, 21, 339–343. [Google Scholar] [CrossRef]
- Zhang, L.; Albon, D.; Jones, M.; Bruschwein, H. Impact of elexacaftor/tezacaftor/ivacaftor on depression and anxiety in cystic fibrosis. Ther. Adv. Respir. Dis. 2022, 16, 175346662211442. [Google Scholar] [CrossRef] [PubMed]
- Arslan, M.; Chalmers, S.; Rentfrow, K.; Olson, J.M.; Dean, V.; Wylam, M.E.; Demirel, N. Suicide attempts in adolescents with cystic fibrosis on Elexacaftor/Tezacaftor/Ivacaftor therapy. J. Cyst. Fibros. 2023. [Google Scholar] [CrossRef] [PubMed]
- Quittner, A.L.; Abbott, J.; Georgiopoulos, A.M.; Goldbeck, L.; Smith, B.; Hempstead, S.E.; Marshall, B.; Sabadosa, K.A.; Elborn, S.; International Committee on Mental Health; et al. International Committee on Mental Health in Cystic Fibrosis: Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus statements for screening and treating depression and anxiety. Thorax 2016, 71, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Gramegna, A.; De Petro, C.; Leonardi, G.; Contarini, M.; Amati, F.; Meazza, R.; Carugo, S.; Blasi, F. Onset of systemic arterial hypertension after initiation of elexacaftor/tezacaftor/ivacaftor in adults with cystic fibrosis: A case series. J. Cyst. Fibros. 2022, 21, 885–887. [Google Scholar] [CrossRef]
- Guo, J.; Wang, J.; Zhang, J.; Fortunak, J.; Hill, A. Current prices versus minimum costs of production for CFTR modulators. J. Cyst. Fibros. 2022, 21, 866–872. [Google Scholar] [CrossRef]
- McGarry, M.E.; Gibb, E.R.; Laguna, T.A.; O’Sullivan, B.P.; Sawicki, G.S.; Zobell, J.T. How many billions is enough? Prioritizing profits over patients with cystic fibrosis. Pediatr. Pulmonol. 2023. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Study/Phase/Follow-Up | Study Population | Results (Primary Endpoints) |
|---|---|---|
| Ramsey et al., 2011 [13] STRIVE—Phase 3 48 weeks | 161 PwCF carrying G551D-CFTR on at least one allele and ≥12 years old Subgroups: 83 with IVA (150 mg every 12 h), and 78 with placebo | Absolute change in ppFEV1 (week 24): +10.6 percentage points |
| Davies et al., 2013 [24] ENVISION—Phase 3 48 weeks | 52 PwCF carrying G551D-CFTR on at least one allele and 6–11 years old Subgroups: 26 with IVA (150 mg every 12 h), and 26 with placebo | Absolute change in ppFEV1 (week 24): +12.5 percentage points |
| Davies et al., 2016 [25] KIWI—Phase 3 24 weeks | 34 PwCF carrying at least one CFTR gating mutation and 2–5 years old Subgroups: 10 with IVA (50 mg every 12 h), and 24 with IVA (75 mg every 12 h) | Pharmacokinetics: Body weight was the most important predictor Safety: two children receiving IVA (50 mg every 12 h) experienced severe adverse effects |
| Wainwright et al., 2015 [14] TRAFFIC/TRANSPORT—Phase 3 24 weeks | 1108 PwCF homozygous for F508del-CFTR and ≥12 years old Subgroups: 368 with LUMA (600 mg/day) + IVA (250 mg every 12 h), 369 with LUMA (400 mg every 12 h) + IVA (250 mg every 12 h), 371 with placebo | Absolute change in ppFEV1 (week 24): +3.3 percentage points for LUMA (600 mg/day) + IVA (250 mg every 12 h), and +2.8 percentage points for LUMA (400 mg every 12 h) + IVA (250 mg every 12 h) |
| Ratjen et al., 2017 [26] Phase 3 24 weeks | 204 PwCF homozygous for F508del-CFTR and 6–11 years old Subgroups: 103 with LUMA (200 mg every 12 h) + IVA (250 mg every 12 h), and 101 with placebo | Average absolute change in LCI2.5 (week 24): −1.09 units for LUMA + IVA vs. placebo |
| Donaldson et al., 2018 [27] Phase 2 25 days of treatment + 25 days of washout (post-treatment observation period) | 172 PwCF homozygous for F508del-CFTR and ≥18 years old Subgroups: 33 with TEZA (10 to 150 mg/day), 106 with TEZA (10 to 150 mg/day) + IVA (150 mg every 12 h), and 33 with placebo 18 PwCF heterozygous for F508del-CFTR and G551D-CFTR and ≥12 years old Subgroups: 14 with TEZA (100 mg/day) + IVA (150 mg every 12 h), and 4 with placebo (IVA monotherapy) (150 mg every 12 h) | Safety (day 56): the majority (81.4%) of adverse effects were mild to moderate Change in SCC (day 28): −6.04 mmol/L after treatment vs. placebo (homozygous for F508del-CFTR), −7.02 mmol/L after TEZA + IVA vs. IVA monotherapy (heterozygous for F508del-CFTR and G551D-CFTR) |
| Taylor-Cousar et al., 2017 [15] EVOLVE—Phase 3 24 weeks | 475 PwCF homozygous for F508del-CFTR and ≥12 years old Subgroups: 235 with TEZA (100 mg/day) + IVA (150 mg every 12 h), and 240 with placebo | Absolute change in ppFEV1 (week 24): +4.0 percentage points |
| Rowe et al., 2017 [16] EXPAND—Phase 3 8 weeks of treatment +8 weeks of washout +8 weeks of treatment | 481 PwCF heterozygous for F508del-CFTR and a residual-function CFTR mutation and ≥12 years old Subgroups: 162 with TEZA (100 mg/day) + IVA (150 mg every 12 h), 157 with IVA (150 mg every 12 h), and 162 with placebo | Absolute change in ppFEV1 (average of weeks 4 and 8): +6.8 percentage points for TEZA + IVA, +4.7 percentage points for IVA monotherapy |
| Study/Phase/Follow-Up | Study Population | Results (Primary Endpoints) |
|---|---|---|
| Davies et al., 2018 [33] Phase 2 4 weeks | 63 PwCF heterozygous for F508del-CFTR and an MF CFTR mutation and ≥18 years old Subgroups: 53 with VX-659 (80, 240, or 400 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 10 with placebo 29 PwCF homozygous for F508del-CFTR and ≥18 years old Subgroups: 18 with VX-659 (400 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 11 with placebo + TEZA (100 mg/day) + IVA (150 mg every 12 h) corresponding to the active control group | Safety and side-effects: Most adverse effects were mild or moderate Absolute change in ppFEV1 (day 29, heterozygous for F508del-CFTR and an MF CFTR mutation): +10.2 percentage points for VX-659 (80 mg/day), +12.0 percentage points for VX-659 (240 mg/day); +13.3 percentage points for VX-659 (400 mg/day) Absolute change in ppFEV1 (day 29, homozygous for F508del-CFTR): +9.7 percentage points |
| Keating et al., 2018 [34] Phase 2 4 weeks | 65 PwCF heterozygous for F508del-CFTR and an MF CFTR mutation and ≥18 years old Subgroups: 53 with ELEXA (50, 100, or 200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 12 with triple placebo 28 PwCF homozygous for F508del-CFTR and ≥18 years old Subgroups: 21 with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 7 with TEZA (100 mg/day) + IVA (150 mg every 12 h) corresponding to the active control group | Absolute change in ppFEV1 (week 4, heterozygous for F508del-CFTR and an MF CFTR mutation): +11.1 percentage points for ELEXA (50 mg/day), +7.9 percentage points for ELEXA (100 mg/day), +13.8 percentage points for ELEXA (200 mg/day) Absolute change in ppFEV1 (week 4, homozygous for F508del-CFTR): +11.0 percentage points for ELEXA (200 mg/day) |
| Heijerman et al., 2019 [17] Phase 3 4 weeks | 107 PwCF homozygous for F508del-CFTR and ≥12 years old Subgroups: 55 with ELEXA (200 mg/day) + TEZA (200 mg/day) + IVA (150 mg every 12 h), and 52 with TEZA (100 mg/day) + IVA (150 mg every 12 h) corresponding to the active control group | Absolute change in ppFEV1 (week 4): +10.0 percentage points |
| Middleton et al., 2019 [18] Phase 3 24 weeks | 107 PwCF heterozygous for F508del-CFTR and an MF CFTR mutation and ≥12 years old Subgroups: 200 with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 203 with placebo | Absolute change in ppFEV1 (week 4): +13.8 percentage points |
| Zemanick et al., 2021 [35] Phase 3 24 weeks | 29 PwCF homozygous for F508del-CFTR and 6–11 years old Subgroups: 16 (<30 kg) with ELEXA (100 mg/day) + TEZA (50 mg/day) + IVA (75 mg every 12 h), 13 (≥30 kg) with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h) 37 PwCF heterozygous for F508del-CFTR and an MF CFTR mutation and 6–11 years old Subgroups: 20 (<30 kg) with ELEXA (100 mg/day) + TEZA (50 mg/day) + IVA (75 mg every 12 h), 17 (≥30 kg) with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h) | Safety and tolerability: the majority (96.9%) of adverse effects were mild or moderate |
| Barry et al., 2021 [36] Phase 3 4-week run-in period 8 weeks | 95 PwCF heterozygous for F508del-CFTR and a gating CFTR mutation and ≥12 years old Subgroups: 50 with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 45 with IVA (150 mg every 12 h) corresponding to the active control group 163 PwCF heterozygous for F508del-CFTR and a residual-function CFTR mutation and ≥12 years old Subgroups: 82 with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 81 with TEZA (100 mg/day) + IVA (150 mg every 12 h) corresponding to the active control group | Absolute change in ppFEV1 (week 8) from baseline (heterozygous for F508del-CFTR and a gating CFTR mutation): +5.8 percentage points Absolute change in ppFEV1 (week 8) from baseline (heterozygous for F508del-CFTR and an MF CFTR mutation): +2.5 percentage points |
| Griese et al., 2021 [37] Phase 3 24 weeks or longer | 107 PwCF homozygous for F508del-CFTR and ≥12 years old Subgroups: ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h) 399 PwCF heterozygous for F508del-CFTR and an MF CFTR mutation and ≥12 years old Subgroups: ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h) | Safety: the majority of adverse effects were mild or moderate |
| Sutharsan et al., 2022 [38] Phase 3b 4-week run-in period 24 weeks | 175 PwCF homozygous for F508del-CFTR, ≥12 years old, and with a baseline ppFEV1 = 40–90% Subgroups: 87 with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), 88 with TEZA (100 mg/day) + IVA (150 mg every 12 h) corresponding to the active control group | Absolute change in CFQ-R respiratory domain score from baseline (week 24): +17.1 (14.1 to 20.1) points for the treatment group, and 15.9 (11.7 to 20.1) points between treatment and control group |
| Mall et al., 2022 [39] Phase 3b 24 weeks | 121 PwCF heterozygous for F508del-CFTR and an MF CFTR mutation, 6–11 years old, and LCI2.5 ≥ 7.5 Subgroups: 39 (<30 kg) with ELEXA (100 mg/day) + TEZA (50 mg/day) + IVA (75 mg every 12 h), 21 (≥30 kg) with ELEXA (200 mg/day) + TEZA (100 mg/day) + IVA (150 mg every 12 h), and 61 with placebo | Absolute change in LCI2.5 (week 24): −2.29 (−2.60 to −1.97) units for ETI, and −2.26 (−2.71 to −1.81) units between treatment and placebo group |
| Nichols et al., 2022 [40] PROMISE—Post-approval study 6 months | 487 PwCF aged ≥12 years with at least one allele of F508del-CFTR starting ETI therapy for the first time Subgroups (modulator use at baseline): 238 PwCF naïve to modulators, 34 PwCF using IVA, 215 PwCF using TEZA + IVA or LUMA + IVA | Absolute change in ppFEV1 from baseline (naïve to modulators): +10.8 percentage points Absolute change in ppFEV1 from baseline (IVA): +6.1 percentage points Absolute change in ppFEV1 from baseline (two-drug combination): +9.2 percentage points |
| Mayer-Hamblett et al., 2022 [41] SIMPLIFY 6 weeks | 594 PwCF aged 12–17 years with ppFEV1 of 70% or more or those aged ≥18 years with ppFEV1 of 60% or more taking ETI and either or both mucoactive therapies (hypertonic saline or dornase alfa) for at least 90 days before screening Subgroups: 370 PwCF were randomly assigned to the hypertonic saline trial, and 477 were assigned to the dornase alfa trial. A subset of 253 participants participated in both trials | Absolute change in ppFEV1 in the hypertonic saline trial: −0.19% (−0.85 to 0.48) in the discontinuation group (n = 133) vs. 0.14% (−0.51 to 0.78) in the continuation group (n = 193) Absolute change in ppFEV1 in the dornase alfa trial: 0.18% (−0.38 to 0.74) in the discontinuation group (n = 199) vs. −0.16% (−0.73 to 0.41) in the continuation group (n = 193) |
| Study | Study Population | Main Results | Conclusion |
|---|---|---|---|
| Burgel et al., 2021 [43] Prospective, observational study | 236 PwCF aged ≥12 years, with advanced lung disease and at least one copy of F508del-CFTR The study was performed between December 2019 and August 2020 | Safety: no PwCF required discontinuation of ETI therapy, and the most prevalent adverse events were mild Effectiveness: Absolute increase in ppFEV1: +15.1; mean increase in weight: +4.2 kg; a significant decrease in the need for long-term oxygen and enteral tube feeding; lung transplantation: 11 of 15 patients that were waitlisted were taken off the candidate list | ETI therapy resulted in a rapid improvement in lung function even in PwCF with advanced lung disease |
| Carnovale et al., 2022 [44] | 26 PwCF (F508del/F508del genotype) with advanced lung disease The study was performed between October 2019 and July 2020 | Safety: No adverse events leading to discontinuation of ETI therapy were reported Effectiveness: The mean absolute improvement in ppFEV1 was 14.48 after 48 weeks. The mean absolute increase in BMI was 2.08. CFQ-R respiratory domain score improved by 32.6 points from baseline after 48 weeks | ETI therapy was safe and effective in PwCF homozygous for F508del-CFTR and advanced lung disease |
| O’Shea et al., 2020 [45] Observational study | 14 PwCF (F508del/F508del or F508del/MF genotypes) with advanced lung disease The study was performed between December 2019 and July 2020 | Safety: ETI therapy was well-tolerated Effectiveness: Improvement in ppFEV1, SCC, BMI, and infective exacerbations requiring hospitalization | ETI therapy improved multiple outcome measures for PwCF with advanced lung disease |
| Terlizzi et al., 2021 [46] Retrospective, observational study | Three adults with CF (F508del/unknown genotype) with advanced lung disease The study was performed between October 2019 and April 2021 | Safety: No adverse events led to the discontinuation of ETI therapy Effectiveness: SCC decreased progressively in all patients; relevant improvements of ppFEV1; all individuals discontinued oxygen therapy after 4–12 weeks of ETI therapy as well as reduced the need for antibiotic therapy | ETI was a feasible therapy for PwCF with F508del/unknown genotype |
| Carnovale et al., 2021 [47] Retrospective cohort study | 47 PwCF (F508del/MF genotypes) with ppFEV1 < 40% or who were considered for lung transplantation The study was performed between October 2019 and May 2020 | Safety: No treatment-related adverse events leading to discontinuation were reported Effectiveness: Improvements in ppFEV1 and 6-min walking test (m) distance were reported, and a significant reduction in the rate of pulmonary exacerbations and the need for oral and intravenous antibiotic therapy | ETI was a safe and effective therapy for PwCF with F508del/MF genotype and advanced pulmonary disease |
| Salvatore et al., 2021 [48] Case report | A 50-year-old woman with CF (F508del/G1244E genotype) with advanced lung disease | Safety: No ETI therapy-related adverse events were reported Effectiveness: Improvements in ppFEV1, SCC, BMI, and CFQ-R respiratory domain scores were reported | ETI therapy may have therapeutic potential for PwCF with advanced lung disease, particularly for whom lung transplantation is contraindicated |
| Gur et al., 2021 [49] Case report | A 24-year-old woman with CF (F508del/R1066C genotype) with severe progressive Mycobacterium abscessus lung disease and nutritional failure | Effectiveness: After 1 year of treatment, ppFEV1 improved from 26% to 45%, BMI improved from 16.4 to 21, and the sputum cultures were negative since the start of ETI | ETI treatment may open a new horizon in continuous efforts to overcome persistent infections in PwCF |
| Migliorisi et al., 2022 [50] Case-Control study | 26 PwCF with at least one F508del mutation and ppFEV1 ≤ 40% | Effectiveness: After 1 year of treatment, respiratory, pancreatic, and sweat function, BMI, and quality of life improved in the case group patients; the rate of airway infections and pulmonary exacerbations were reduced; sputum samples collected progressively resulted in no detection of relevant pathogenic microorganisms | Long-term treatment with ETI can give rise to changes in pulmonary microbiota and may reduce the need for antibiotics |
| Macconi et al., 2022 [51] Prospective, observational study | 3 PwCF with genotypes F508del/N1303K, F508del/R553Q, and F508del L065P with advanced lung disease | Effectiveness: MRI performed 1 month before and 3 months after the start of ETI therapy showed a significant reduction in mucus plugging and bronchial wall thickening | Chest MRI could be a useful tool to evaluate disease progression in PwCF |
| Stylemans et al., 2022 [52] Real-life follow-up study | 14 adults with CF (F508del/F508del and F508del/MF genotypes) The study was performed between December 2019 and November 2020 | Safety: ETI therapy was well-tolerated; one patient had to interrupt treatment due to liver injury Effectiveness: Marked improvements in peripheral lung function | Marked effects on ppFEV1 and pulmonary exacerbations could be obtained in real life under ETI therapy, even in severely obstructive PwCF |
| Kos et al., 2022 [53] Longitudinal, real-life, observational study | 19 PwCF with at least one F508del-CFTR allele and advanced lung disease | Safety: ETI therapy was well-tolerated, and only mild adverse effects were reported Effectiveness: BMI and mean absolute FEV1 improved; there was a marked reduction in the frequency of pulmonary exacerbations | Clinical benefits of ETI therapy were maintained for 12 months in PwCF with advanced lung disease |
| Salvatore et al., 2022 [54] Observational study | Nine children with CF aged 6–11 years with at least one copy of F508del-CFTR and advanced lung disease | Safety: ETI therapy was safe and there was no need for treatment discontinuation Effectiveness: After 24 weeks, the mean absolute changes in ppFEV1, BMI, and SCC were +22.4 points, +0.60 kg/m2, and −79.2 mmol/L, respectively; the CFQ-R respiratory domain score increased from the median baseline score of 25 to 100 after 24 weeks; treatment led to a reduction of the rate of antibiotic treatment to 80% over the 24 weeks of the study | ETI therapy improved lung function, nutrition status, and quality of life of children with CF aged 6–11 years with advanced lung disease and at least one F508del-CFTR allele |
| Dhote et al., 2023 [55] Prospective study | 79 adults with CF carrying at least one F508del-CFTR allele and advanced lung disease The study was performed between December 2019 and July 2021 | Effectiveness: 12 months of ETI therapy was associated with a significant decrease in circulating neutrophils, monocytes, and platelets, but not lymphocytes, to values within the laboratory reference range | ETI therapy may normalize neutrophil counts in PwCF with advanced lung disease |
| McCoy et al., 2023 [56] Retrospective study | 18 PwCF with at least one F508del-CFTR allele and advanced lung disease The study was performed between July 2019 and September 2022 | Safety: ETI was well-tolerated, without the need for treatment discontinuation Effectiveness: After 24 months, SCC decreased from 84–140 mmol/L to 15–63 mmol/L; ppFEV1 improved from a median of 27.5 to 45.0; and BMI significantly increased from 19.1 kg/m2 to 22.8 kg/m2 | ETI was safe with positive changes in nutrition and respiratory symptoms, CFQ-R, and lung function, and a reduction in therapy burden was maintained for 2 years after the start of ETI therapy in PwCF with advanced lung disease |
| Study | Study Population | Main Results | Conclusion |
|---|---|---|---|
| Wucherpfenning et al., 2022 [64] Retrospective, observational study | 43 adults with CF and at least one F508del-CFTR allele The study was performed between June 2020 and August 2021 | Effectiveness: MRI revealed improvements in chest MRI morphology score and chronic rhinosinusitis–MRI scores after the start of ETI therapy | MRI studies indicated reversibility of structural lung and paranasal sinus abnormalities in PwCF who received ETI therapy. These improvements correlated well with proportionally improved spirometry |
| Graeber et al., 2022 [65] Prospective, observational study | 91 PwCF aged ≥12 years; 46 homozygous for F508del-CFTR and 45 heterozygous (F508del/MF genotype) | Effectiveness: In individuals homozygous for F508del-CFTR, LCI improved by 15.3%, and MRI global improved by 29.3% after the start of ETI therapy In individuals heterozygous (F508del/MF), ETI therapy led to improvements of LCI of 23.4% from baseline and MRI global of 25.6% from baseline | ETI therapy improved lung ventilation and abnormalities in lung morphology in PwCF with at least one copy of F508del-CFTR in a real-world setting |
| Streibel et al., 2023 [66] Retrospective, observational study | 24 children with CF | Effectiveness: Evaluation of structural and functional MRI parameters before and two weeks after the start of ETI therapy showed a significant improvement in lung function | Functional and structural MRI are promising tools to complement information obtained using lung function measures (spirometry and LCI) |
| Stapleton et al., 2022 [67] | 34 PwCF with ≥12 years old (28 participants completed both study visits) The study was performed between November 2019 and March 2020 | Effectiveness: Sinonasal symptoms improved rapidly by day 7 of ETI therapy, and the improvement was persistent for up to 189 days | ETI therapy improved the sinonasal quality of life scores, nasal endoscopy scores, and sinonasal CT scans and led to the regression of nasal polyps |
| Lee et al., 2022 [68] A score-matched historical cohort study | Total of 468 PwCF treated with ETI (n = 367 F508del/MF genotype; n = 101 homozygous for F508del) Total of 1714 PwCF untreated (control group) (n = 1242 F508del/MF genotype; n = 472 homozygous for F508del) | Effectiveness: Participants treated with ETI had, on average, no loss of pulmonary function over 2 years in comparison with a decline in ppFEV1 of 1.92% annually in untreated controls | ETI therapy showed sustained lung function for an extended period |
| Bec et al., 2022 [69] Retrospective, observational study | 12 adults with CF homozygous or heterozygous for F508del-CFTR The study was performed between April 2018 and November 2021 | Effectiveness: After one year, ETI therapy showed improvement in lung damage on chest CT and a decrease in the visual Brody-II score of 21% due to lower mucus plugging and peribronchial thickening | The study supports the start of ETI therapy early in life to avoid lung sequelae |
| DiMango et al., 2021 [70] Observational study | 43 adults with CF with at least one F508del-CFTR allele; 23 participants were taking a CFTR modulator at the time of participation | Effectiveness: ETI therapy significantly improved both SNOT and CFQ-R scores at 3 months | PwCF who had been taking other CFTR modulators before the start of ETI therapy had more pronounced benefits (higher SNOT score at baseline) compared to those who were not taking any CFTR modulators |
| Causer et al., 2022 [71] Case series | Three adolescents with CF homozygous for F508del-CFTR | Effectiveness: ETI therapy improved exercise capacity with a VO2 peak observed in all three cases (+17.65%, +52.4%, and +32.9%, respectively) | ETI therapy may improve exercise capacity in PwCF |
| Martin et al., 2021 [72] Mixed method study through an online questionnaire | 101 PwCF aged 12 years or older with advanced lung disease receiving ETI The online questionnaire was available from July to August 2020 | Effectiveness: Participants reported a rapid impact on sleep quality, general well-being, respiratory symptoms, and a reduction in treatment burden | After the start of ETI therapy, PwCF reported rapid and positive physical, psychological, and social effects with an improvement in quality of life |
| Bacon et al., 2022 [73] Observational study | 34 PwCF aged ≥12 years (28 participants completed the study) The study was performed between November 2019 to March 2020 | Effectiveness: There was no significant difference in the olfactory test (UPSIT) after the start of ETI therapy | Larger studies and longer follow-up periods are needed given that the small cohort did not show improvement in UPSIT score |
| Beswick et al., 2022 [74] Prospective, observational study | 25 adults with CF (F508del/F508del and F508del/MF genotypes) and with chronic rhinosinusitis The study was performed between August 2019 to October 2020 | Effectiveness: After 6 months of follow-up, sinus CT opacification improved by a mean of 22.9% with ETI therapy | ETI therapy was associated with clinical improvements in sinus disease; however, it did not fully resolve sinus disease after 6 months of treatment |
| Castellanos et al., 2022 [75] Observational study | 23 children with CF | Effectiveness: ETI therapy improved sinonasal symptoms and CFQ-R score | ETI therapy was associated with an improvement in sinonasal function and quality of life in children with CF |
| Sheikh et al., 2022 [76] Observational study | 48 adults with CF carrying at least one F508del-CFTR allele and 20 healthy adult controls | Effectiveness: ETI therapy improved clinical outcomes (ppFEV1 and BMI), reduced bacterial infection in respiratory cultures, and significantly reduced circulating neutrophils and levels of pro-inflammatory cytokines (IL-6, IL-8, and IL-17A) | ETI therapy may reduce neutrophilic inflammation and neutrophil-mediated lung disease |
| Bode et al., 2023 [77] A cross-sectional, retrospective study | 43 PwCF (six children) who started ETI therapy and 20 controls (PwCF naïve to modulator therapy) | Effectiveness: Overall, a reduction in the SNOT-22 score and objective clinical improvement were observed in the PwCF receiving ETI therapy | ETI may promote clinical benefits on upper airway symptoms in PwCF |
| Giallongo et al., 2023 [78] Retrospective study | Nine PwCF aged ≥12 years and ppFEV1 < 40% | Effectiveness: After 12 months of ETI therapy, the absolute change from baseline in nocturnal cardiorespiratory polygraphy parameters showed a significant improvement in nocturnal oxygenation, time spent with SpO2 ≤ 90%, and respiratory rate | ETI therapy resulted in improved nocturnal SpO2 at month 3 that was sustained up to 12 months after the start of ETI |
| Beck et al., 2023 [79] Retrospective study | 124 PwCF aged ≥12 years The study was performed between October 2019 and October 2021 | Effectiveness: Culture positivity for Pseudomonas aeruginosa, methicillin-resistant, and methicillin-susceptible Staphylococcus aureus was 54%, 33%, and 31%, respectively, before the start of ETI therapy; the prevalence of detection decreased to 30%, 32%, and 24%, respectively | ETI therapy results in a significant reduction in the detection of common bacterial pathogens in CF respiratory cultures, after 12 months of treatment |
| Study | Study Population | Main Results | Conclusion |
|---|---|---|---|
| Bermingham et al., 2021 [87] Retrospective study | 50 adults with CF with advanced lung disease | Safety: ETI therapy was well-tolerated, and no participant required discontinuation Effectiveness: 64% of patients experienced an improvement of ≥5% in absolute ppFEV1 | Clinical improvements after ETI therapy resulted in adjustments to lung transplantation planning using CFF guidelines |
| Martin et al., 2022 [88] Prospective observational study | 65 PwCF with advanced lung disease (lung transplant candidates at the time of starting ETI therapy) | Safety: Adverse events were mild and transient Effectiveness: Most participants experienced rapid and clinically meaningful improvements in lung function, pulmonary exacerbations, gas exchange, and nutritional status | ETI therapy improved multiple outcome measures for PwCF with advanced lung disease |
| Benniger et al., 2021 [89] Retrospective observational study | 9 PwCF, homozygous for F508del, who underwent bilateral lung transplantation | Effectiveness: BMI, sinus, and GI symptoms improved after the start of ETI therapy | ETI therapy did not cause graft function decline or significant impact on immunosuppressive drug regimens or doses, in post-transplant recipients |
| Doligalski et al., 2022 [90] Observational study | 13 PwCF with potential benefits for lung transplant recipients and at least one copy of F508del-CFTR The study was performed between November 2019 and July 2021 | Safety: Five participants discontinued therapy due to declining pulmonary function, mood disturbances, or lack of benefit; four participants reported adverse events; and three interrupted treatment temporarily Effectiveness: Six participants reported improvement in sinus symptoms and four reported improvement in gastrointestinal symptoms; the tacrolimus dose declined by 50% after the start of ETI therapy | ETI therapy was poorly tolerated and showed modest extra-pulmonary benefit |
| Ramos et al., 2022 [91] Observational study | 94 PwCF who started ETI therapy after a lung transplant | Safety: ≥40% of participants stopped ETI therapy due to adverse events or lack of perceived benefit Effectiveness: Frequency of antibiotic prescriptions decreased, hemoglobin A1c improved, BMI did not show a significant difference | The risks and benefits of ETI therapy after a lung transplant should be further determined in a greater CF population with a lung transplant |
| McKinzie et al., 2022 [92] Case series | Two PwCF who had a liver transplant: Subject 1— F508del/Nt1652delCTT; Subject 2— F508del/F508del | Safety: Neither individual required dose adjustment in their baseline immunosuppression regimens: Subject 1—significant elevations in liver function tests were drug-associated; after dose correction, liver profiles remain acceptable Subject 2—Mild acute cellular rejection episode that was successfully treated with corticosteroids Effectiveness: Improvement in lung function and nutritional status | There may be a role for ETI therapy in liver transplant recipients with close monitoring for adverse effects |
| Ragan et al., 2022 [93] Retrospective case series | 10 PwCF who had liver transplant: Subject 1—F508del/F508del Subject 2—F508del/W1282X Subject 3—F508del/F508del Subject 4—F508del/F508del Subject 5—F508del/F508del Subject 6—F508del/F508del-Y301C Subject 7—F508del/W1282X Subject 8—F508del/F508del Subject 9—F508del/Q493X Subject 10—F508del/1216+1G->A | Safety and Effectiveness of ETI therapy: Subject 1—no significant adverse events were observed; quality of life improved Subject 2—treatment was tolerated with no marked adverse events; quality of life improved from baseline Subject 3—individuals had abdominal pain with evidence of drug-induced liver disease; therapy was discontinued Subject 4—individuals had symptoms of Tacrolimus toxicity, namely severe gastrointestinal onset, and acute kidney injury; therapy was discontinued Subject 5—there was no evidence of drug-induced liver injury; quality of life improved Subject 6—there was no evidence of drug-induced adverse events; respiratory symptoms improved Subject 7—treatment was tolerated with no marked adverse events; ppFEV1 increased to 100% after 2 weeks Subject 8—treatment was tolerated after some drugs dose adjustments; improvement in symptoms and quality of life Subject 9—there was no evidence of drug-induced adverse events; respiratory symptoms and quality of life improved Subject 10—treatment was tolerated after the reduction of tacrolimus dose; respiratory symptoms and quality of life improved | PwCF who underwent to liver transplant may initiate ETI therapy with a close therapeutic drug monitoring of immunosuppression |
| Ørum et al., 2022 [94] | Four PwCF who had solid organ transplants: Two had a bilateral lung transplant and two had a liver transplant | Safety: ETI therapy was well-tolerated with no adverse events that led to discontinuation Effectiveness: pulmonary symptoms improved in all subjects but the increase in FEV1 was significantly larger in liver transplant recipients with native lungs | All participants experienced subjective pulmonary and extra-pulmonary improvements after the start of ETI therapy |
| Study | Study Population | Main Results | Conclusion |
|---|---|---|---|
| Schwarzenberg et al., 2022 [95] PROMISE Prospective observational study | 438 PwCF aged ≤ 12 years and at least one F508del-CFTR allele | Effectiveness: Evaluation of GI symptoms using validated questionnaires (PAGI-SYM, PAC-SYM, and PAC-QOL) showed improvement; the fecal marker of inflammation (calprotectin) decreased, while pancreatic insufficiency measured by fecal elastase did not improve | After 6 months of ETI therapy, changes in GI symptoms were clinically unimportant, emphasizing the need for continued attention to GI disease |
| Petersen et al., 2021 [96] Retrospective observational study | 134 adults with CF and at least one F508del-CFTR allele | Effectiveness: BMI substantially increased, and systolic and diastolic blood pressure also increased; in individuals with CFRD, total cholesterol, LDL-c, and HDL-c increased; in individuals without CFRD, random blood glucose, and hemoglobin A1c decreased | PwCF receiving ETI therapy should be monitored for evidence of overnutrition and related complications, such as hypertension and hypercholesterolemia |
| Scully et al., 2021 [97] Prospective observational study | 33 adults with CF and at least one F508del-CFTR allele | Effectiveness: The start of ETI therapy was associated with significant improvements in continuous glucose monitoring-derived measures of average glucose, hyperglycemia, and glycemic variability | ETI therapy in adults with CF was associated with improvement in continuous glucose monitoring-derived measures of hyperglycemia and glycemic variability with no effect on hypoglycemia |
| Wright et al., 2021 [98] Retrospective observational study | 76 PwCF with at least one F508del-CFTR allele | Effectiveness: ETI therapy increased vitamin D absorption as measured by serum 25-hydroxyvitamin D | The increase in serum 25-hydroxyvitamin D may reduce the dose of cholecalciferol and/or the need for multivitamin products |
| Shakir et al., 2022 [99] Prospective observational study | 32 PwCF with at least one F508del-CFTR allele and advanced lung disease | Effectiveness: Sinonasal symptoms, gastroesophageal reflux, and extraesophageal reflux improved substantially within three months after the start of ETI therapy | ETI therapy improved upper GI symptoms, lung function, BMI, and quality of life in PwCF with advanced lung disease |
| Safirstein et al., 2020 [100] Case series | Seven adults with CF with different CF genotypes who developed biliary colic after the start of ETI therapy | The approximate number of days on ETI therapy before biliary symptoms and pathology report findings post-cholecystectomy: Subject 1—27 days, chronic cholecystitis with cholelithiasis and serosal fibrous adhesions; Subject 2—1 day, acute cholecystitis with mucosal necrosis and cholelithiasis; Subject 3—3 days, chronic cholecystitis with cholelithiasis, extensive mucosal erosion, and wall fibrosis and serositis; Subject 4—1 day, chronic cholecystitis with cholelithiasis; Subject 5—one day before treatment, not performed; Subject 6—14 days, acute cholecystitis with cholelithiasis with obstruction of the neck; Subject 7—1 day, chronic cholecystitis with cholelithiasis. | Asymptomatic gallbladder disease may be exacerbated after the start of ETI therapy in adults with CF |
| Lowry et al., 2021 [101] Case report | One individual with CF (F508del/c4077-4080delTGTTinsAA) with no known history of liver disease | Safety: Transaminase values increased after 5 months of ETI therapy; liver enzyme values normalized after 6 months of ETI therapy discontinuation; other identified sources of injury were excluded Effectiveness: Over ETI therapy, ppFEV1 improved from a baseline of 35% predicted to 53% predicted | Attempts to prevent liver injury will be important for the long-term use of CFTR modulators |
| Korten et al., 2022 [102] Observational study | 16 adolescents with CF with at least one F508del-CFTR allele | Effectiveness: Glucose tolerance measured by oral glucose tolerance tests improved after the start of ETI therapy | ETI therapy shows beneficial effects on endocrine pancreatic function |
| Chan et al., 2022 [103] Prospective study | 20 PwCF with at least one F508del-CFTR allele | Effectiveness: BMI z-score, insulin secretion, and resistance increased; no significant differences were observed in glucose tolerance and β-cell function | One year after the start of ETI therapy, insulin secretion increased but insulin resistance also increased, and there was no significant change in β-cell function |
| Mainz et al., 2022 [104] | 107 PwCF with at least one F508del-CFTR allele | Effectiveness: Abdominal symptoms before and during ETI therapy were assessed using CFAbd-Score: the mean total CFAbd-Score decreased significantly by 29% during therapy | ETI therapy results in a significant reduction of abdominal symptoms |
| Steinack et al., 2023 [105] Retrospective, observational study | 33 PwCF carrying at least one F508del CFTR allele | Effectiveness: At least 3 months after the start of ETI therapy, 48.5% of PwCF improved their glucose tolerance, 39.4% remained unchanged and 12.1% deteriorated | ETI therapy likely improves glucose tolerance without increasing insulin secretion |
| Tewkesbury et al., 2023 [106] | 255 PwCF (30.6% had a diagnosis of CF-related liver disease) | Safety: Overall, ETI therapy led to a significant rise in AST, ALT, and bilirubin values at 3 months (median values remained in the normal range), which was sustained at 12 months with no further significant increase; no differences were observed between PwCF with or without liver disease | Pre-existing CF-related liver disease does not appear to correlate with alterations in liver tests and should not prevent the start of ETI therapy |
| Francalanci et al., 2023 [107] Cross-section, retrospective study | 318 PwCF (3 children aged <2 years, 135 children aged 2–18 years, and 180 adults) | Effectiveness: One year of ETI therapy increased BMI and the levels of all circulating fat-soluble vitamins more consistently compared with previous modulators | ETI therapy promotes a beneficial effect on nutritional status and circulating levels of fat-soluble vitamins |
| Schnell et al. 2023 [108] Prospective observational study | 20 PwCF (10 aged < 20 years) The study was performed between September 2020 and November 2021 | Safety: In PwCF < 20 years, ARFI SWV increased after 6 months of ETI therapy; bile acid profiles revealed an increase in glycine-conjugated derivatives; there was a positive correlation between ARFI SWV values and glycine-conjugated derivatives | ETI therapy may lead to an early increase in liver stiffness and altered bile metabolism in children with CF. Evaluation of bile acid profile and liver stiffness can be useful tools to monitor hepatic alterations |
| Wood et al., 2023 [109] Retrospective study | 83 adult PwCF | Safety: Transaminase elevations of >3 times the upper limit of normal were experienced by 11% of PwCF and an elevation of ≥25% above baseline was experienced by 75% of participants, during ETI therapy | Transaminase value elevation is a common feature in PwCF receiving ETI therapy. |
| Study | Study Population | Main Results | Conclusion |
|---|---|---|---|
| O’Connor et al., 2021 [111] Case series | 14 women with CF with childbearing potential The study was performed between October 2019 and May 2020 | 14 women got pregnant 8 weeks after the start of ETI therapy; 7 individuals were attempting conception and had a history of subfertility/infertility; 7 participants were not attempting to conceive, using methods of contraception | There is a need for further studies to clarify the reproductive implications of ETI therapy and women’s pre-conception counseling |
| Chamagne et al., 2022 [112] Case study | A 32-year-old woman with CF, homozygous for F508del-CFTR | During the first pregnancy, she was not using ETI therapy and she got pregnant after 7 years of different in vitro fertilization cycles; in the second pregnancy, she was receiving ETI therapy for 20 days and became spontaneously pregnant | This case report shows a real benefit for the mother without the detection of any adverse fetal outcomes |
| Collins et al., 2022 [113] Observational study | 3 women with CF, homozygous for F508del-CFTR, and their infants | Results demonstrated a relatively high concentration of ETI in cord blood and low levels in breast milk and infant blood | These findings suggest that ETI can cross the placenta leading to fetal exposure in women continuing ETI therapy during pregnancy |
| Fortner et al., 2021 [114] Case report | A woman with CF, heterozygous for F508del-CFTR | Six weeks after starting ETI therapy, she became pregnant; an infant with CF (F508del/F508del genotype) was born without any apparent organ dysfunction and the follow-up revealed high SCC | For this infant, exposure to ETI therapy in utero and through maternal breastmilk may have helped delay organ damage |
| Balmpouzis et al., 2022 [115] Case report | A 30-year-old woman with CF (F508del/Y1092X genotype) and advanced lung disease | Four months after the start of ETI therapy, an unplanned pregnancy occurred; no complications occurred up to the 31st week of pregnancy when she presented symptoms of threatened preterm labor and a rhinovirus infection; after childbirth, oxygen therapy was stopped; no further complications were reported over the one-year follow-up | ETI therapy has changed family planning and childbearing decisions for PwCF |
| Szentpetery et al., 2022 [116] | A woman (F508del carrier) pregnant with a fetus with CF (F508del/F508del genotype) | The mother started to receive ETI therapy to resolve bowel dilation in the fetus | Maternal ETI therapy likely resulted in the resolution of meconium ileus |
| Rotolo et al., 2020 [117] Case series | Seven men with CF and at least one F508del-CFTR allele | Individuals reported testicular pain or discomfort within the first two weeks of ETI therapy | Further study is required to determine the impact of this therapy on male fertility |
| Study | Study Population | Main Results | Conclusion |
|---|---|---|---|
| Stashower et al., 2021 [123] Case report | 24-year-old woman | One week after the start of ETI therapy, she presented with blanching, annular, pruritic plaques with dusky centers, and swelling in the hands, feet, and face | She cleared within one week of discontinuation of ETI therapy, suggesting the diagnosis of urticaria multiforme secondary to ETI |
| Goldberg et al., 2021 [124] | 12-year-old boy (F508del/R347P) | One week after the start of ETI therapy, he presented with a rash consisting of pink-to-erythematous, edematous, round papules coalescing into plaques | Based on the clinical and histologic features, a diagnosis of Urticaria multiforme-like drug eruption due to ETI therapy was made |
| Bhaskaran et al., 2022 [127] | 21-year-old woman (F508del/F508del) | Eight days after the start of ETI therapy, she developed a maculopapular rash that was violaceous and red over her shins, progressing to her thighs; the rash worsened in the next 36 h to progress centripetally affecting her back, anterior chest wall, anterior abdominal wall, neck, and arms | She needed supportive therapy for pruritus but never used steroids; the rash resolved spontaneously without stopping ETI therapy after four weeks, leaving no residual scars or discoloration |
| Diseroad et al., 2022 [128] | Subject 1—12-year-old girl (F508del/5T-M470V) Subject 2—14-year-old boy (Y569D/Y569D) | Subject 1—She developed a pruritic, maculopapular rash on her lower back, eyelids, ears, knees, hands, and head 30 min after the first dose of ETI that progressively worsened throughout the day Case 2—Eight days after starting ETI, he developed a pruritic, maculopapular rash on his hands that spread to his feet, palms, knee, and face | Both subjects completed ETI rechallenge with a practical titration schedule; the use of antihistamines with or without a short course of glucocorticosteroids might have aided in the resolution of the rash |
| Hudson et al., 2022 [129] | 18 PwCF ranging from 19 to 38 years old | 18 individuals reported either new or worsening acne symptoms within 8 months of ETI therapy, with 9 individuals reporting this manifestation within the first 3 months of ETI therapy | The underlying mechanism responsible for this adverse effect remains unknown |
| Pomi et al., 2022 [130] | Subject 1—a 19-year-old woman (F508del/F508del) Subject 2—a 24-year-old woman | Subject 1—Three days after starting ETI, she developed a widespread follicular papulopustular itchy rash localized to the abdomen and upper limbs progressively involving the buttocks and lower limbs; Malassezia folliculitis developed after ten days from re-administration of ETI Subject 2—An itchy papulopustular rash located on the back was observed 9 days after starting ETI, and the presence of Malassezia in the follicular pustule was confirmed | ETI therapy may induce changes in the skin microbiome, potentially able to favor colonization and proliferation of Malassezia sp; given the therapeutic benefit of ETI and its increasing use, oral desensitization may be considered as an option to avoid the full discontinuation of this therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacalhau, M.; Camargo, M.; Magalhães-Ghiotto, G.A.V.; Drumond, S.; Castelletti, C.H.M.; Lopes-Pacheco, M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals 2023, 16, 410. https://doi.org/10.3390/ph16030410
Bacalhau M, Camargo M, Magalhães-Ghiotto GAV, Drumond S, Castelletti CHM, Lopes-Pacheco M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals. 2023; 16(3):410. https://doi.org/10.3390/ph16030410
Chicago/Turabian StyleBacalhau, Mafalda, Mariana Camargo, Grace A. V. Magalhães-Ghiotto, Sybelle Drumond, Carlos Henrique M. Castelletti, and Miquéias Lopes-Pacheco. 2023. "Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis" Pharmaceuticals 16, no. 3: 410. https://doi.org/10.3390/ph16030410
APA StyleBacalhau, M., Camargo, M., Magalhães-Ghiotto, G. A. V., Drumond, S., Castelletti, C. H. M., & Lopes-Pacheco, M. (2023). Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals, 16(3), 410. https://doi.org/10.3390/ph16030410