
The STAT3-Regulated Autophagy Pathway in Glioblastoma



{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. GBM
1.2. Clinical Therapy for GBM
2. The STAT3 Signaling Pathway
2.1. The STAT3 Signaling Pathway in GBM
2.2. Therapeutic Targeting of STAT3
3. Autophagy
3.1. General Review of Autophagy
3.2. Overview of Macroautophagy
3.3. Chaperone-Mediated Autophagy (CMA)
3.4. Microautophagy
4. Autophagy in GBM
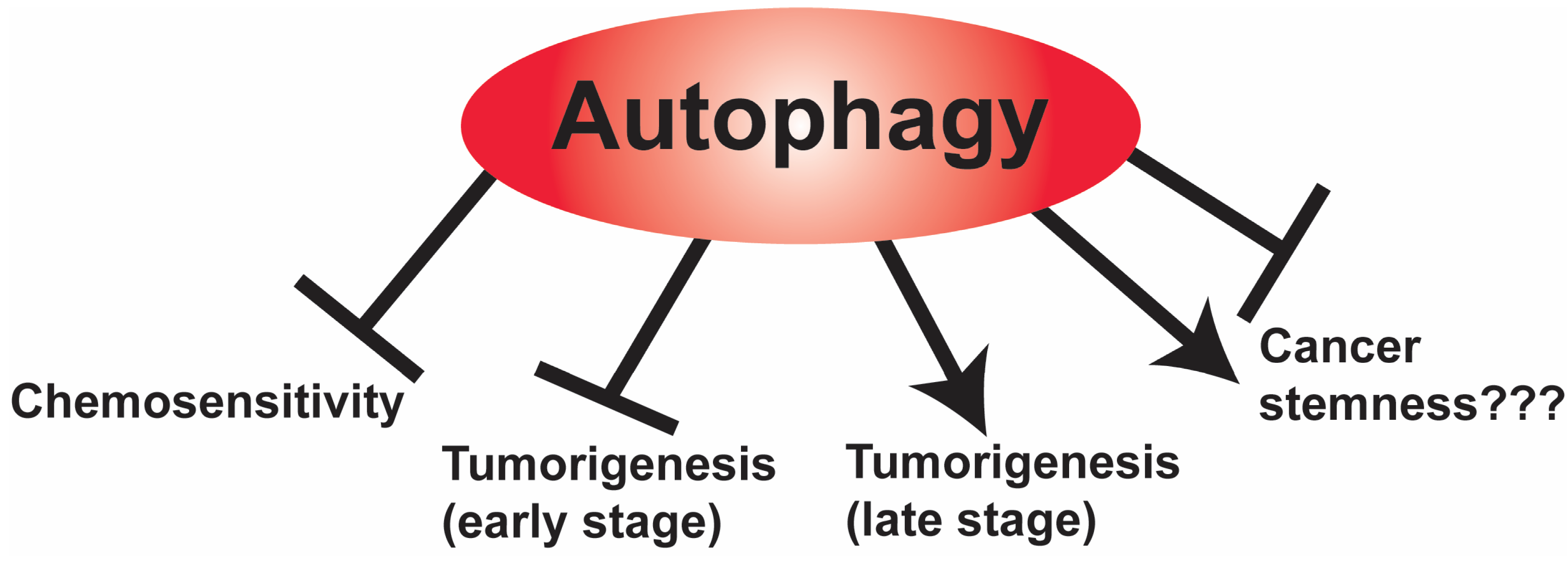
4.1. The Role of Autophagy
4.2. The Role of Autophagy in Cancer Stem Cells
4.3. Role of mTORC1-Dependent Autophagy Control in GSCs
4.4. Recent Studies of Autophagy in GSC Regulation
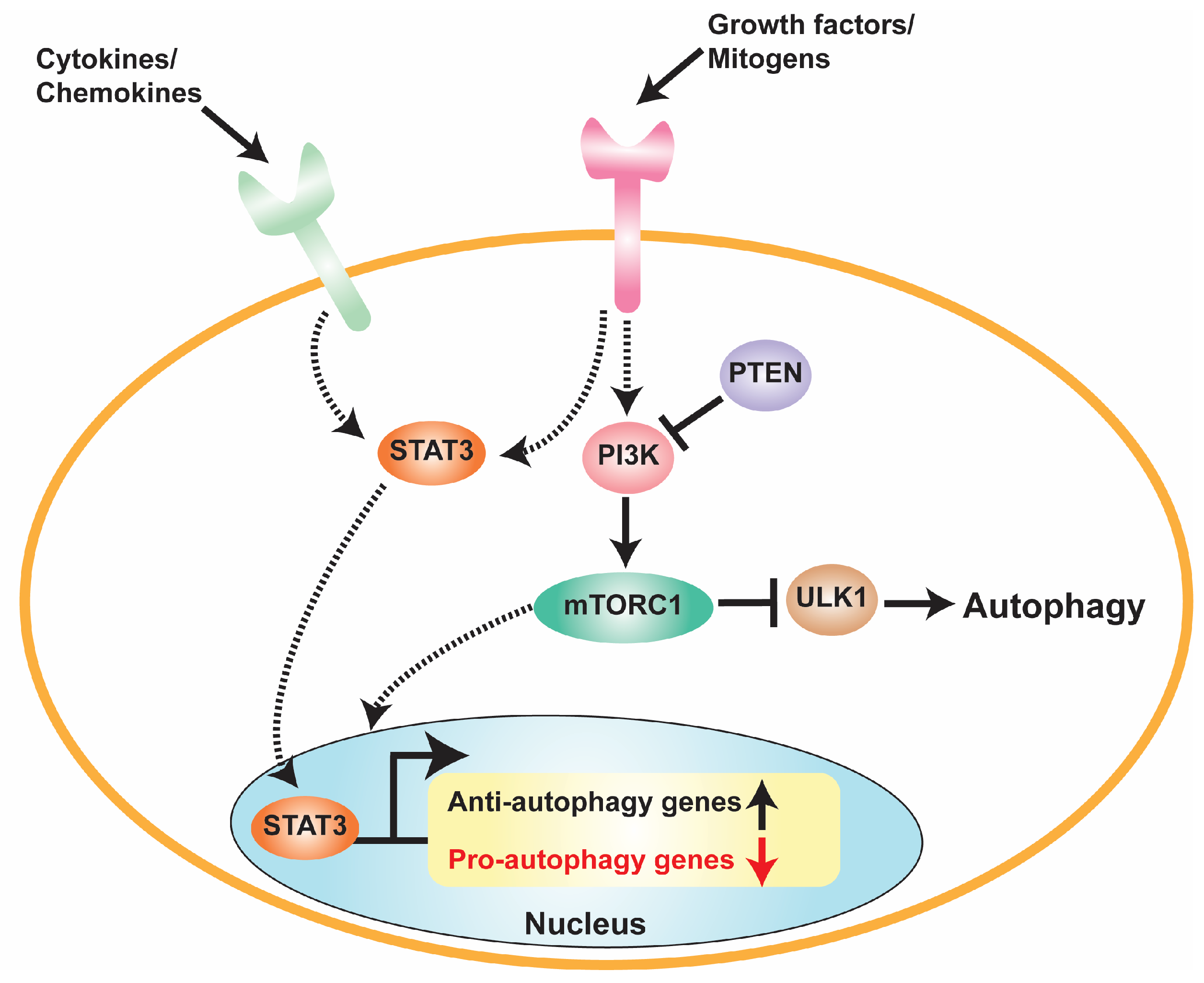
5. Crosstalk between STAT3 and Autophagy in GBM
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Surawicz, T.S.; Davis, F.; Freels, S.; Laws, E.R., Jr.; Menck, H.R. Brain tumor survival: Results from the National Cancer Data Base. J. Neuro-Oncol. 1998, 40, 151–160. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Wong, E.T.; Hess, K.R.; Gleason, M.J.; Jaeckle, K.A.; Kyritsis, A.P.; Prados, M.D.; Levin, V.A.; Yung, W.K. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J. Clin. Oncol. 1999, 17, 2572–2578. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumor Pathol. 2011, 28, 177–183. [Google Scholar] [CrossRef]
- Appin, C.L.; Gao, J.; Chisolm, C.; Torian, M.; Alexis, D.; Vincentelli, C.; Schniederjan, M.J.; Hadjipanayis, C.; Olson, J.J.; Hunter, S.; et al. Glioblastoma with oligodendroglioma component (GBM-O): Molecular genetic and clinical characteristics. Brain Pathol. 2013, 23, 454–461. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Zong, H.; Verhaak, R.G.; Canoll, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef]
- Ganguly, D.; Fan, M.; Yang, C.H.; Zbytek, B.; Finkelstein, D.; Roussel, M.F.; Pfeffer, L.M. The critical role that STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma. Oncotarget 2018, 9, 22095–22112. [Google Scholar] [CrossRef]
- Lauko, A.; Lo, A.; Ahluwalia, M.S.; Lathia, J.D. Cancer cell heterogeneity & plasticity in glioblastoma and brain tumors. Semin. Cancer Biol. 2022, 82, 162–175. [Google Scholar] [CrossRef]
- Ganguly, D.; Cai, C.; Sims, M.M.; Yang, C.H.; Thomas, M.; Cheng, J.; Saad, A.G.; Pfeffer, L.M. APELA Expression in Glioma, and Its Association with Patient Survival and Tumor Grade. Pharmaceuticals 2019, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef]
- Saikali, S.; Avril, T.; Collet, B.; Hamlat, A.; Bansard, J.Y.; Drenou, B.; Guegan, Y.; Quillien, V. Expression of nine tumour antigens in a series of human glioblastoma multiforme: Interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J. Neuro-Oncol. 2007, 81, 139–148. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Pazdur, R. Food and Drug Administration Drug approval summary: Temozolomide plus radiation therapy for the treatment of newly diagnosed glioblastoma multiforme. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6767–6771. [Google Scholar] [CrossRef]
- Ronning, P.A.; Helseth, E.; Meling, T.R.; Johannesen, T.B. A population-based study on the effect of temozolomide in the treatment of glioblastoma multiforme. Neuro-Oncology 2012, 14, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Dietrich, P.Y.; Ostermann Kraljevic, S.; Pica, A.; Maillard, I.; Maeder, P.; Meuli, R.; Janzer, R.; Pizzolato, G.; Miralbell, R.; et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J. Clin. Oncol. 2002, 20, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Hashimoto, N.; Matsutani, M. Management of glioblastoma. Expert Opin. Pharmacother. 2007, 8, 3133–3146. [Google Scholar] [CrossRef]
- Rajaratnam, V.; Islam, M.M.; Yang, M.; Slaby, R.; Ramirez, H.M.; Mirza, S.P. Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments. Cancers 2020, 12, 937. [Google Scholar] [CrossRef]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R.; Fan, M.; Du, Z.; Yang, C.H.; Pfeffer, L.M. Unphosphorylated STAT3 regulates the antiproliferative, antiviral, and gene-inducing actions of type I interferons. Biochem. Biophys. Res. Commun. 2017, 490, 739–745. [Google Scholar] [CrossRef]
- Du, Z.; Cai, C.; Sims, M.; Boop, F.A.; Davidoff, A.M.; Pfeffer, L.M. The effects of type I interferon on glioblastoma cancer stem cells. Biochem. Biophys. Res. Commun. 2017, 491, 343–348. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Pfeffer, S.R.; Handorf, C.R.; Pfeffer, L.M. MicroRNA miR-21 regulates the metastatic behavior of B16 melanoma cells. J. Biol. Chem. 2011, 286, 39172–39178. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Fan, M.; Pfeffer, L.M. IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer Res. 2010, 70, 8108–8116. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Wei, L.; Pfeffer, S.R.; Du, Z.; Murti, A.; Valentine, W.J.; Zheng, Y.; Pfeffer, L.M. Identification of CXCL11 as a STAT3-dependent gene induced by IFN. J. Immunol. 2007, 178, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Valentine, W.J.; Du, Z.; Pfeffer, L.M. Interferon alpha activates NF-kappaB in JAK1-deficient cells through a TYK2-dependent pathway. J. Biol. Chem. 2005, 280, 25849–25853. [Google Scholar] [CrossRef]
- Yang, C.H.; Murti, A.; Pfeffer, L.M. STAT3 complements defects in an interferon-resistant cell line: Evidence for an essential role for STAT3 in interferon signaling and biological activities. Proc. Natl. Acad. Sci. USA 1998, 95, 5568–5572. [Google Scholar] [CrossRef]
- Pfeffer, L.M.; Mullersman, J.E.; Pfeffer, S.R.; Murti, A.; Shi, W.; Yang, C.H. STAT3 as an adapter to couple phosphatidylinositol-3-kinase to the IFNAR-1 chain of the type I IFN receptor. Science 1997, 276, 1418–1420. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Shi, W.; Basu, L.; Murti, A.; Constantinescu, S.N.; Blatt, L.; Croze, E.; Mullersman, J.E.; Pfeffer, L.M. Direct association of STAT3 with the IFNAR1 signal transducing chain of the type I IFN receptor. J. Biol. Chem. 1996, 271, 8057–8061. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Jove, R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Garner, J.M.; Ellison, D.W.; Finkelstein, D.; Ganguly, D.; Du, Z.; Sims, M.; Yang, C.H.; Interiano, R.B.; Davidoff, A.M.; Pfeffer, L.M. Molecular heterogeneity in a patient-derived glioblastoma xenoline is regulated by different cancer stem cell populations. PLoS ONE 2015, 10, e0125838. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Roeser, J.C.; Leach, S.D.; McAllister, F. Emerging strategies for cancer immunoprevention. Oncogene 2015, 34, 6029–6039. [Google Scholar] [CrossRef]
- Garner, J.M.; Fan, M.; Yang, C.H.; Du, Z.; Sims, M.; Davidoff, A.M.; Pfeffer, L.M. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J. Biol. Chem. 2013, 288, 26167–26176. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Sims, M.; Cai, C.; Fan, M.; Pfeffer, L.M. Chromatin Remodeling Factor BRG1 Regulates Stemness and Chemosensitivity of Glioma Initiating Cells. Stem Cells 2018, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C.; Sims, M.M.; Sacher, J.R.; Raje, M.; Deokar, H.; Yue, P.; Turkson, J.; Buolamwini, J.K.; Pfeffer, L.M. SS-4 is a highly selective small molecule inhibitor of STAT3 tyrosine phosphorylation that potently inhibits GBM tumorigenesis in vitro and in vivo. Cancer Lett. 2022, 533, 215614. [Google Scholar] [CrossRef] [PubMed]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Guc, E.; Kapourani, C.A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470. [Google Scholar] [CrossRef] [PubMed]
- Iglesia, M.D.; Parker, J.S.; Hoadley, K.A.; Serody, J.S.; Perou, C.M.; Vincent, B.G. Genomic Analysis of Immune Cell Infiltrates Across 11 Tumor Types. J. Natl. Cancer Inst. 2016, 108, djw144. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Zemek, R.M.; De Jong, E.; Chin, W.L.; Schuster, I.S.; Fear, V.S.; Casey, T.H.; Forbes, C.; Dart, S.J.; Leslie, C.; Zaitouny, A.; et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med. 2019, 11, eaav7816. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, L.H.M.; Garcia, C.; da Fonseca, A.C.C.; Dubois, L.G.F.; de Sampaio, E.S.T.C.L.; Matias, D.; de Camargo Magalhaes, E.S.; do Amaral, R.F.; da Rosa, B.G.; Grimaldi, I.; et al. Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer 2019, 5, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Lei, Y.; Klionsky, D.J. The Emerging Roles of Autophagy in Human Diseases. Biomedicines 2021, 9, 1651. [Google Scholar] [CrossRef]
- Ariosa, A.R.; Lahiri, V.; Lei, Y.; Yang, Y.; Yin, Z.; Zhang, Z.; Klionsky, D.J. A perspective on the role of autophagy in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166262. [Google Scholar] [CrossRef]
- Yu, G.; Klionsky, D.J. Life and Death Decisions-The Many Faces of Autophagy in Cell Survival and Cell Death. Biomolecules 2022, 12, 866. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef]
- Lei, Y.; Huang, Y.; Wen, X.; Yin, Z.; Zhang, Z.; Klionsky, D.J. How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems. Antioxidants 2022, 11, 304. [Google Scholar] [CrossRef]
- An, H.; Harper, J.W. Ribosome Abundance Control Via the Ubiquitin-Proteasome System and Autophagy. J. Mol. Biol. 2020, 432, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Klionsky, D.J.; Shen, H.M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Cuervo, A.M.; Taylor, M.R.; Nishino, I.; Blum, J.S.; Dice, J.F.; Sandoval, I.V.; Lippincott-Schwartz, J.; August, J.T.; Saftig, P. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic 2005, 6, 1058–1061. [Google Scholar] [CrossRef]
- Into, T.; Horie, T.; Inomata, M.; Gohda, J.; Inoue, J.I.; Murakami, Y.; Niida, S. Basal autophagy prevents autoactivation or enhancement of inflammatory signals by targeting monomeric MyD88. Sci. Rep. 2017, 7, 1009. [Google Scholar] [CrossRef] [PubMed]
- Vevea, J.D.; Garcia, E.J.; Chan, R.B.; Zhou, B.; Schultz, M.; Di Paolo, G.; McCaffery, J.M.; Pon, L.A. Role for Lipid Droplet Biogenesis and Microlipophagy in Adaptation to Lipid Imbalance in Yeast. Dev. Cell 2015, 35, 584–599. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of Glioblastoma Multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression of the invasive potential of Glioblastoma cells by mTOR inhibitors involves modulation of NFkappaB and PKC-alpha signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Guo, H.; Zhang, R.; Liu, H.; Wu, L.; Wu, Q.; Liu, J.; Liu, T.; Zhang, Q. The PI3K inhibitor GDC-0941 enhances radiosensitization and reduces chemoresistance to temozolomide in GBM cell lines. Neuroscience 2017, 346, 298–308. [Google Scholar] [CrossRef]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Aoki, H.; Kondo, Y.; Aldape, K.; Yamamoto, A.; Iwado, E.; Yokoyama, T.; Hollingsworth, E.F.; Kobayashi, R.; Hess, K.; Shinojima, N.; et al. Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy 2008, 4, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Tini, P.; Belmonte, G.; Toscano, M.; Miracco, C.; Palumbo, S.; Pastina, P.; Battaglia, G.; Nardone, V.; Butorano, M.A.; Masucci, A.; et al. Combined epidermal growth factor receptor and Beclin1 autophagic protein expression analysis identifies different clinical presentations, responses to chemo- and radiotherapy, and prognosis in glioblastoma. BioMed Res. Int. 2015, 2015, 208076. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rahim, S.A.; Dirkse, A.; Oudin, A.; Schuster, A.; Bohler, J.; Barthelemy, V.; Muller, A.; Vallar, L.; Janji, B.; Golebiewska, A.; et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br. J. Cancer 2017, 117, 813–825. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008, 4, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wang, Q.; Li, B.; Xie, B.; Wang, W. Temozolomide induces autophagy via ATM-AMPK-ULK1 pathways in glioma. Mol. Med. Rep. 2014, 10, 411–416. [Google Scholar] [CrossRef]
- Goncalves, R.M.; Agnes, J.P.; Delgobo, M.; de Souza, P.O.; Thome, M.P.; Heimfarth, L.; Lenz, G.; Moreira, J.C.F.; Zanotto-Filho, A. Late autophagy inhibitor chloroquine improves efficacy of the histone deacetylase inhibitor SAHA and temozolomide in gliomas. Biochem. Pharmacol. 2019, 163, 440–450. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The Challenge of Developing Autophagy Inhibition as a Therapeutic Strategy. Cancer Res. 2016, 76, 5610–5614. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lee, J.; Xie, C. Autophagy Regulation on Cancer Stem Cell Maintenance, Metastasis, and Therapy Resistance. Cancers 2022, 14, 381. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Vieira de Castro, J.; Goncalves, C.S.; Hormigo, A.; Costa, B.M. Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. Int. J. Mol. Sci. 2020, 21, 5287. [Google Scholar] [CrossRef]
- LiCausi, F.; Hartman, N.W. Role of mTOR Complexes in Neurogenesis. Int. J. Mol. Sci. 2018, 19, 1544. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Los, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef]
- Galan-Moya, E.M.; Le Guelte, A.; Lima Fernandes, E.; Thirant, C.; Dwyer, J.; Bidere, N.; Couraud, P.O.; Scott, M.G.; Junier, M.P.; Chneiweiss, H.; et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011, 12, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef]
- Hartman, N.W.; Lin, T.V.; Zhang, L.; Paquelet, G.E.; Feliciano, D.M.; Bordey, A. mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 2013, 5, 433–444. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Shin, H.R.; Zoncu, R. The Lysosome at the Intersection of Cellular Growth and Destruction. Dev. Cell 2020, 54, 226–238. [Google Scholar] [CrossRef]
- Dechant, R.; Saad, S.; Ibanez, A.J.; Peter, M. Cytosolic pH regulates cell growth through distinct GTPases, Arf1 and Gtr1, to promote Ras/PKA and TORC1 activity. Mol. Cell 2014, 55, 409–421. [Google Scholar] [CrossRef]
- Dechant, R.; Binda, M.; Lee, S.S.; Pelet, S.; Winderickx, J.; Peter, M. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 2010, 29, 2515–2526. [Google Scholar] [CrossRef]
- Brunel, A.; Hombourger, S.; Barthout, E.; Battu, S.; Kogel, D.; Antonietti, P.; Deluche, E.; Saada, S.; Durand, S.; Lalloue, F.; et al. Autophagy inhibition reinforces stemness together with exit from dormancy of polydisperse glioblastoma stem cells. Aging 2021, 13, 18106–18130. [Google Scholar] [CrossRef] [PubMed]
- Appin, C.L.; Brat, D.J. Molecular genetics of gliomas. Cancer J. 2014, 20, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Benitez, J.A.; Finlay, D.; Castanza, A.; Parisian, A.D.; Ma, J.; Longobardi, C.; Campos, A.; Vadla, R.; Izurieta, A.; Scerra, G.; et al. PTEN deficiency leads to proteasome addiction: A novel vulnerability in glioblastoma. Neuro-Oncology 2021, 23, 1072–1086. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Yin, J.; Yang, C.; Wang, Y.; Sims, M.; Pfeffer, L.M.; Chaum, E. STAT3 suppresses the AMPKalpha/ULK1-dependent induction of autophagy in glioblastoma cells. J. Cell. Mol. Med. 2022, 26, 3873–3890. [Google Scholar] [CrossRef]
- Lv, D.; Gimple, R.C.; Zhong, C.; Wu, Q.; Yang, K.; Prager, B.C.; Godugu, B.; Qiu, Z.; Zhao, L.; Zhang, G.; et al. PDGF signaling inhibits mitophagy in glioblastoma stem cells through N(6)-methyladenosine. Dev. Cell 2022, 57, 1466–1481 e1466. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Zhang, Y.; Celiku, O.; Zhang, W.; Song, H.; Williams, B.J.; Giles, A.J.; Rich, J.N.; Abounader, R.; Gilbert, M.R.; et al. Mitochondrial NIX Promotes Tumor Survival in the Hypoxic Niche of Glioblastoma. Cancer Res. 2019, 79, 5218–5232. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shan, S.; Yuan, Z.; Wu, F.; Zheng, M.; Wang, Y.; Gui, J.; Xu, W.; Wang, C.; Ren, T.; et al. Mitophagy bridges DNA sensing with metabolic adaption to expand lung cancer stem-like cells. EMBO Rep. 2023, 24, e54006. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Otaegi-Ugartemendia, M.; Carrasco-Garcia, E.; Azkargorta, M.; Diaz, A.; Saenz-Antonanzas, A.; Andermatten, J.A.; Garcia-Puga, M.; Garcia, I.; Elua-Pinin, A.; et al. Chaperone-Mediated Autophagy Controls Proteomic and Transcriptomic Pathways to Maintain Glioma Stem Cell Activity. Cancer Res. 2022, 82, 1283–1297. [Google Scholar] [CrossRef]
- Mukthavaram, R.; Ouyang, X.; Saklecha, R.; Jiang, P.; Nomura, N.; Pingle, S.C.; Guo, F.; Makale, M.; Kesari, S. Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. J. Transl. Med. 2015, 13, 269. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Kondo, Y.; Kondo, S. Roles of mTOR and STAT3 in autophagy induced by telomere 3′ overhang-specific DNA oligonucleotides. Autophagy 2007, 3, 496–498. [Google Scholar] [CrossRef]
- Yuan, X.; Du, J.; Hua, S.; Zhang, H.; Gu, C.; Wang, J.; Yang, L.; Huang, J.; Yu, J.; Liu, F. Suppression of autophagy augments the radiosensitizing effects of STAT3 inhibition on human glioma cells. Exp. Cell Res. 2015, 330, 267–276. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laribee, R.N.; Boucher, A.B.; Madireddy, S.; Pfeffer, L.M. The STAT3-Regulated Autophagy Pathway in Glioblastoma. Pharmaceuticals 2023, 16, 671. https://doi.org/10.3390/ph16050671
Laribee RN, Boucher AB, Madireddy S, Pfeffer LM. The STAT3-Regulated Autophagy Pathway in Glioblastoma. Pharmaceuticals. 2023; 16(5):671. https://doi.org/10.3390/ph16050671
Chicago/Turabian StyleLaribee, Ronald Nicholas, Andrew B. Boucher, Saivikram Madireddy, and Lawrence M. Pfeffer. 2023. "The STAT3-Regulated Autophagy Pathway in Glioblastoma" Pharmaceuticals 16, no. 5: 671. https://doi.org/10.3390/ph16050671
APA StyleLaribee, R. N., Boucher, A. B., Madireddy, S., & Pfeffer, L. M. (2023). The STAT3-Regulated Autophagy Pathway in Glioblastoma. Pharmaceuticals, 16(5), 671. https://doi.org/10.3390/ph16050671