

Application of Ligand- and Structure-Based Prediction Models for the Design of Alkylhydrazide-Based HDAC3 Inhibitors as Novel Anti-Cancer Compounds

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
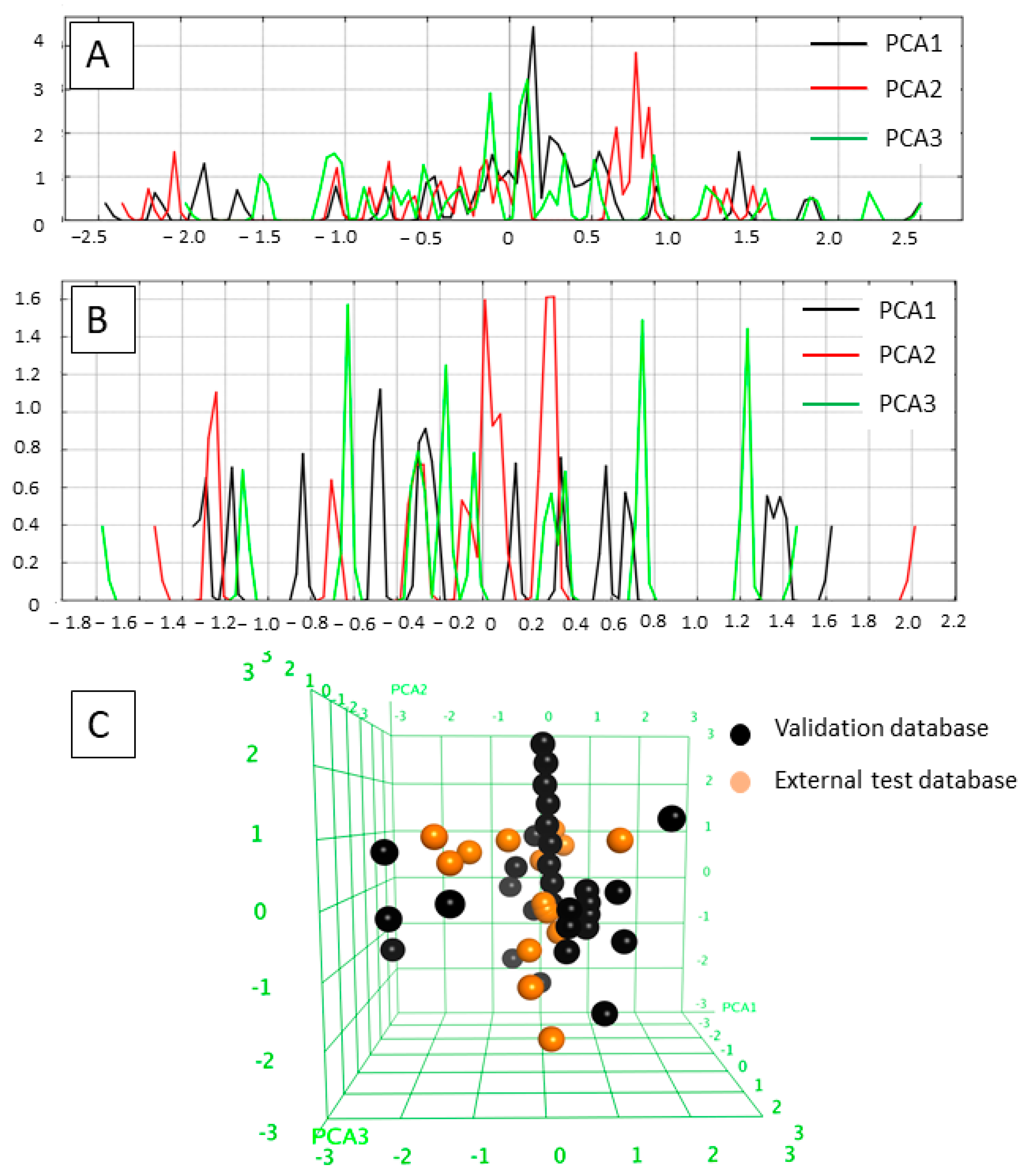
2.1. Diversity Analysis of the Datasets
- Highly active inhibitors showing pIC50 > 7
- Moderately active inhibitors showing pIC50 between 5.30 and 7
- Inactive inhibitors showing pIC50 < 5.30
2.2. Analysis of the Pharmacophore Model
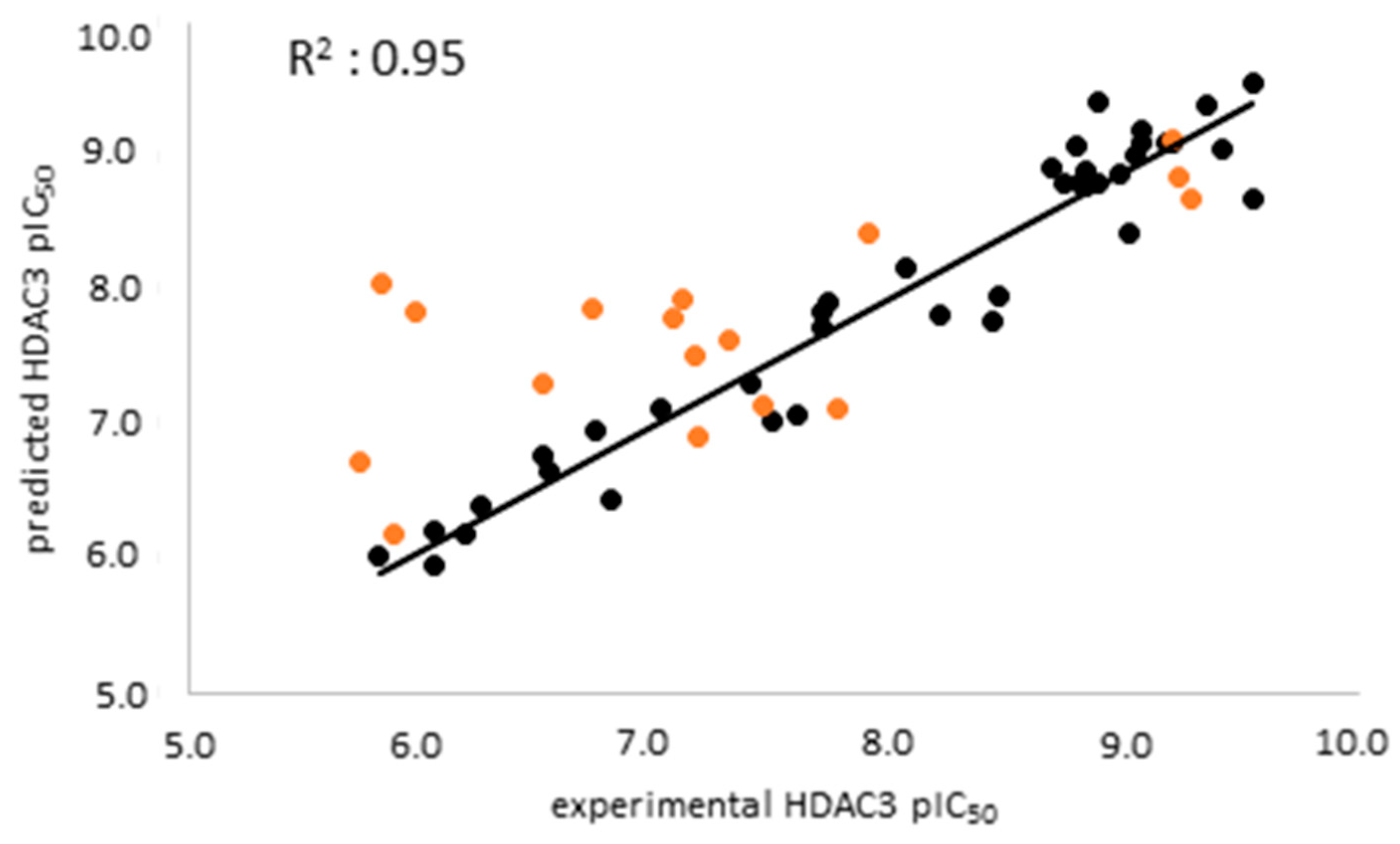
2.3. Analysis of the Atom-Based 3D-QSAR Model
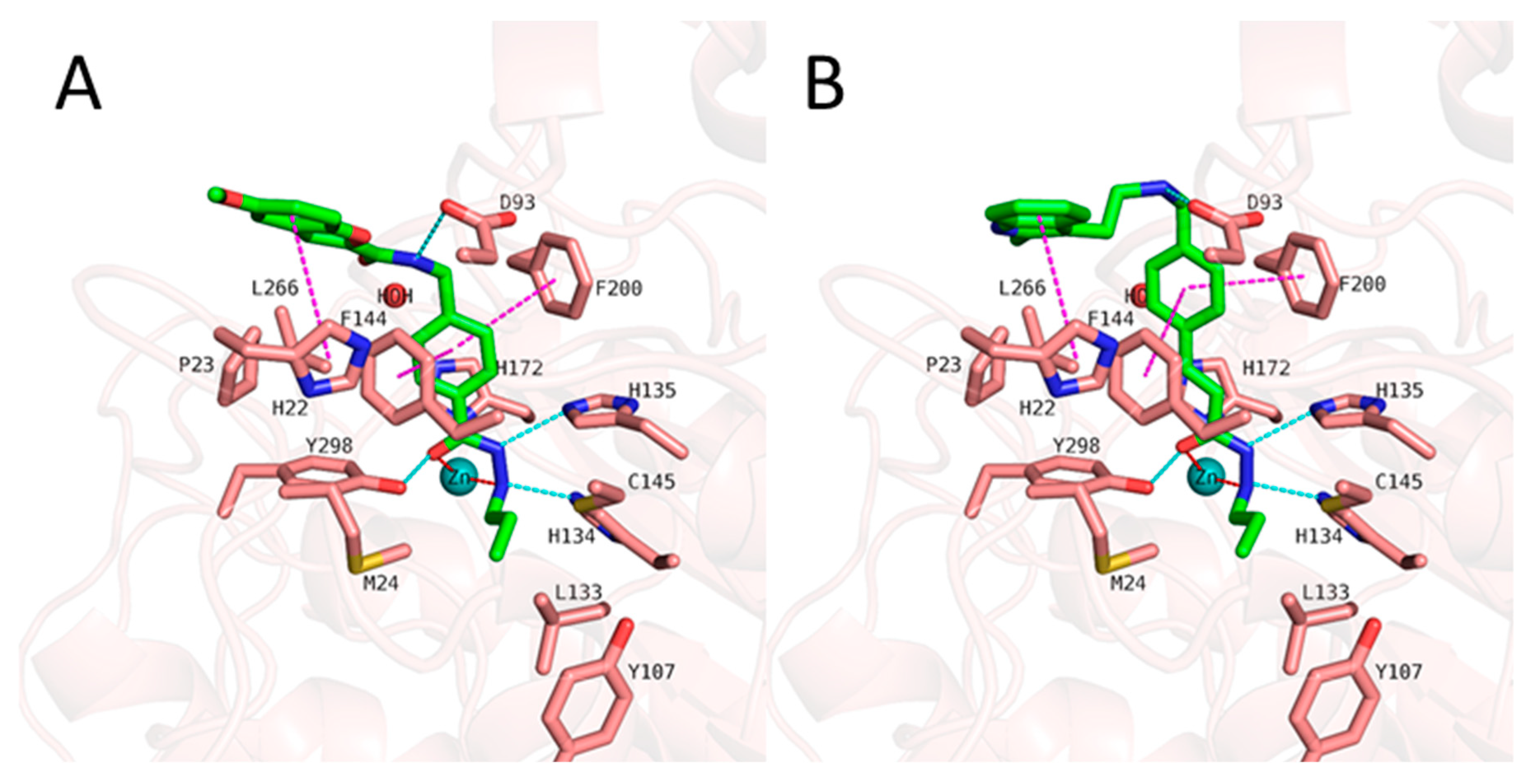
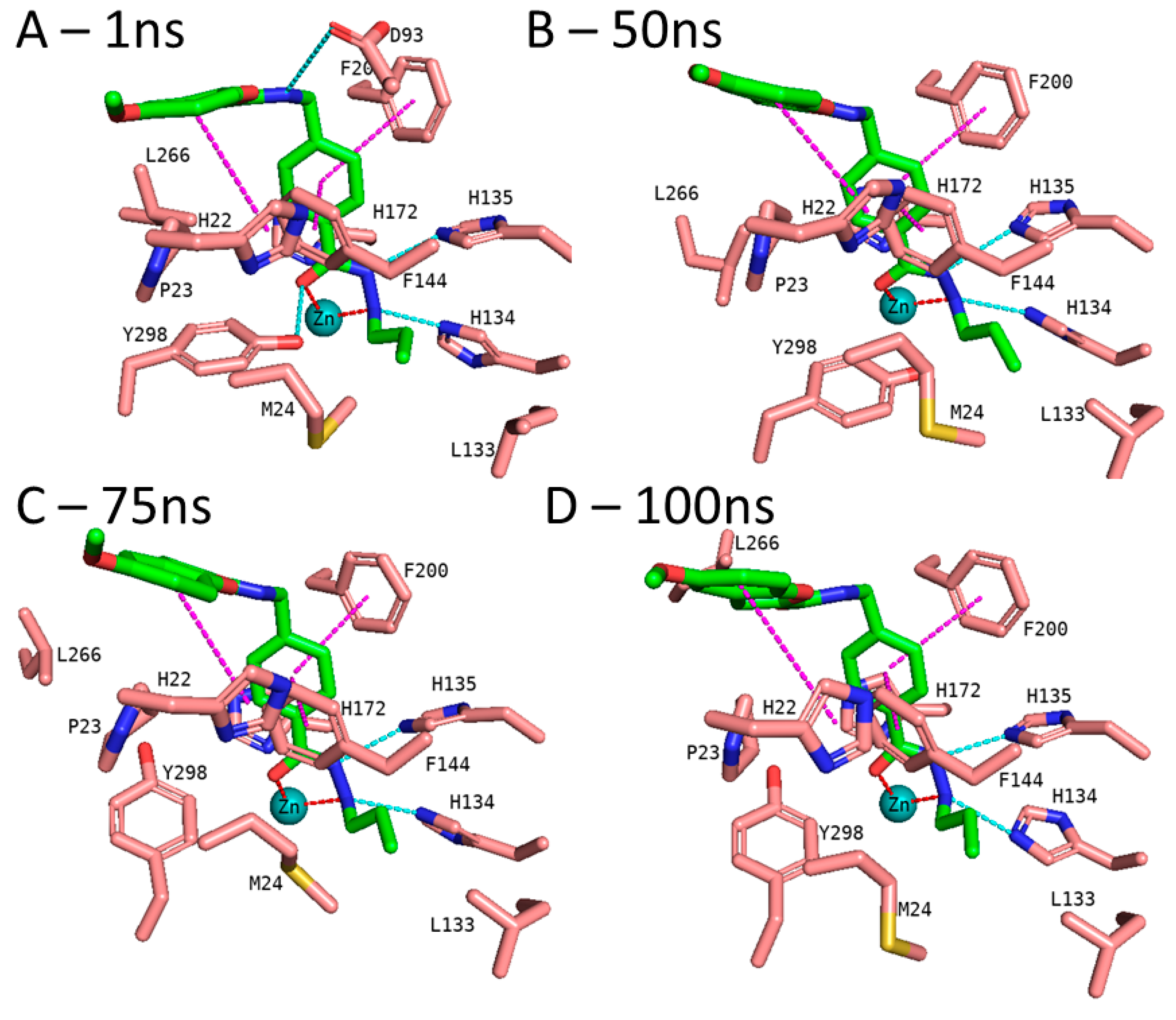
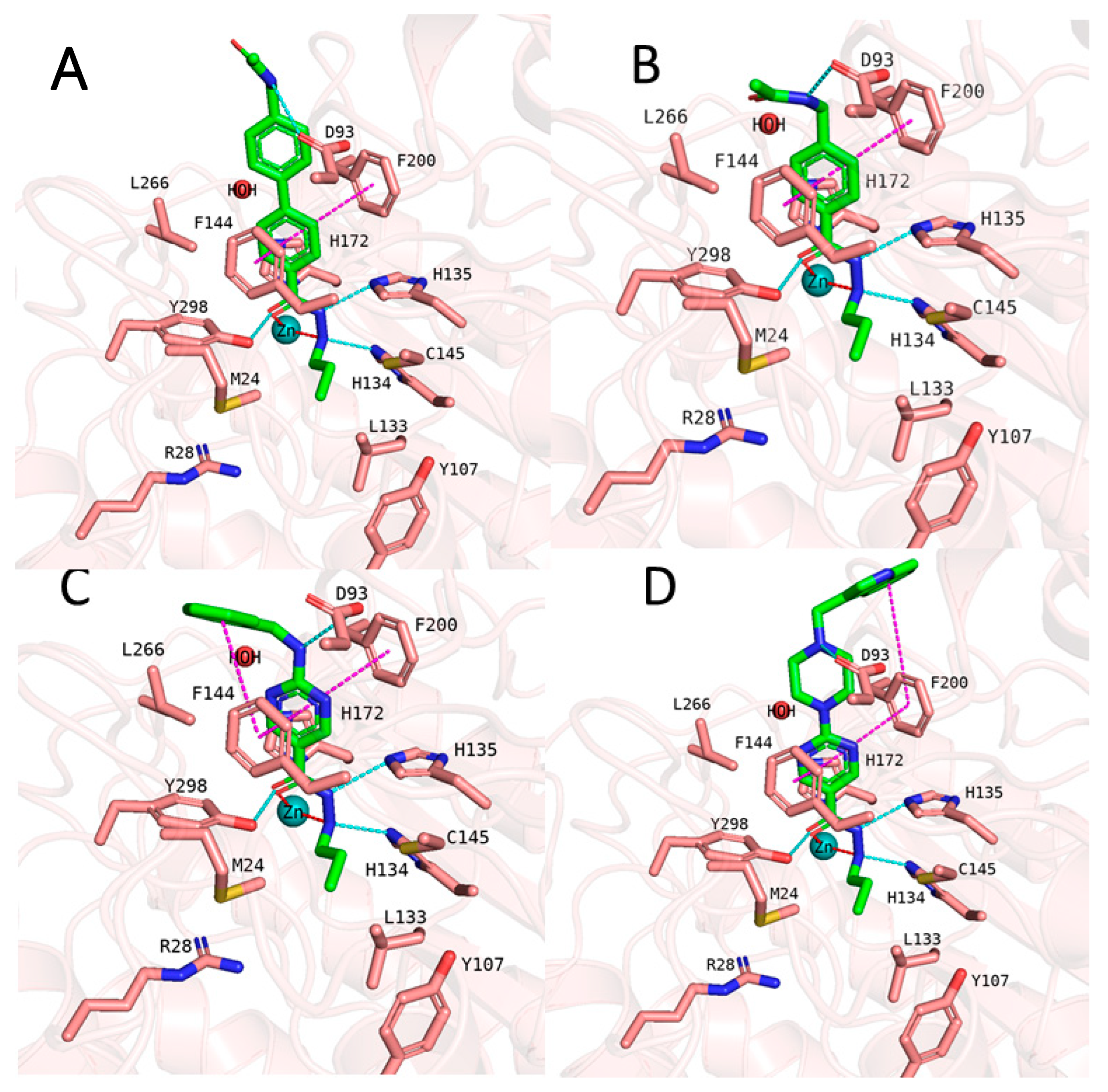
2.4. Analyzing the Binding Mode of Alkylhydrazides in HDAC3
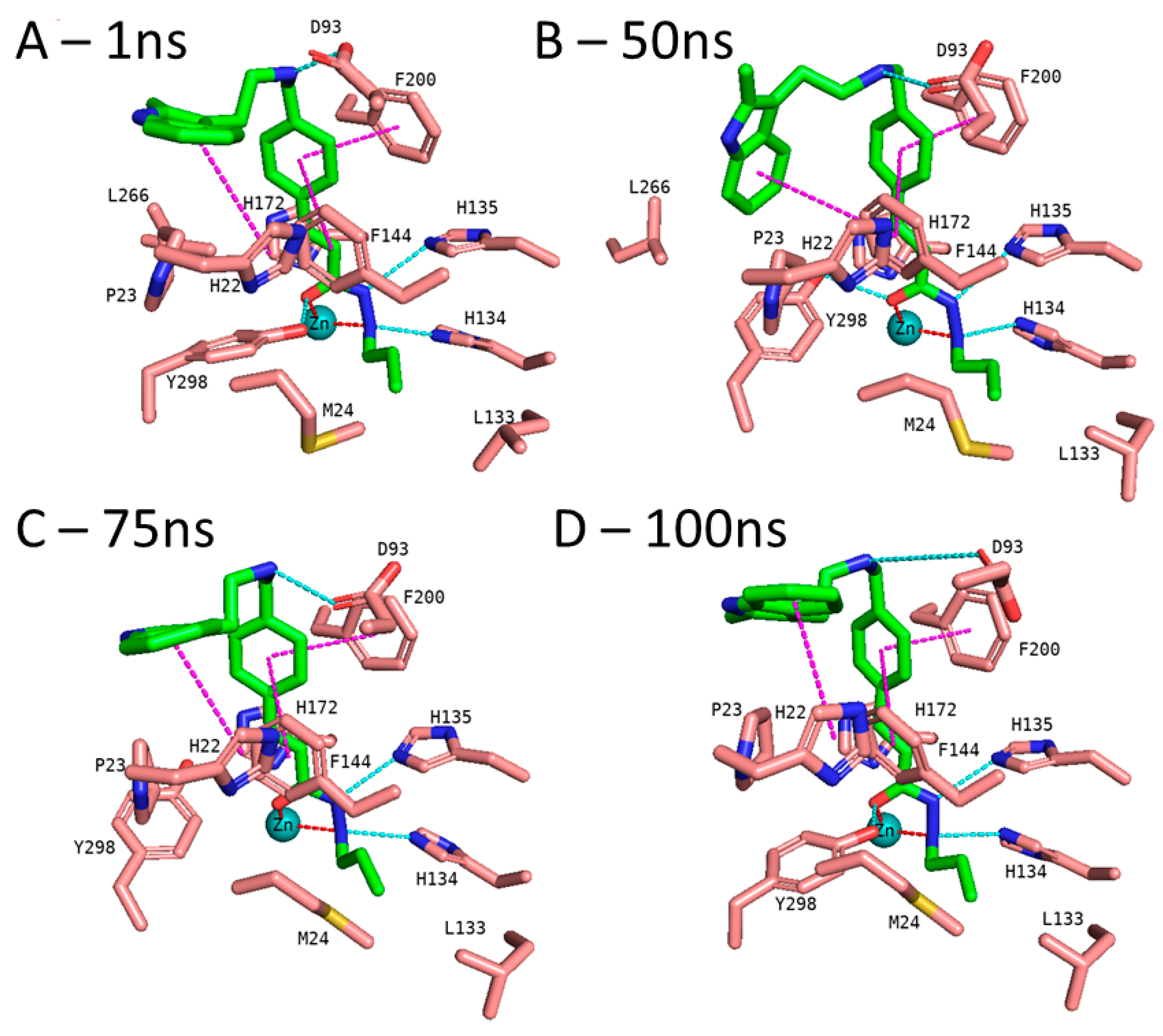
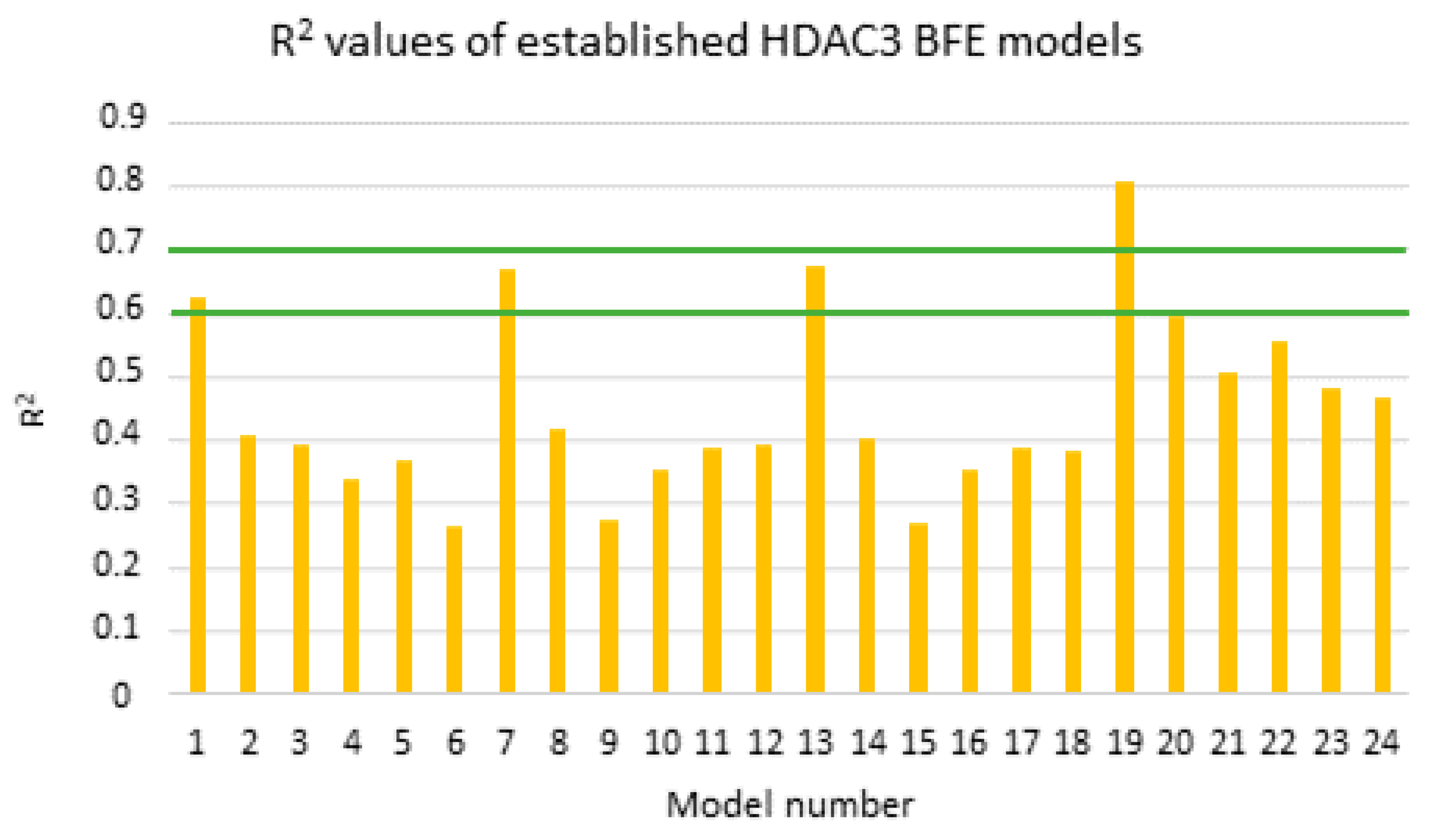
2.5. Binding Free Energy Calculation
2.6. Evaluation of the Generated Models on Newly Designed Compounds
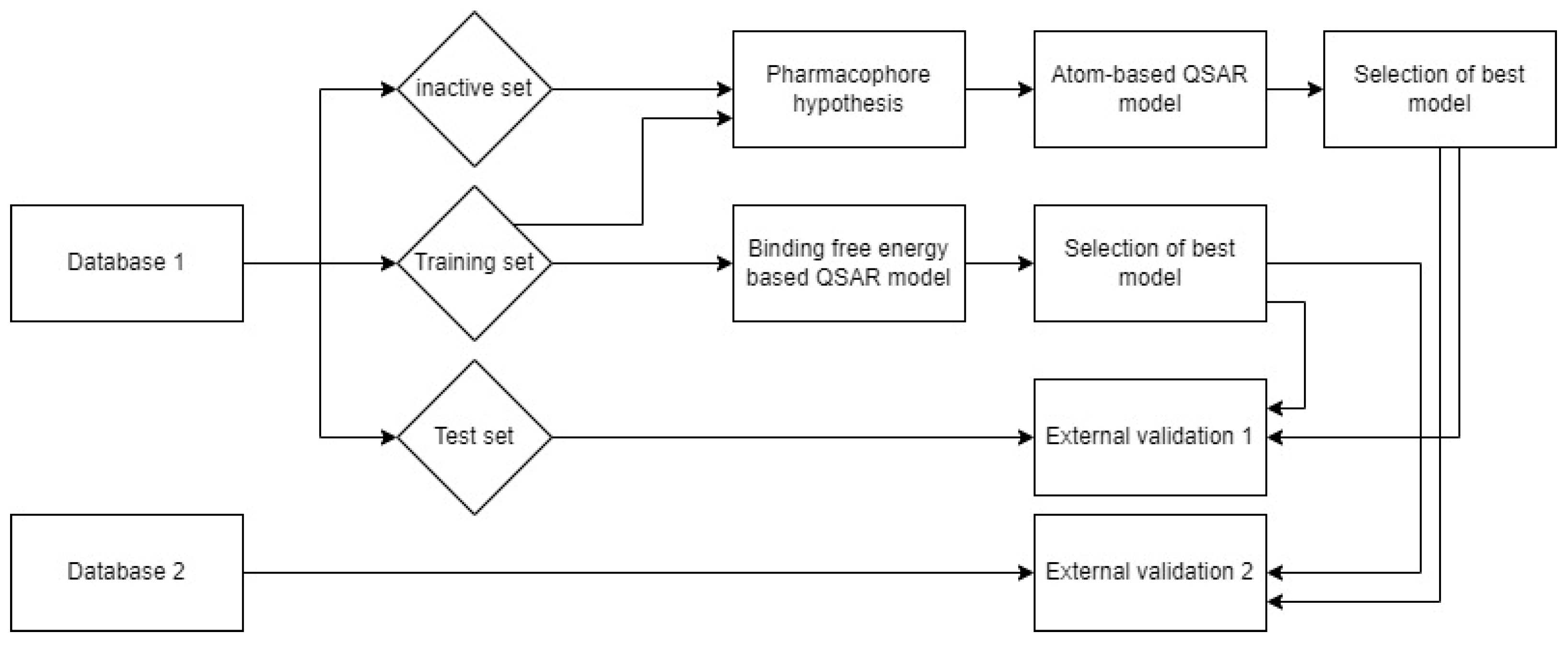
3. Materials and Methods
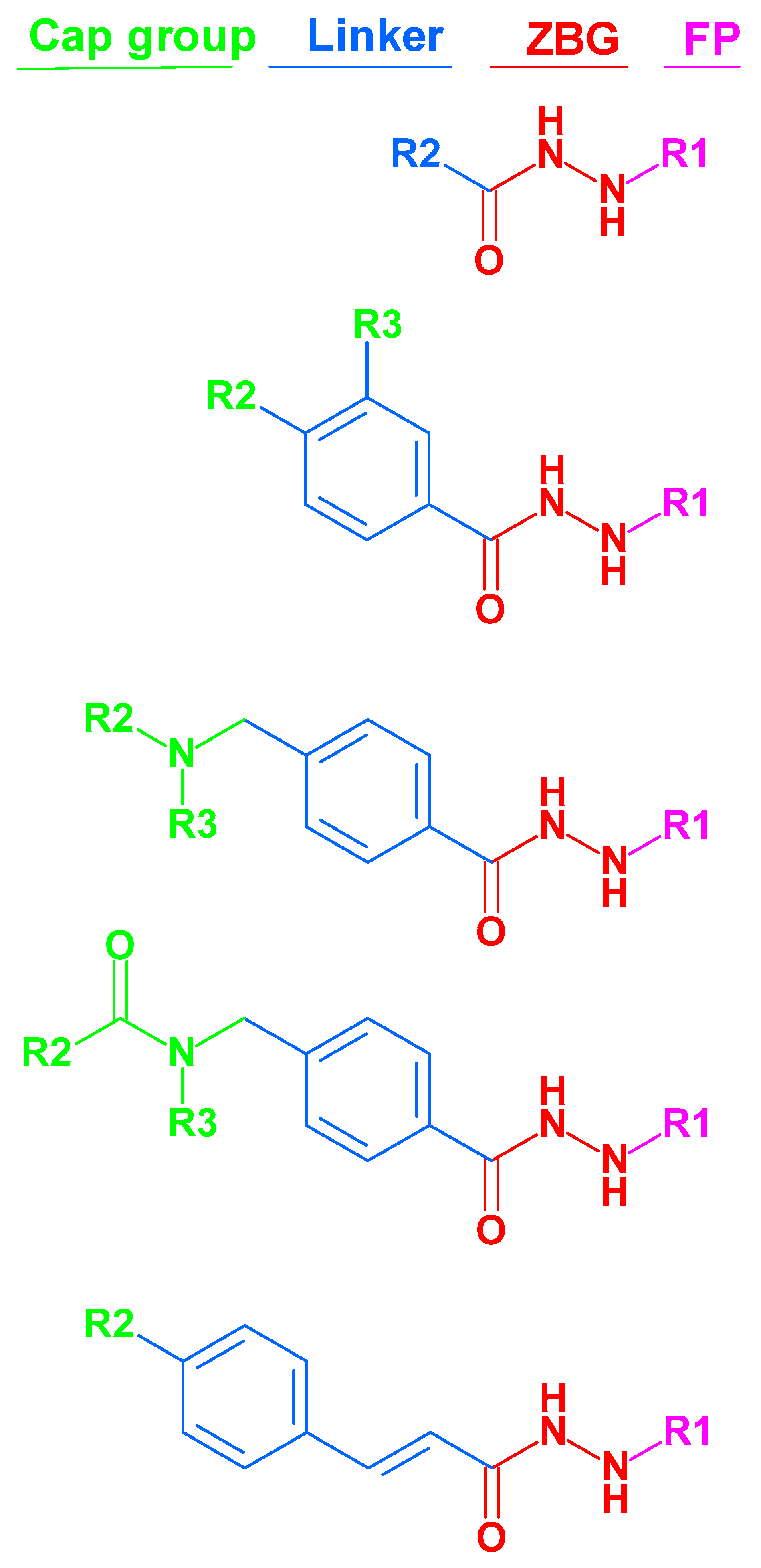
3.1. Ligand Database Preparation
3.2. Pharmacophore Model
3.3. Atom-Based 3D-QSAR Model
3.4. Docking Study
3.5. Molecular Dynamics Simulation
3.6. Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraczek, J.; van Grunsven, L.A.; Vinken, M.; Snykers, S.; Deleu, S.; Vanderkerken, K.; Vanhaecke, T.; Rogiers, V. Histone deacetylase inhibition and the regulation of cell growth with particular reference to liver pathobiology. J. Cell. Mol. Med. 2009, 13, 2990–3005. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.J.; Huang, C.; Meng, X.M.; Li, J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 2015, 116, 61–69. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bulbul, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. Chemmedchem 2021, 16, 1336–1359. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Hildmann, C.; Riester, D.; Schwienhorst, A. Histone deacetylases--an important class of cellular regulators with a variety of functions. Appl. Microbiol. Biotechnol. 2007, 75, 487–497. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Denslow, S.A.; Wade, P.A. The human Mi-2/NuRD complex and gene regulation. Oncogene 2007, 26, 5433–5438. [Google Scholar] [CrossRef] [Green Version]
- Grozinger, C.M.; Schreiber, S.L. Deacetylase enzymes: Biological functions and the use of small-molecule inhibitors. Chem. Biol. 2002, 9, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laherty, C.D.; Yang, W.M.; Sun, J.M.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone deacetylases associated with the mSin3 corepressor mediate Mad transcriptional repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, R.E.; Fairall, L.; Saleh, A.; Kelsall, E.; Morris, K.L.; Ragan, T.J.; Savva, C.G.; Chandru, A.; Millard, C.J.; Makarova, O.V.; et al. The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nat. Commun. 2020, 11, 3252. [Google Scholar] [CrossRef]
- Xue, Y.T.; Wong, J.M.; Moreno, G.T.; Young, M.K.; Cote, J.; Wang, W.D. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 1998, 2, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Wang, J.; Wang, J.X.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J.M. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [Green Version]
- Oberoi, J.; Fairall, L.; Watson, P.J.; Yang, J.C.; Czimmerer, Z.; Kampmann, T.; Goult, B.T.; Greenwood, J.A.; Gooch, J.T.; Kallenberger, B.C.; et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 2011, 18, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Hu, E.; Chen, Z.X.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.G.; Winkler, J. Cloning and characterization of a novel human. Class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Vahid, F.; Zand, H.; Nosrat-Mirshekarlou, E.; Najafi, R.; Hekmatdoost, A. The role dietary of bioactive compounds on the regulation of histone acetylases and deacetylases: A review. Gene 2015, 562, 8–15. [Google Scholar] [CrossRef]
- Chien, W.W.; Lee, D.H.; Zheng, Y.; Wuensche, P.; Alvarez, R.; Wen, D.L.; Aribi, A.M.; Thean, S.M.; Doan, N.B.; Said, J.W.; et al. Growth Inhibition of Pancreatic Cancer Cells by Histone Deacetylase Inhibitor Belinostat Through Suppression of Multiple Pathways Including HIF, NFkB, and mTOR Signaling In Vitro and In Vivo. Mol. Carcinogen. 2014, 53, 722–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, N.; Nakajima, I.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Mann, B.S.; Johnson, J.R.; He, K.; Sridhara, R.; Abraham, S.; Booth, B.P.; Verbois, L.; Morse, D.E.; Jee, J.M.; Pope, S.; et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. 2007, 13, 2318–2322. [Google Scholar] [CrossRef] [Green Version]
- Sivaraj, D.; Green, M.M.; Gasparetto, C. Panobinostat for the management of multiple myeloma. Future Oncol. 2017, 13, 477–488. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [Green Version]
- Hailu, G.S.; Robaa, D.; Forgione, M.; Sippl, W.; Rotili, D.; Mai, A. Lysine Deacetylase Inhibitors in Parasites: Past, Present, and Future Perspectives. J. Med. Chem. 2017, 60, 4780–4804. [Google Scholar] [CrossRef]
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, 780. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Brosch, G.; Kolle, D.; Scherf, H.; Gerhauser, C.; Loidl, P. Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J. Med. Chem. 1999, 42, 4669–4679. [Google Scholar] [CrossRef]
- Bulbul, E.F.; Melesina, J.; Ibrahim, H.S.; Abdelsalam, M.; Vecchio, A.; Robaa, D.; Zessin, M.; Schutkowski, M.; Sippl, W. Docking, Binding Free Energy Calculations and In Vitro Characterization of Pyrazine Linked 2-Aminobenzamides as Novel Class I Histone Deacetylase (HDAC) Inhibitors. Molecules 2022, 27, 2526. [Google Scholar] [CrossRef]
- Burli, R.W.; Luckhurst, C.A.; Aziz, O.; Matthews, K.L.; Yates, D.; Lyons, K.A.; Beconi, M.; McAllister, G.; Breccia, P.; Stott, A.J.; et al. Design, Synthesis, and Biological Evaluation of Potent and Selective Class IIa Histone Deacetylase (HDAC) Inhibitors as a Potential Therapy for Huntington’s Disease. J. Med. Chem. 2013, 56, 9934–9954. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-Based Design and Synthesis of Novel Inhibitors Targeting HDAC8 from Schistosoma mansoni for the Treatment of Schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, H.S.; Abdelsalam, M.; Zeyn, Y.; Zessin, M.; Mustafa, A.M.; Fischer, M.A.; Zeyen, P.; Sun, P.; Bulbul, E.F.; Vecchio, A.; et al. Synthesis, Molecular Docking and Biological Characterization of Pyrazine Linked 2-Aminobenzamides as New Class I Selective Histone Deacetylase (HDAC) Inhibitors with Anti-Leukemic Activity. Int. J. Mol. Sci. 2022, 23, 369. [Google Scholar] [CrossRef]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [Green Version]
- Luckhurst, C.A.; Breccia, P.; Stott, A.J.; Aziz, O.; Birch, H.L.; Burli, R.W.; Hughes, S.J.; Jarvis, R.E.; Lamers, M.; Leonard, P.M.; et al. Potent, Selective, and CNS-Penetrant Tetrasubstituted Cyclopropane Class Ila Histone Deacetylase (HDAC) Inhibitors. ACS Med. Chem. Lett. 2016, 7, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Marek, M.; Ramos-Morales, E.; Picchi-Constante, G.F.A.; Bayer, T.; Norstrom, C.; Herp, D.; Sales, P.A.; Guerra-Slompo, E.P.; Hausmann, K.; Chakrabarti, A.; et al. Species-selective targeting of pathogens revealed by the atypical structure and active site of Trypanosoma cruzi histone deacetylase DAC2. Cell Rep. 2021, 37, 110129. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef] [PubMed]
- Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules 2018, 23, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.F.; Wiest, O.; Helquist, P.; Lan-Hargest, H.Y.; Wiech, N.L. On the function of the 14 angstrom long internal cavity of histone deacetylase-like protein: Implications for the design of histone deacetylase inhibitors. J. Med. Chem. 2004, 47, 3409–3417. [Google Scholar] [CrossRef]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.Q.; Melkus, R.; De Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef]
- Liu, J.; Kelly, J.; Yu, W.S.; Clausen, D.; Yu, Y.N.; Kim, H.; Duffy, J.L.; Chung, C.C.; Myers, R.W.; Carroll, S.; et al. Selective Class I HDAC Inhibitors Based on Aryl Ketone Zinc Binding Induce HIV-1 Protein for Clearance. ACS Med. Chem. Lett. 2020, 11, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Weiwer, M.; Steinbacher, S.; Schomburg, A.; Reinemer, P.; Gale, J.P.; Campbell, A.J.; Fisher, S.L.; Zhao, W.N.; Reis, S.A.; et al. Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors. Bioorgan. Med. Chem. 2016, 24, 4008–4015. [Google Scholar] [CrossRef]
- Yu, W.S.; Liu, J.; Yu, Y.N.; Zhang, V.; Clausen, D.; Kelly, J.; Wolkenberg, S.; Beshore, D.; Duffy, J.L.; Chung, C.C.; et al. Discovery of ethyl ketone-based HDACs 1, 2, and 3 selective inhibitors for HIV latency reactivation. Bioorg. Med. Chem. Lett. 2020, 30, 127197. [Google Scholar] [CrossRef]
- Liu, J.; Yu, Y.N.; Kelly, J.; Sha, D.Y.; Alhassan, A.B.; Yu, W.S.; Maletic, M.M.; Duffy, J.L.; Klein, D.J.; Holloway, M.K.; et al. Discovery of Highly Selective and Potent HDAC3 Inhibitors Based on a 2-Substituted Benzamide Zinc Binding Group. ACS Med. Chem. Lett. 2020, 11, 2476–2483. [Google Scholar] [CrossRef]
- Wang, Y.F.; Stowe, R.L.; Pinello, C.E.; Tian, G.M.; Madoux, F.; Li, D.W.; Zhao, L.S.Y.; Li, J.L.; Wang, Y.R.; Wang, Y.; et al. Identification of Histone Deacetylase Inhibitors with Benzoylhydrazide Scaffold that Selectively Inhibit Class I Histone Deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.Q.; Xu, J.; Yue, K.R.; Huang, C.; Qin, M.T.; Chi, D.Y.; Yu, Q.X.; Zhu, Y.; Hou, X.H.; Xu, T.Q.; et al. Potent Hydrazide-Based HDAC Inhibitors with a Superior Pharmacokinetic Profile for Efficient Treatment of Acute Myeloid Leukemia In Vivo. J. Med. Chem. 2022, 65, 285–302. [Google Scholar] [CrossRef]
- Kozlov, M.V.; Konduktorov, K.A.; Shcherbakova, A.S.; Kochetkov, S.N. Synthesis of N′-propylhydrazide analogs of hydroxamic inhibitors of histone deacetylases (HDACs) and evaluation of their impact on activities of HDACs and replication of hepatitis C virus (HCV). Bioorg. Med. Chem. Lett. 2019, 29, 2369–2374. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Jiang, Y.Q.; Peterson, Y.K.; Xu, T.Q.; Himes, R.A.; Luo, X.; Yin, G.L.; Inks, E.S.; Dolloff, N.; Halene, S.; et al. Design of Hydrazide-Bearing HDACIs Based on Panobinostat and Their p53 and FLT3-ITD Dependency in Antileukemia Activity. J. Med. Chem. 2020, 63, 5501–5525. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Peterson, Y.K.; Inks, E.S.; Himes, R.A.; Li, J.Y.; Zhang, Y.J.; Kong, X.J.; Chou, C.J. Class I HDAC Inhibitors Display Different Antitumor Mechanism in Leukemia and Prostatic Cancer Cells Depending on Their p53 Status. J. Med. Chem. 2018, 61, 2589–2603. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.J.; Zhang, C.; Inks, E.S.; Peterson, Y.K.; Li, J.Y.; Chou, C.J. Development of Allosteric Hydrazide-Containing Class I Histone Deacetylase Inhibitors for Use in Acute Myeloid Leukemia. J. Med. Chem. 2016, 59, 9942–9959. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.F.; Wang, J.; Zhao, L.S.Y.; Chen, X.Y.; Zheng, G.R.; Zhang, X.; Liao, D.Q. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef]
- Sun, P.; Wang, J.; Khan, K.S.; Yang, W.; Ng, B.W.-L.; Ilment, N.; Zessin, M.; Bülbül, E.F.; Robaa, D.; Erdmann, F.; et al. Development of alkylated hydrazides as highly potent and selective class I HDAC inhibitors with T cell modulatory properties. J. Med. Chem. 2022, 65, 16313–16337. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Jha, T.; Ghosh, B. Dissecting Histone Deacetylase 3 in Multiple Disease Conditions: Selective Inhibition as a Promising Therapeutic Strategy. J. Med. Chem. 2021, 64, 8827–8869. [Google Scholar] [CrossRef]
- You, S.H.; Lim, H.W.; Sun, Z.; Broache, M.; Won, K.J.; Lazar, M.A. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 2013, 20, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, R.; Banerjee, S.; Amin, S.A.; Adhikari, N.; Jha, T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur. J. Med. Chem. 2020, 192, 112171. [Google Scholar] [CrossRef]
- Janczura, K.J.; Volmar, C.H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambert, G.; Brothers, S.P.; Wahlestedt, C. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E11148–E11157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.P.; Yu, X.H.; Chen, J.Z.; Hu, M.; Zhang, Y.K.; Lin, H.L.; Tang, W.Y.; He, P.P.; Ouyang, X.P. Histone Deacetylase 3: A Potential Therapeutic Target for Atherosclerosis. Aging Dis. 2022, 13, 773–786. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, W. Histone deacetylase 3 (HDAC3) as an important epigenetic regulator of kidney diseases. J. Mol. Med. 2022, 100, 43–51. [Google Scholar] [CrossRef]
- Carey, R.N.; Wold, S.; Westgard, J.O. Principal component analysis: An alternative to “referee” methods in method comparison studies. Anal. Chem. 1975, 47, 1824–1829. [Google Scholar] [CrossRef]
- Eichner, L.J.; Curtis, S.D.; Brun, S.N.; McGuire, C.K.; Gushterova, I.; Baumgart, J.T.; Trefts, E.; Ross, D.S.; Rymoff, T.J.; Shaw, R.J. HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non-small cell lung cancer. Sci. Adv. 2023, 9, eadd3243. [Google Scholar] [CrossRef]
- Jia, H.; Wang, Y.; Morris, C.D.; Jacques, V.; Gottesfeld, J.M.; Rusche, J.R.; Thomas, E.A. The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington’s Disease Mice. PLoS ONE 2016, 11, e0152498. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group (CCG). Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group (CCG): Montreal, QC, Canada, 2019. [Google Scholar]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A 2016, 374, 20150202. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC. Release 2019-1: Phase; Schrödinger LLC: New York, NY, USA, 2019. [Google Scholar]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput.-Aid. Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Okur, A.; Wickstrom, L.; Hornak, V.; Simmerling, C. Secondary structure bias in generalized Born solvent models: Comparison of conformational ensembles and free energy of solvent polarization from explicit and implicit solvation. J. Phys. Chem. B 2007, 111, 1846–1857. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC. Release 2019-1: Maestro, Protein Preparation Wizard, Prime, Epik, Ligprep, Confgen, Glide; Schrödinger LLC: New York, NY, USA, 2019. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.J.; Reboul, M.; Xiang, J.Y.; Wang, L.L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Li, P.F.; Song, L.F.; Merz, K.M. Parameterization of Highly Charged Metal Ions Using the 12-6-4 LJ-Type Nonbonded Model in Explicit Water. J. Phys. Chem. B 2015, 119, 883–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins-Struct. Funct. Bioinform. 2004, 55, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.M.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesan equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins-Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A.; Brooks, C.L. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Karaman, B.; Sippl, W. Docking and binding free energy calculations of sirtuin inhibitors. Eur. J. Med. Chem. 2015, 93, 584–598. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Dis. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDAC3 Dataset | Number of Compounds | 7 < pIC50 Highly Active | 5.3 < pIC50 < 7 Moderately Active | pIC50 < 5.3 Inactive |
|---|---|---|---|---|
| Training | 39 | 30 | 9 | - |
| Test | 17 | 11 | 6 | - |
| Inactive | 7 | - | - | 7 |
| Total | 63 | 41 | 15 | 7 |
| Abbreviations | Molecular Descriptors |
|---|---|
| PEOE_VSA_HYD | The partial equalization of orbital electronegativity (PEOE). Total hydrophobic van der Waals surface area |
| GCUT_SLOGP_0 | The GCUT descriptors using atomic contribution to logP |
| TPSA | Polar surface area |
| b_single | Number of single bonds (including implicit hydrogens). Aromatic bonds are not considered to be single bonds |
| lip_acc | The number of O and N atoms |
| lip_don | The number of OH and NH atoms |
| vsa_hyd | Approximation of the sum of VDW surface areas of hydrophobic atoms. |
| HYPO ID | Survival Score | Inactive Score |
|---|---|---|
| ADDDHRR | 6.923 | 1.688 |
| DDDHRR | 6.464 | 1.711 |
| DDHR | 5.405 | 2.069 |
| HDAC3 Model | N | SD | R2 | RMSE | Q2 |
|---|---|---|---|---|---|
| 1 | 39 | 0.27 | 0.95 | 0.39 | 0.88 |
| Compound Number | pIC50 HDAC3 | Prediction by Atom-Based QSAR | Difference (Experimental—Predicted Activity) Atom-Based | References |
|---|---|---|---|---|
| 40 | 9.29 | 8.69 | 0.60 | [47] |
| 41 | 9.24 | 8.87 | 0.37 | [47] |
| 42 | 9.21 | 9.14 | 0.07 | [47] |
| 43 | 7.90 | 8.43 | 0.53 | [49] |
| 44 | 7.78 | 7.13 | 0.64 | [49] |
| 45 | 7.46 | 7.17 | 0.29 | [49] |
| 46 | 7.31 | 7.65 | 0.34 | [49] |
| 47 | 7.17 | 6.92 | 0.24 | [51] |
| 48 | 7.16 | 7.54 | 0.38 | [51] |
| 49 | 7.11 | 7.96 | 0.85 | [49] |
| 50 | 7.07 | 7.81 | 0.74 | [51] |
| 51 | 6.73 | 7.88 | 1.16 | [51] |
| 52 | 6.51 | 7.32 | 0.81 | [51] |
| 53 | 5.96 | 7.85 | 1.89 | [51] |
| 54 | 5.87 | 6.19 | 0.33 | [51] |
| 55 | 5.81 | 8.08 | 2.26 | [51] |
| 56 | 5.72 | 6.74 | 1.01 | [51] |
| LOO CV | 3-Fold CV | 10-Fold CV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model Number | N | Method | Frame | 2D Descriptor | R2 | RMSE | Q2 | QMSE | Q2 | QMSE | Q2 | QMSE |
| MODEL1 | 39 | GB1 | Emin1 | - | 0.63 | 0.69 | 0.58 | 0.73 | 0.60 | 0.74 | 0.60 | 0.73 |
| MODEL7 | 39 | GB2 | Emin1 | - | 0.66 | 0.65 | 0.63 | 0.69 | 0.64 | 0.69 | 0.64 | 0.69 |
| MODEL13 | 39 | GB5 | Emin1 | - | 0.67 | 0.65 | 0.64 | 0.68 | 0.65 | 0.69 | 0.65 | 0.68 |
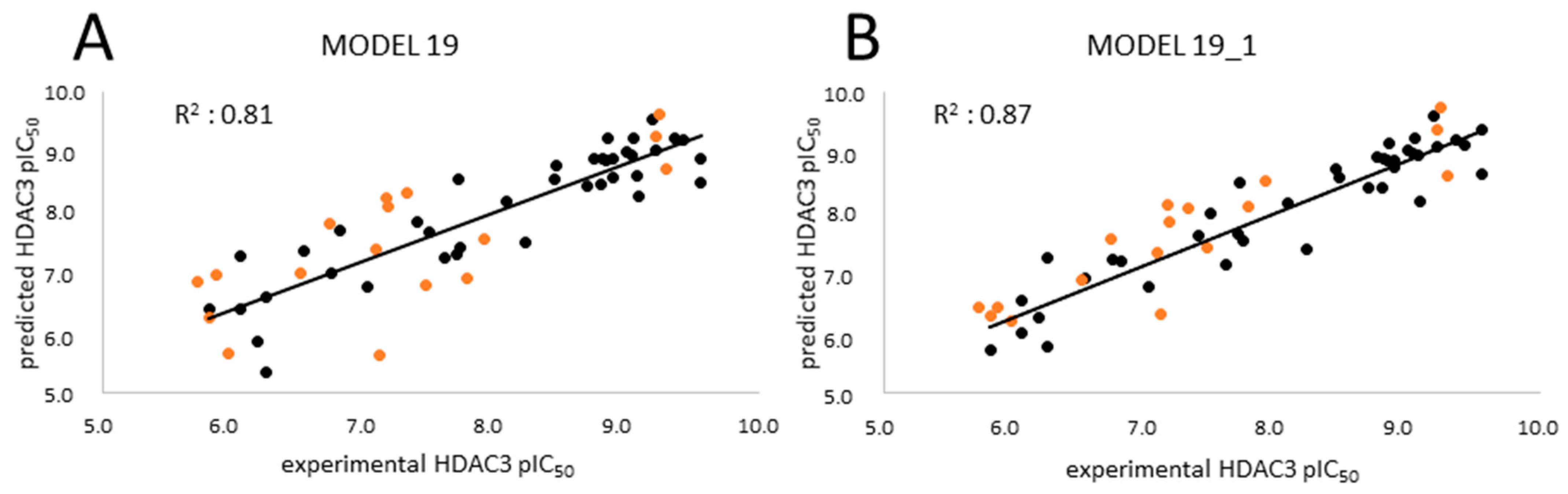
| MODEL19 | 39 | GB8 | Emin1 | - | 0.81 | 0.49 | 0.78 | 0.52 | 0.78 | 0.54 | 0.77 | 0.53 |
| MODEL19_1 | 39 | GB8 | Emin1 | PEOE_VSA_HYD | 0.87 | 0.40 | 0.84 | 0.44 | 0.85 | 0.45 | 0.83 | 0.45 |
| Compound Number | pIC50 HDAC3 | Prediction of BFE | Difference (Experimental—Predicted Activity) | References |
|---|---|---|---|---|
| 40 | 9.29 | 8.60 | 0.70 | [47] |
| 41 | 9.24 | 9.75 | 0.51 | [47] |
| 42 | 9.21 | 9.37 | 0.17 | [47] |
| 43 | 7.90 | 8.53 | 0.63 | [49] |
| 44 | 7.78 | 8.10 | 0.33 | [49] |
| 45 | 7.46 | 7.42 | 0.03 | [49] |
| 46 | 7.31 | 8.06 | 0.75 | [49] |
| 47 | 7.17 | 7.85 | 0.68 | [51] |
| 48 | 7.16 | 8.12 | 0.96 | [51] |
| 49 | 7.11 | 6.30 | 0.81 | [49] |
| 50 | 7.07 | 7.32 | 0.25 | [51] |
| 51 | 6.73 | 7.56 | 0.83 | [51] |
| 52 | 6.51 | 6.89 | 0.38 | [51] |
| 53 | 5.96 | 6.19 | 0.22 | [51] |
| 54 | 5.87 | 6.41 | 0.55 | [51] |
| 55 | 5.81 | 6.27 | 0.46 | [51] |
| 56 | 5.72 | 6.42 | 0.69 | [51] |
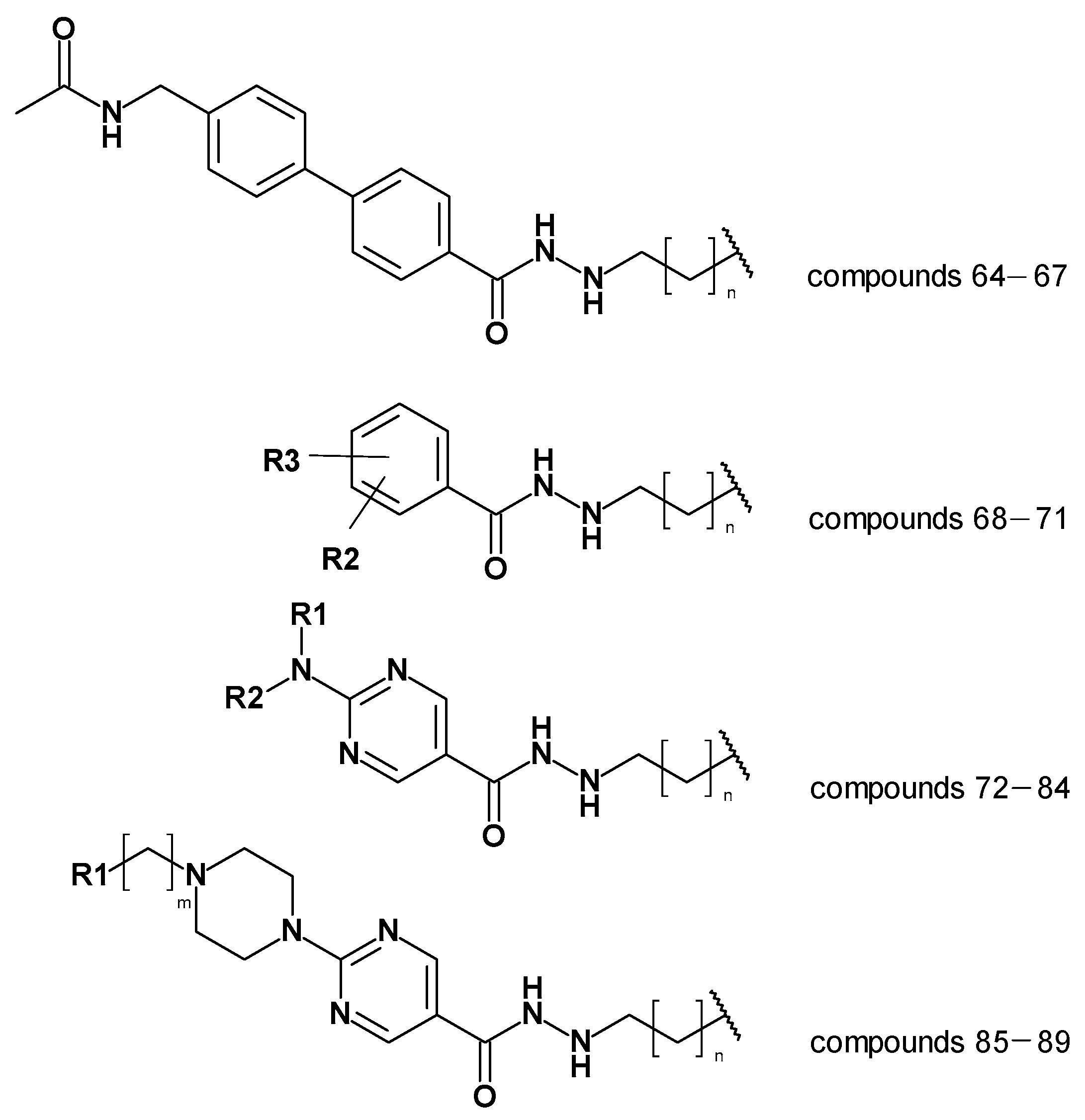
| Compound Number | pIC50 HDAC3 | Prediction by Atom-Based QSAR | Difference (Experimental—Predicted Activity) Atom-Based | Prediction by BFE Model | Difference (Experimental—Predicted Activity) BFE |
|---|---|---|---|---|---|
| 64 | 7.04 | 7.03 | 0.01 | 7.16 | −0.12 |
| 65 | 6.46 | 6.59 | −0.13 | 6.89 | −0.43 |
| 66 | 5.82 | 6.57 | −0.75 | 6.36 | −0.54 |
| 67 | <5.00 | 6.43 | <−1.43 | 5.41 | - |
| 68 | 5.80 | 7.25 | −1.46 | 6.69 | −0.89 |
| 69 | <5.00 | 7.05 | <−2.05 | 5.07 | - |
| 70 | <5.00 | 6.98 | <−1.98 | 3.92 | - |
| 71 | 9%@1 µM | 6.82 | - | 0.24 | - |
| 72 | 7.37 | 7.85 | −0.49 | 7.60 | −0.23 |
| 73 | 6.70 | 7.29 | −0.59 | 6.68 | 0.02 |
| 74 | 41%@1 µM | 7.39 | - | 7.29 | - |
| 75 | 7.09 | 7.82 | −0.73 | 8.06 | −0.97 |
| 76 | 7.22 | 7.12 | 0.10 | 6.86 | 0.36 |
| 77 | 7.43 | 7.96 | −0.53 | 7.06 | 0.38 |
| 78 | 7.24 | 7.03 | 0.20 | 7.98 | −0.75 |
| 79 | 6.92 | 6.80 | 0.12 | 6.32 | 0.60 |
| 80 | 6.96 | 8.10 | −1.14 | 7.42 | −0.46 |
| 81 | <5.00 | 6.54 | <−1.54 | 1.65 | - |
| 82 | <5.00 | 6.86 | <1.86 | 1.61 | - |
| 83 | 5.52 | 7.25 | −1.73 | 3.26 | 2.26 |
| 84 | <5.00 | 7.61 | <−1.67 | 2.69 | - |
| 85 | 7.52 | 7.63 | −0.10 | 7.64 | −0.11 |
| 86 | 7.00 | 7.18 | −0.18 | 7.29 | −0.29 |
| 87 | 6.52 | 6.52 | 0.00 | 6.86 | −0.33 |
| 88 | 6.00 | 7.40 | −1.40 | 6.13 | −0.13 |
| 89 | 5.85 | 7.13 | −1.28 | 6.11 | −0.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bülbül, E.F.; Robaa, D.; Sun, P.; Mahmoudi, F.; Melesina, J.; Zessin, M.; Schutkowski, M.; Sippl, W. Application of Ligand- and Structure-Based Prediction Models for the Design of Alkylhydrazide-Based HDAC3 Inhibitors as Novel Anti-Cancer Compounds. Pharmaceuticals 2023, 16, 968. https://doi.org/10.3390/ph16070968
Bülbül EF, Robaa D, Sun P, Mahmoudi F, Melesina J, Zessin M, Schutkowski M, Sippl W. Application of Ligand- and Structure-Based Prediction Models for the Design of Alkylhydrazide-Based HDAC3 Inhibitors as Novel Anti-Cancer Compounds. Pharmaceuticals. 2023; 16(7):968. https://doi.org/10.3390/ph16070968
Chicago/Turabian StyleBülbül, Emre F., Dina Robaa, Ping Sun, Fereshteh Mahmoudi, Jelena Melesina, Matthes Zessin, Mike Schutkowski, and Wolfgang Sippl. 2023. "Application of Ligand- and Structure-Based Prediction Models for the Design of Alkylhydrazide-Based HDAC3 Inhibitors as Novel Anti-Cancer Compounds" Pharmaceuticals 16, no. 7: 968. https://doi.org/10.3390/ph16070968
APA StyleBülbül, E. F., Robaa, D., Sun, P., Mahmoudi, F., Melesina, J., Zessin, M., Schutkowski, M., & Sippl, W. (2023). Application of Ligand- and Structure-Based Prediction Models for the Design of Alkylhydrazide-Based HDAC3 Inhibitors as Novel Anti-Cancer Compounds. Pharmaceuticals, 16(7), 968. https://doi.org/10.3390/ph16070968