
A Tale of Two Proteases: MPro and TMPRSS2 as Targets for COVID-19 Therapies


Abstract
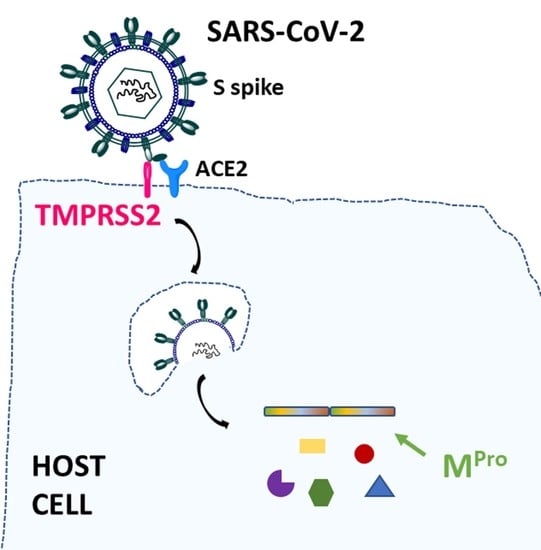
:
1. Introduction
2. Proteases as Targets
2.1. Proteases as Targets in Viral Infections
2.2. Viral MPro and Host TMPRSS2 Proteases in COVID-19: Overview of Reported Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Company/Research Group | Approval State |
|---|---|---|---|
| Remdesevir | RNA polymerase (inhibitor) | Gilead | FDA and EMA approved, 2020 |
| Molnupiravir | RNA polymerase (inhibitor) | Merck | FDA approved, 2021 Pending EMA approval |
| Paxlovid (Nirmatrelvir/Ritonavir) | Viral proteases, MPro and HIV protease (inhibitors) | Pfizer | FDA approved, 2021 EMA/UK/Canada approved, 2022 |
| Ensitrelvir | Viral protease MPro (inhibitor) | Shionogy | Emergency approval Japan, 2022 |
| Xiannuoxin (Simnotrelvir/Ritonavir) | Viral proteases, MPro and HIV protease (inhibitors) | approved in China in January 2023 | |
| Myricetin | Viral protease MPro (inhibitor) | [50] | Preclinical phase |
| Ebselen | Viral protease MPro (inhibitor) | [51] | Preclinical phase |
 | Viral protease MPro (inhibitor) | [52] | Preclinical phase |
| GC-376 | Viral protease MPro (inhibitor) | Anivive Lifesciences | Preclinical phase |
| Boceprevir | Viral protease MPro (inhibitor) | [53] | Preclinical phase |
| Y180 | Viral protease MPro (inhibitor) | [54] | Preclinical phase |
| MG-101, | Viral protease MPro (inhibitor) | [55] | Preclinical phase |
| Camostat | Host protease TMPRSS2 (inhibitor) | Ono Pharmaceutical | Approved for pancreatitis, Japan 1985. Phase 3 for COVID-19, 2021 |
| Nafamostat | Host protease TMPRSS2 (inhibitor) | [56,57] | Approved as anticoagulant, Japan and Korea 2003 Phase 3 COVID-19, 2020 |
| Gabexate | Host protease TMPRSS2 (inhibitor) | [58] | Preclinical phase |
| BC-11 | Host protease TMPRSS2 (inhibitor) | [59] | Preclinical phase |
| Otamixaban | Host protease TMPRSS2 (inhibitor) | [60,61] | Preclinical phase |
| MI-432 and MI-1900 | Host protease TMPRSS2 (inhibitor) | [62,63] | Preclinical phase |
| α1-antitrypsin | Host protease TMPRSS2 (inhibitor) | [64,65] | Preclinical phase |
2.2.1. Inhibitors of Viral MPro
2.2.2. Inhibitors of Host TMPRSS2
3. Computational Studies and Modelling
3.1. Computational Studies with viral MPro as a Target
3.2. Computational Studies with Host TMPRSS2 as a Target
4. Dual-Action Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ning, Z.; Chen, Y.; Guo, M.; Liu, Y.; Kumar Gali, N.; Sun, L.; Duan, Y.; Cai, J.; Westerdahl, D.; et al. Aerodynamic analysis of SARS-CoV-2 in two Wuhan hospitals. Nature 2020, 582, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Wu, N.C.; Tsang, O.T.; Yuan, M.; Perera, R.A.P.M.; Leung, W.S.; So, R.T.Y.; Chun Chan, J.M.C.; Yip, G.K.; Chik, T.S.; et al. Cross-reactive antibody response between SARS-CoV-2 and SARS-CoV infections. Cell Rep. 2020, 31, 107725. [Google Scholar] [CrossRef]
- Estimates of Mortality Vary Regionally between 0.8% and 14.5%. Available online: https://coronavirus.jhu.edu/data/mortality (accessed on 20 April 2023).
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 18 May 2023).
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Yang, X.; Dong, N.; Chan, E.W.; Chen, S. Genetic cluster analysis of SARS-CoV-2 and the identification of those responsible for the major outbreaks in various countries. Emerg. Microbes Infect. 2020, 9, 1287–1299. [Google Scholar] [CrossRef]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, B.; Piller, C. Emails offer look into whistleblower charges of cronyism behind potential COVID-19 drug. Science 2020. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Kim, Y.; Liu, H.; Kankanamalage, A.C.G.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.-O. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. [Google Scholar] [CrossRef]
- Kempf, D.J.; Sham, H.L.; Marsh, K.C.; Flentge, C.A.; Betebenner, D.; Green, B.E.; McDonald, E.; Vasavanonda, S.; Saldivar, A.; Wideburg, N.E.; et al. Discovery of ritonavir, a potent inhibitor of HIV protease with high oral bioavailability and clinical efficacy. J. Med. Chem. 1998, 41, 602–617. [Google Scholar] [CrossRef]
- Mehta, N.; Mazer-Amirshahi, M.; Alkindi, N.; Pourmand, A. Pharmacotherapy in COVID-19; A narrative review for emergency providers. Am. J. Emerg. Med. 2020, 38, 1488–1493. [Google Scholar] [CrossRef]
- Veklury. European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/veklury (accessed on 23 June 2020).
- FDA Approves First Treatment for COVID-19. U.S. Food and Drug Administration (FDA). Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19 (accessed on 22 October 2020).
- Cully, M. A tale of two antiviral targets—And the COVID-19 drugs that bind them. Nat. Rev. Drug Discov. 2022, 21, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Masyeni, S.; Iqhrammullah, M.; Frediansyah, A.; Nainu, F.; Tallei, T.; Bin Emran, T.; Ophinni, Y.; Dhama, K.; Harapan, H. Molnupiravir: A lethal mutagenic drug against rapidly mutating severe acute respiratory syndrome coronavirus 2—A narrative review. J. Med. Virol. 2022, 94, 3006–3016. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: Molnupiravir reduces risk of hospital admission or death by 50% in patients at risk, MSD reports. BMJ 2021, 375, n2422. [Google Scholar] [CrossRef]
- Akinosoglou, K.; Schinas, G.; Gogos, C. Oral Antiviral Treatment for COVID-19: A Comprehensive Review on Nirmatrelvir/Ritonavir. Viruses 2022, 14, 2540. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An overview of severe acute respiratory syndrome-Coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19 (accessed on 25 May 2023).
- Available online: https://www.gov.uk/government/news/oral-covid-19-antiviral-paxlovid-approved-by-uk-regulator (accessed on 25 May 2023).
- Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/paxlovid (accessed on 25 May 2023).
- Available online: https://www.canada.ca/en/health-canada/news/2022/01/health-canada-authorizes-paxlovidtm-for-patients-with-mild-to-moderate-covid-19-at-high-risk-of-developing-serious-disease.html (accessed on 25 May 2023).
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Non-hospitalized Adults with COVID-19. NEJM 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.shionogi.com/us/en/news/2022/11/xocova-ensitrelvir-fumaric-acid-tablets-125mg-approved-in-japan-for-the-treatment-of-sars-cov-2-infection,-under-the-emergency-regulatory-approval-system.html (accessed on 25 May 2023).
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Fang, Z.; Hou, G.; Han, M.; Xu, X.; Dong, J.; Zheng, J. The psychological impact of the COVID-19 epidemic on college students in China. Psychiatry Res. 2020, 287, 112934. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gupta, S.P. Fundamentals of Viruses and Their Proteases. Viral Proteases Inhib. 2017, 1–24. [Google Scholar] [CrossRef]
- Steinkühler, C. Viral Proteases. In Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar] [CrossRef]
- Delre, P.; Caporuscio, F.; Saviano, M.; Mangiatordi, G.F. Repurposing Known Drugs as Covalent and Non-covalent Inhibitors of the SARS-CoV-2 Papain-Like Protease. Front. Chem. 2020, 8, 594009. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Gordon, L.A.; Fung, H.B. Boceprevir: A protease inhibitor for the treatment of hepatitis C. Clin. Ther. 2012, 34, 2021–2038. [Google Scholar] [CrossRef]
- Raboisson, P.; de Kock, H.; Rosenquist, Å.; Nilsson, M.; Salvador-Oden, L.; Lin, T.-I.; Roue, N.; Ivanov, V.; Wähling, H.; Wickström, K.; et al. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg. Med. Chem. Lett. 2008, 18, 4853–4858. [Google Scholar] [CrossRef]
- Ohta, Y.; Shinkai, I. New drugs-reports of new drugs recently approved by the FDA. Lamivudine. Bioorg. Med. Chem. 1997, 5, 639–640. [Google Scholar] [CrossRef]
- Kalu, N.N.; Desai, P.V.; Shirley, C.M.; Gibson, W.; Dennis, P.A.; Ambinder, R.F. Nelfinavir inhibits maturation and export of Herpes Simplex Virus 1. J. Virol. 2014, 88, 5455–5461. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, C.; Passioura, T.; Varava, P.; Mahawaththa, M.C.; Leuthold, M.M.; Klein, C.; Suga, H.; Otting, G. De novo discovery of nonstandard macrocyclic peptides as non-competitive inhibitors of the Zika Virus NS2B-NS3 protease. ACS Med. Chem. Lett. 2019, 10, 168–174. [Google Scholar] [CrossRef]
- Xiao, T.; Cui, M.; Zheng, C.; Wang, M.; Sun, R.; Gao, D.; Bao, J.; Ren, S.; Yang, B.; Lin, J.; et al. Myricetin Inhibits SARS-CoV-2 Viral Replication by Targeting Mpro and Ameliorates Pulmonary Inflammation. Front. Pharmacol. 2021, 12, 669642. [Google Scholar] [CrossRef]
- Sahoo, P.; Lenka, D.R.; Batabyal, M.; Pain, P.K.; Kumar, S.; Manna, D.; Kumar, A. Detailed Insights into the Inhibitory Mechanism of New Ebselen Derivatives against Main Protease (Mpro) of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). ACS Pharmacol. Transl. Sci. 2023, 6, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M.; Tomar, S.; et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013, 24, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, R.; Ruiz-Moreno, A.J.; Cong, Y.; Dinesh Kumar, N.; Velasco-Velazquez, M.A.; Neochoritis, C.G.; Smith, J.; Reggiori, F.; Groves, M.R.; Dömling, A. Repurposing the HCV NS3–4A protease drug boceprevir as COVID-19 therapeutics. RSC Med. Chem. 2021, 12, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Quan, B.-X.; Shuai, H.; Xia, A.-J.; Hou, Y.; Zeng, R.; Liu, X.-L.; Lin, G.-F.; Qiao, J.-X.; Li, W.-P.; Wang, F.-L.; et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol. 2022, 7, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef]
- Akizawa, T.; Koshikawa, S.; Ota, K.; Kazama, M.; Mimura, N.; Hirasawa, Y. Nafamostat mesilate: A regional anticoagulant for hemodialysis in patients at high risk for bleeding. Nephron 1993, 64, 376–381. [Google Scholar] [CrossRef]
- Minakata, D.; Fujiwara, S.I.; Ikeda, T.; Kawaguchi, S.I.; Toda, Y.; Ito, S.; Ochi, S.I.; Nagayama, T.; Mashima, K.; Umino, K.; et al. Comparison of gabexate mesilate and nafamostat mesilate for disseminated intravascular coagulation associated with hematological malignancies. Int. J. Hematol. 2019, 109, 141–146. [Google Scholar] [CrossRef]
- Oh, S.H.; Lee, H.Y.; Ki, Y.J.; Kim, S.H.; Lim, K.J.; Jung, K.T. Gabexate mesilate ameliorates the neuropathic pain in a rat model by inhibition of proinflammatory cytokines and nitric oxide pathway via suppression of nuclear factor-κB. Korean J. Pain 2020, 33, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Moumbock, A.F.A.; Tran, H.T.T.; Lamy, E.; Günther, S. BC-11 is a covalent TMPRSS2 fragment inhibitor that impedes SARS-CoV-2 host cell entry. Arch. Pharm. 2023, 356, e2200371. [Google Scholar] [CrossRef]
- Hu, X.; Shrimp, J.H.; Guo, H.; Xu, M.; Chen, C.Z.; Zhu, W.; Zakharov, A.V.; Jain, S.; Shinn, P.; Simeonov, A.; et al. Discovery of TMPRSS2 Inhibitors from Virtual Screening as a Potential Treatment of COVID-19. ACS Pharmacol. Translat. Sci. 2021, 4, 1124–1135. [Google Scholar] [CrossRef]
- Shrimp, J.H.; Janiszewski, J.; Chen, C.Z.; Xu, M.; Wilson, K.M.; Kales, S.C.; Sanderson, P.E.; Shinn, P.; Schneider, R.; Itkin, Z.; et al. Suite of TMPRSS2 Assays for Screening Drug Repurposing Candidates as Potential Treatments of COVID-19. ACS Infect. Dis. 2022, 8, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; van Lam, T.V.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Liu, Y.; Liang, C.; Xin, L.; Xie, X.; Zhang, D.; Wan, M.; Li, H.; Fu, X.; Liu, H.; et al. An update review of emerging small-molecule therapeutic options for COVID-19. Biomed. Pharmacother. 2021, 137, 111313. [Google Scholar] [CrossRef]
- Essa, R.Z.; Wu, Y.-S.; Batumalaie, K.; Sekar, M.; Poh, C.-L. Antiviral peptides against SARS-CoV-2: Therapeutic targets, mechanistic antiviral activity, and efficient delivery. Pharmacol. Rep. 2022, 74, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Prelli Bozzo, C.; et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.; Zhang, Y.; Zhang, Y.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef]
- Qiao, J.; Li, Y.S.; Zeng, R.; Liu, F.L.; Luo, R.H.; Huang, C.; Wang, Y.F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H.; et al. The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef]
- Boozari, M.; Hosseinzadeh, H. Natural products for COVID-19 prevention and treatment regarding to previous coronavirus infections and novel studies. Phytother. Res. 2021, 35, 864–876. [Google Scholar] [CrossRef]
- Available online: http://english.scio.gov.cn/pressroom/2023-01/30/content_85079258.htm (accessed on 25 May 2023).
- Available online: https://www.pharmaceutical-technology.com/news/china-nmpa-oral-drugs-covid-19/ (accessed on 25 May 2023).
- Ramajayam, R.; Tan, K.-P.; Liu, H.-G.; Liang, P.-H. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg. Med. Chem. 2010, 18, 7849–7854. [Google Scholar] [CrossRef]
- Ohnishi, K.; Hattori, Y.; Kobayashi, K.; Akaji, K. Evaluation of a non-prime site substituent and warheads combined with a decahydroisoquinolin scaffold as a SARS 3CL protease inhibitor. Bioorg. Med. Chem. 2019, 27, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bao, B.-B.; Qing Song, G.; Chen, C.; Zhang, X.; Lu, W.; Wang, Z.; Cai, Y.; Li, S.; Fu, S.; et al. Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: Chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem. 2017, 137, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, S.; Hattori, Y.; Kobayashi, K.; Akaji, K. Evaluation of an octahydroisochromene scaffold used as a novel SARS 3CL protease inhibitor. Bioorg. Med. Chem. 2020, 28, 115273. [Google Scholar] [CrossRef]
- Galasiti Kankanamalage, A.C.; Kim, Y.; Damalanka, V.C.; Rathnayake, A.D.; Fehr, A.R.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Lushington, G.H.; Perlman, S.; et al. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 2018, 150, 334–346. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, S.; Xue, F.; Zou, Y.; Chen, C.; Bartlam, M.; Rao, Z. Structure of the main protease from a global infectious human coronavirus, HCoV-HKU1. J. Virol. 2008, 82, 8647–8655. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 11, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Belovodskiy, A.; Hena, M.; Kandadai, A.S.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Khan, M.B.; Arutyunova, E.; Lu, J.; et al. Peptidomimetic α-Acyloxymethylketone Warheads with Six-Membered Lactam P1 Glutamine Mimic: SARS-CoV-2 3CL Protease Inhibition, Coronavirus Antiviral Activity, and in Vitro Biological Stability. J. Med. Chem. 2022, 65, 2905–2925. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef] [PubMed]
- Gant, T.G. Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 2014, 57, 3595–3611. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, I.S.; Jarrar, Y.B. Targeting the intestinal TMPRSS2 protease to prevent SARS-CoV-2 entry into enterocytes-prospects and challenges. Mol. Biol. Rep. 2021, 48, 4667–4675. [Google Scholar] [CrossRef]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat. Chem. Biol. 2022, 18, 963–971. [Google Scholar] [CrossRef]
- Breining, P.; Frølund, A.L.; Højen, J.F.; Gunst, J.D.; Staerke, N.B.; Saedder, E.; Cases-Thomas, M.; Little, P.; Nielsen, L.P.; Søgaard, O.S.; et al. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin. Pharmacol. Toxicol. 2021, 128, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Ohkoshi, M. Inhibition of growth of 3-methylcholanthrene-induced mouse skin tumor by protease inhibitor [N,N-dimethylcarbamoylmethyl 4-(4-guanidinobenzoyloxy)-phenylacetate] methanesulfate. Gan 1981, 72, 959–964. Available online: https://pubmed.ncbi.nlm.nih.gov/7341342/ (accessed on 26 May 2023).
- Göke, B.; Printz, H.; Koop, I.; Rausch, U.; Richter, G.; Arnold, R.; Adler, G. Endogenous CCK release and pancreatic growth in rats after feeding a proteinase inhibitor (camostate). Pancreas 1986, 1, 509–515. [Google Scholar] [CrossRef]
- Kitamura, K.; Tomita, K. Proteolytic activation of the epithelial sodium channel and therapeutic application of a serine protease inhibitor for the treatment of salt-sensitive hypertension. Clin. Exp. Nephrol. 2012, 16, 44–48. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Ueda, M.; Uchimura, K.; Narita, Y.; Miyasato, Y.; Mizumoto, T.; Morinaga, J.; Hayata, M.; Kakizoe, Y.; Adachi, M.; Miyoshi, T.; et al. The serine protease inhibitor camostat mesilate attenuates the progression of chronic kidney disease through its antioxidant effects. Nephron 2015, 129, 223–232. [Google Scholar] [CrossRef]
- Takahashi, W.; Yoneda, T.; Koba, H.; Ueda, T.; Tsuji, N.; Ogawa, H.; Asakura, H. Potential mechanisms of nafamostat therapy for severe COVID-19 pneumonia with disseminated intravascular coagulation. Int. J. Infect. Dis. 2021, 102, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Zhuravel, S.V.; Khmelnitskiy, O.K.; Burlaka, O.O.; Gritsan, A.I.; Goloshchekin, B.M.; Kim, S.; Hong, K.Y. Nafamostat in hospitalized patients with moderate to severe COVID-19 pneumonia: A randomised Phase II clinical trial. EClinicalMedicine 2021, 41, 101169. [Google Scholar] [CrossRef]
- Gunst, J.D.; Staerke, N.B.; Pahus, M.H.; Kristensen, L.H.; Bodilsen, J.; Lohse, N.; Dalgaard, L.S.; Brønnum, D.; Fröbert, O.; Hønge, B.; et al. Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with COVID-19—A double-blind randomized controlled trial. EClinicalMedicine 2021, 35, 100849. [Google Scholar] [CrossRef] [PubMed]
- Shrimp, J.H.; Kales, S.C.; Sanderson, P.E.; Simeonov, A.; Shen, M.; Hall, M.D. An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19. ACS Pharmacol. Translat. Sci. 2020, 3, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, C.Z.; Xu, M.; Hu, Z.; Guo, H.; Itkin, Z.; Shinn, P.; Ivin, P.; Leek, M.; Liang, T.J.; et al. Discovery of Small Molecule Entry Inhibitors Targeting the Fusion Peptide of SARS-CoV-2 Spike Protein. ACS Med. Chem. Lett. 2021, 12, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Shapira, T.; Monreal, I.A.; Dion, S.P.; Buchholz, D.W.; Imbiakha, B.; Olmstead, A.D.; Jager, M.; Désilets, A.; Gao, G.; Martins, M.; et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature 2022, 605, 340–348. [Google Scholar] [CrossRef]
- Metzdorf, K.; Jacobsen, H.; Greweling-Pils, M.C.; Hoffmann, M.; Lüddecke, T.; Miller, F.; Melcher, L.; Kempf, A.; Nehlmeier, I.; Bruder, D.; et al. TMPRSS2 Is Essential for SARS-CoV-2 Beta and Omicron Infection. Viruses 2023, 15, 271. [Google Scholar] [CrossRef]
- Bojkova, D.; Widera, M.; Ciesek, S.; Wass, M.N.; Michaelis, M.; Cinatl, J. Reduced interferon antagonism but similar drug sensitivity in Omicron variant compared to Delta variant of SARS-CoV-2 isolates. Cell Res. 2022, 32, 319–321. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Worrall, L.J.; Vuckovic, M.; Rosell, F.I.; Gentile, F.; Ton, A.-T.; Caveney, N.A.; Ban, F.; Cherkasov, A.; Paetzel, M.; et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020, 11, 5877. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank (RCSB PDB). Available online: https://www.rcsb.org/ (accessed on 27 February 2023).
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, S.T.; Quynh Anh Pham, N.; Thi Le, L.; Pham, D.-H.; Vu, V.V. Computational Determination of Potential Inhibitors of SARS-CoV-2 Main Protease. J. Chem. Inf. Model. 2020, 60, 5771–5780. [Google Scholar] [CrossRef] [PubMed]
- Semenov, V.A.; Krivdin, L.B. Combined Computational NMR and Molecular Docking Scrutiny of Potential Natural SARS-CoV-2 Mpro Inhibitors. J. Phys. Chem. B 2022, 126, 2173–2187. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wu, X.; Zhao, Y.; Chen, C.; Yang, Z.; Zhang, X.; Ren, J.; Wang, Y.; Wu, C.; Li, C.; et al. Computational Simulation of HIV Protease Inhibitors to the Main Protease (Mpro) of SARS-CoV-2: Implications for COVID-19 Drugs Design. Molecules 2021, 26, 7385. [Google Scholar] [CrossRef] [PubMed]
- De Souza Gomes, I.; Santana, C.C.; Marcolino, L.S.; de Lima, L.H.F.; de Melo-Minardi, R.C.; Dias, R.S.; de Paula, S.O.; de Azevedo Silveira, S. Computational prediction of potential inhibitors for SARS-COV-2 main protease based on machine learning, docking, MM-PBSA calculations, and metadynamics. PLoS ONE 2022, 17, e0267471. [Google Scholar] [CrossRef]
- Patel, C.J.; Jani, S.P.; Kumar, S.P.; Modi, K.; Kumar, Y. Computational investigation of natural compounds as potential main protease (Mpro) inhibitors for SARS-CoV-2 virus. Comput. Biol. Med. 2022, 151, 106318. [Google Scholar] [CrossRef]
- El Khoury, L.; Jing, Z.; Cuzzolin, A.; Deplano, A.; Loco, D.; Sattarov, B.; Hédin, F.; Wendeborn, S.; Ho, C.; El Ahdab, D.; et al. Computationally driven discovery of SARS-CoV-2 Mpro inhibitors: From design to experimental validation. Chem. Sci. 2022, 13, 3674–3687. [Google Scholar] [CrossRef]
- Piplani, S.; Singh, P.; Petrovsky, N.; Winkler, D.A. Computational repurposing of drugs and natural products against SARS-CoV-2 main protease (Mpro) as potential COVID-19 therapies. Front. Mol. Biosci. 2022, 9, 781039. [Google Scholar] [CrossRef]
- Singh, N.; Decroly, E.; Khatib, A.-M.; Villoutreix, B.O. Structure-based drug repositioning over the human TMPRSS2 protease domain: Search for chemical probes able to repress SARS-CoV-2 Spike protein cleavages. Eur. J. Pharm. Sci. 2020, 153, 105495. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Amanullah, A.; Baig, A.A.; Aziz, B.; Shabbir, S.; Raza, F.; Uddin, N. Molecular docking between human TMPRSS2 and SARS-CoV-2 spike protein: Conformation and intermolecular interactions. AIMS Microbiol. 2020, 6, 350–360. [Google Scholar] [CrossRef]
- Manandhar, S.; Pai, K.S.R.; Krishnamurthy, P.T.; Kiran, A.V.V.V.R.; Kumari, G.K. Identification of novel TMPRSS2 inhibitors against SARS-CoV-2 infection: A structure-based virtual screening and molecular dynamics study. Struct. Chem. 2022, 33, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.; Hassan Baig, M.; Imran Khan, M.; Alotaibi, S.S.; Alorabi, M.; Dong, J.-J. Computational screening of camostat and related compounds against human TMPRSS2: A potential treatment of COVID-19. Saudi Pharm. J. 2022, 30, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Salleh, M.Z.; Deris, Z.Z. In Silico Molecular Characterization of Human TMPRSS2 Protease Polymorphic Variants and Associated SARS-CoV-2 Susceptibility. Life 2022, 12, 231. [Google Scholar] [CrossRef]
- Serra, A.; Fratello, M.; Federico, A.; Ojha, R.; Provenzani, R.; Tasnadi, E.; Cattelani, L.; del Giudice, G.; Kinaret, P.A.S.; Saarimäki, L.A.; et al. Computationally prioritized drugs inhibit SARS-CoV-2 infection and syncytia formation. Brief. Bioinformatics 2022, 23, bbab507. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Fujimoto, K.J.; Yanai, T. A Quantum Chemical Study on the Anti-SARS-CoV-2 Activity of TMPRSS2 Inhibitors. ChemRxiv Camb. Camb. Open Engag. 2023. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, H.; Liu, J.; He, L.; Yu, R.; Kang, C. Design of SARS-CoV-2 Mpro, PLpro dual-target inhibitors based on deep reinforcement learning and virtual screening. Future Med. Chem. 2022, 14, 393–405. [Google Scholar] [CrossRef]
- Kumari, A.; Rajput, V.S.; Nagpal, P.; Kukrety, H.; Grover, S.; Grover, A. Dual inhibition of SARS-CoV-2 spike and main protease through a repurposed drug, rutin. J. Biomol. Struct. Dyn. 2022, 40, 4987–4999. [Google Scholar] [CrossRef]
- Huang, S.-T.; Chen, Y.; Chang, W.C.; Chen, H.-F.; Lai, H.-C.; Lin, Y.-C.; Wang, W.-J.; Wang, Y.; Yang, C.-S.; Wang, S.C.; et al. Scutellaria barbata D. Don Inhibits the Main Proteases (Mpro and TMPRSS2) of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. Viruses 2021, 13, 826. [Google Scholar] [CrossRef]
- Wang, S.-C.; Chen, Y.; Wang, Y.-C.; Wang, W.-J.; Yang, C.-S.; Tsai, C.-L.; Hou, M.-H.; Chen, H.-F.; Shen, Y.-C.; Hung, M.-C. Tannic acid suppresses SARS-CoV-2 as a dual inhibitor of the viral main protease and the cellular TMPRSS2 protease. Am. J. Cancer Res. 2020, 10, 4538–4546. Available online: https://pubmed.ncbi.nlm.nih.gov/33415017 (accessed on 26 May 2023).
- Mahgoub, M.A.; Alnaem, A.; Fadlelmola, M.; Abo-Idris, M.; Makki, A.A.; Abdelgadir, A.A.; Alzain, A.A. Discovery of novel potential inhibitors of TMPRSS2 and Mpro of SARS-CoV-2 using E-pharmacophore and docking-based virtual screening combined with molecular dynamic and quantum mechanics. J. Biomol. Struct. Dyn. 2022, 1–14. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farkaš, B.; Minneci, M.; Misevicius, M.; Rozas, I. A Tale of Two Proteases: MPro and TMPRSS2 as Targets for COVID-19 Therapies. Pharmaceuticals 2023, 16, 834. https://doi.org/10.3390/ph16060834
Farkaš B, Minneci M, Misevicius M, Rozas I. A Tale of Two Proteases: MPro and TMPRSS2 as Targets for COVID-19 Therapies. Pharmaceuticals. 2023; 16(6):834. https://doi.org/10.3390/ph16060834
Chicago/Turabian StyleFarkaš, Barbara, Marco Minneci, Matas Misevicius, and Isabel Rozas. 2023. "A Tale of Two Proteases: MPro and TMPRSS2 as Targets for COVID-19 Therapies" Pharmaceuticals 16, no. 6: 834. https://doi.org/10.3390/ph16060834
APA StyleFarkaš, B., Minneci, M., Misevicius, M., & Rozas, I. (2023). A Tale of Two Proteases: MPro and TMPRSS2 as Targets for COVID-19 Therapies. Pharmaceuticals, 16(6), 834. https://doi.org/10.3390/ph16060834