
Research in the Field of Drug Design and Development

 , , and
, , and 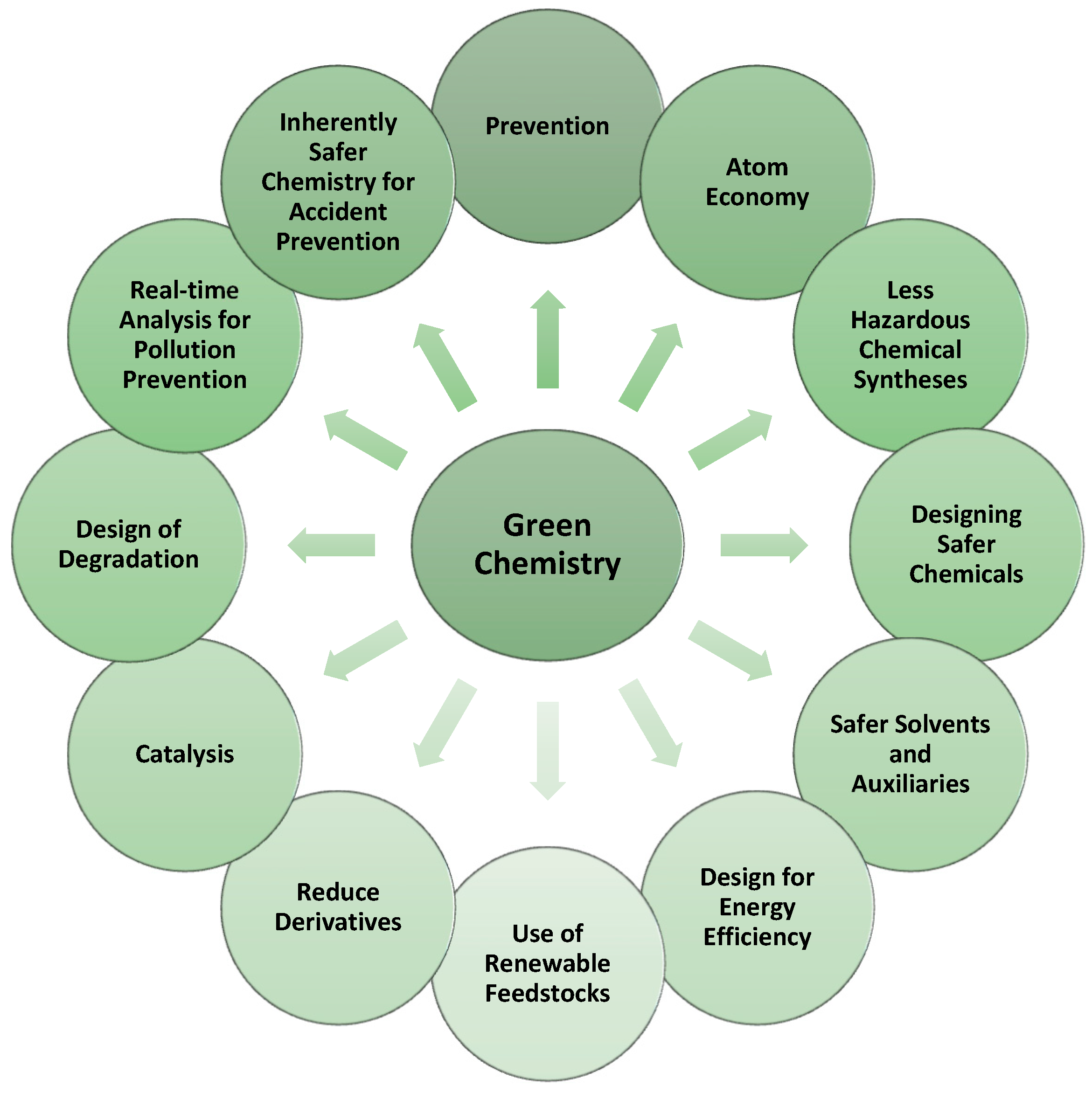{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Principles of Green Chemistry—A New Approach to the Synthesis of Drugs
3. Brief Insight into Development and Optimization in Drug Discovery, and Fundamental Roles and Importance of Computer-Aided Drug Design in This Process
- (a)
- rapidly cross biological membranes;
- (b)
- gain access to the relevant intracellular biological targets (the DNA or RNA of proteins, for example).
- (a)
- peptidomimetics design (de novo design, peptide-driven pharmacophoric method, geometry-similarity method, sequence-based method, fragment-based method, hybrid peptide-driven shape, and pharmacophoric method);
- (b)
- peptide design (ligand-based design, target-based design, and de novo design);
- (c)
- the designing of therapeutic proteins (template-based design and de novo design).
- (a)
- the target protein ligand (warhead)—the ligand (structural scaffold of a molecule of natural origin or a synthetic compound) targets proteins of interest (POIs); i.e., several nuclear receptors, various protein kinases, proteins involved in transcriptional regulation, neurodegenerative-related proteins, or fusion proteins;
- (b)
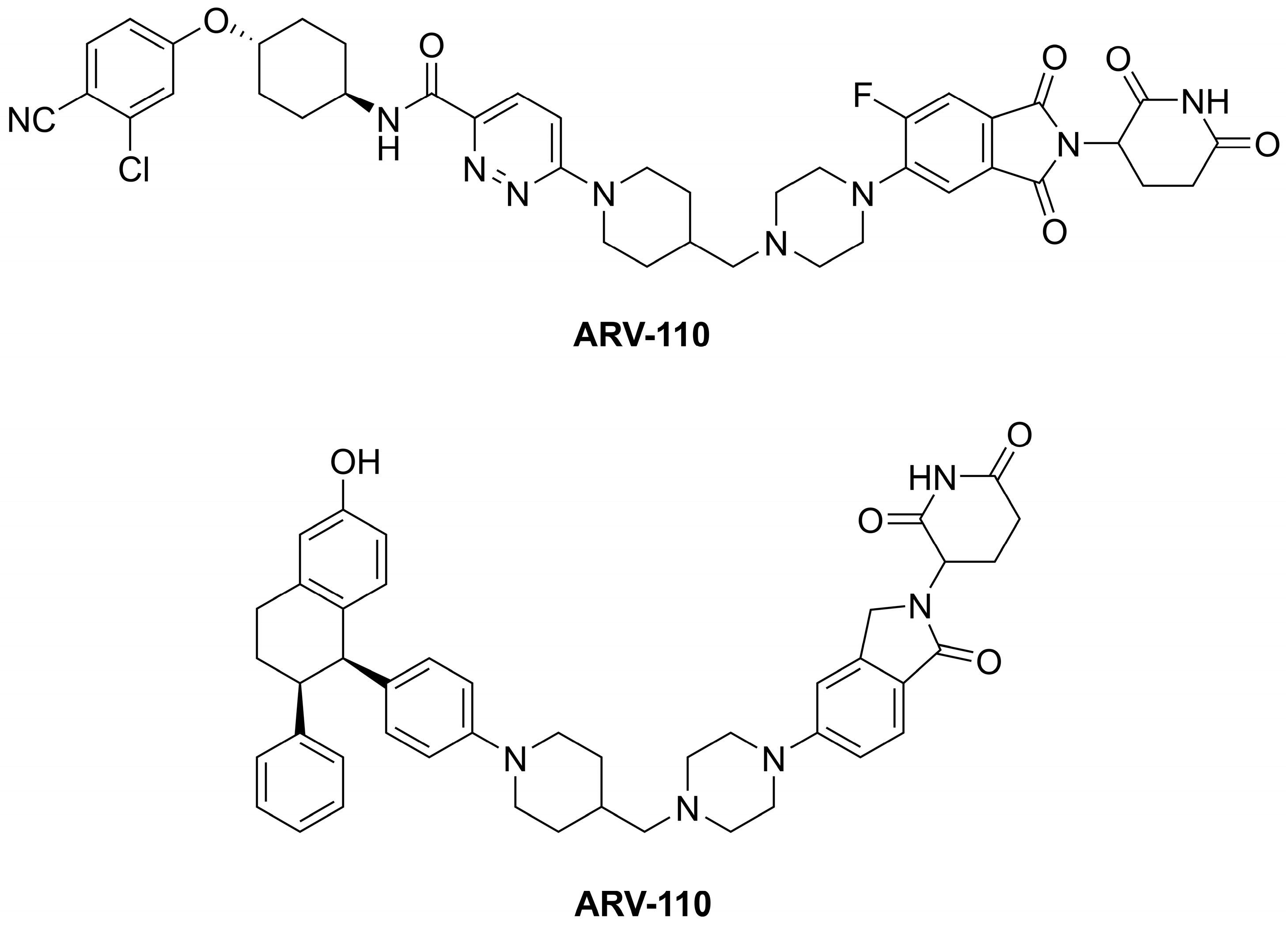
- the E3 ubiquitin ligase ligand (E3-binder)—the ligand can be a structural scaffold of lenalidomide, thalidomide, or pomalidomide (so-called LTP agents), for example. These binders target E3 ubiquitin ligases, such as the Von Hippel–Lindau or cereblon (CRBN);
- (c)
- a linker of varying size connecting the warhead and E3-binder—the linker contains a so-called anchor point influencing the length and steric properties (spatial arrangement) of a PROTAC therapeutic.
- (a)
- structural and physicochemical properties—MW, lipophilicity defined via a logarithm of a partition coefficient value estimated/calculated for an octan-1-ol/water partition system (log P), effective lipophilicity, acid–base properties (acid–base dissociation constant; pKa), size, flexibility (number of aromatic and non-aromatic cyclic systems, number of double and triple bonds, number of rotatable bonds (nrotb)), number of carbon atoms, fraction of sp3 carbon atoms (number of sp3 hybridized carbons/total carbon count; Fsp3), distribution of electrons, polar surface area value (PSA; expressed in Å2 units), number of hydrogen bond donors (nOHNH) and acceptors (nON), shape and stereochemical characteristics, reactivity, number of so-called heavy atoms, presence and number/absence of stereogenic centers, solubility, permeability, or chemical stability;
- (b)
- biochemical properties—biotransformation, affinity to proteins, tissue binding, transport properties (connected with PSA);
- (c)
- pharmacodynamics—the proper characteristics of the pharmacophore; the pharmacodynamic profile of a drug is notably influenced by structural and physicochemical properties, as well as its ADMET;
- (d)
- pharmacokinetics and toxicity (ADMET) features—biological availability, drug–drug interactions, half-life, lethal dose values, proper characteristics of the toxicophore (the qualitative structural feature of a drug that is assumed to be primarily responsible for its toxic properties).
- (a)
- their chemical modifications—particular changes are made according to empirical processes (PEGylation, glycosylation, the use of adjuvant molecules to enhance delivery and lipidization), as well as outputs from CADD (the optimization of aqueous solubility, for example);
- (b)
- their modifications in formulation design—various permeation or absorption enhancers are used, for example;
- (c)
- the modulation of a pH value of the environment;
- (d)
- the direct inhibition of the enzymes responsible for therapeutics’ degradation (cleavage of peptide bond(s)).
4. Several Innovations within In Vitro Screening Approaches for Drug Candidates or Drugs
5. Pre-Clinical In Vitro and In Vivo Studies
5.1. Rodent Experiments, the 3R Rule
- replacement,
- reduction,
- refinement.
5.2. Zebrafish Model
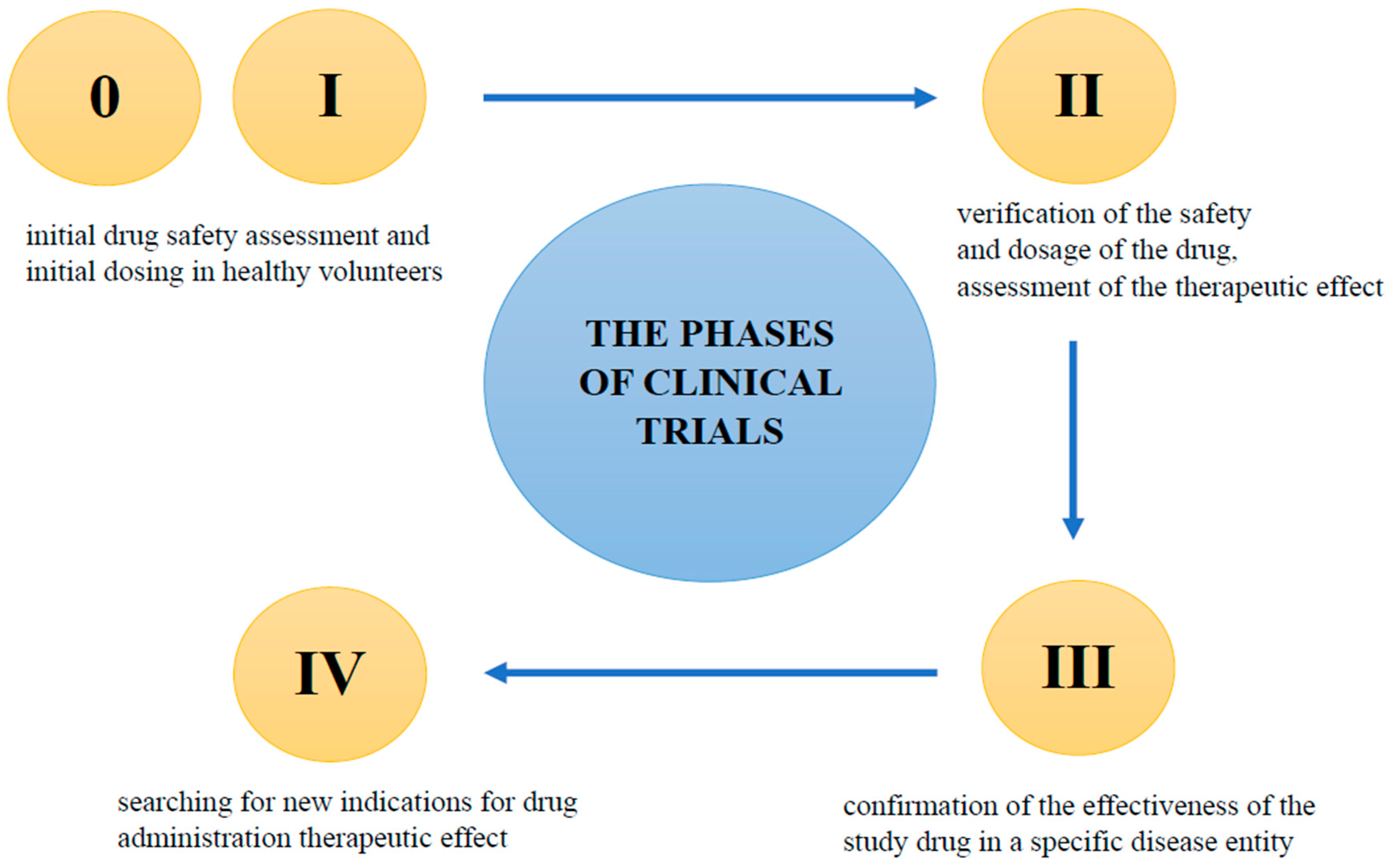
6. Clinical Trials—General Information
- Phase 0
- Phase I
- Phase II
- Phase III
- Phase IV
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doytchinova, I. Drug design–past, present, future. Molecules 2022, 27, 1496. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Fogel, D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp. Clin. Trials Commun. 2018, 7, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Smith, A. Screening for drug discovery: The leading question. Nature 2002, 418, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.T.; Warner, J.C. Principles of green chemistry. In Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998; Volume 29. [Google Scholar]
- O’Neil, N.J.; Scott, S.; Relph, R.; Ponnusamy, E. Approaches to incorporating green chemistry and safety into laboratory culture. J. Chem. Educ. 2020, 98, 84–91. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Sheldon, R.A. Fundamentals of green chemistry: Efficiency in reaction design. Chem. Soc. Rev. 2012, 41, 1437–1451. [Google Scholar] [CrossRef]
- Sheldon, R.A. E Factors, green chemistry and catalysis: An odyssey. Chem. Commun. 2008, 3352–3365. [Google Scholar] [CrossRef]
- Holbach, M.; Weck, M. Modular approach for the development of supported, monofunctionalized, salen catalysts. J. Org. Chem. 2006, 71, 1825–1836. [Google Scholar] [CrossRef]
- Madhavan, N.; Jones, C.W.; Weck, M. Rational approach to polymer-supported catalysts: Synergy between catalytic reaction mechanism and polymer design. Acc. Chem. Res. 2008, 41, 1153–1165. [Google Scholar] [CrossRef]
- Gates, B.C. Supported metal clusters: Synthesis, structure, and catalysis. Chem. Rev. 1995, 95, 511–522. [Google Scholar] [CrossRef]
- Copéret, C.; Chabanas, M.; Saint-Arroman, R.P.; Basset, J.M. Homogeneous and heterogeneous catalysis: Bridging the gap through surface organometallic chemistry. Angew. Chem. Int. Ed. 2003, 42, 156–181. [Google Scholar] [CrossRef] [PubMed]
- Samantaray, M.K.; Pump, E.; Bendjeriou-Sedjerari, A.; D’Elia, V.; Pelletier, J.D.A.; Guidotti, M.; Psaro, R.; Basset, J.M. Surface organometallic chemistry in heterogeneous catalysis. Chem. Soc. Rev. 2018, 47, 8403–8437. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lian, X.; Fang, Y.; Zhou, H.C. Applications of immobilized bio-catalyst in metal-organic frameworks. Catalysts 2018, 8, 166. [Google Scholar] [CrossRef]
- Fraile, J.M.; García, J.I.; Mayoral, J.A.; Fraile, J.M.; Garcia, J.I.; Mayoral, J.A. Noncovalent immobilization of enantioselective catalysts. Chem. Rev. 2009, 109, 360–417. [Google Scholar] [CrossRef]
- Sabater, S.; Mata, J.A.; Peris, E. Catalyst enhancement and recyclability by immobilization of metal complexes onto graphene surface by noncovalent interactions. ACS Catal. 2014, 4, 2038–2047. [Google Scholar] [CrossRef]
- Sánchez-López, P.; Kotolevich, Y.; Yocupicio-Gaxiola, R.I.; Antúnez-García, J.; Chowdari, R.K.; Petranovskii, V.; Fuentes-Moyado, S. Recent advances in catalysis based on transition metals supported on zeolites. Front. Chem. 2021, 9, 716745. [Google Scholar] [CrossRef]
- Federsel, H.J.; Moody, T.S.; Taylor, S.J.C. Recent trends in enzyme immobilization—Concepts for expanding the biocatalysis toolbox. Molecules 2021, 26, 2822. [Google Scholar] [CrossRef]
- Klein, W.P.; Thomsen, R.P.; Turner, K.B.; Walper, S.A.; Vranish, J.; Kjems, J.; Ancona, M.G.; Medintz, I.L. Enhanced catalysis from multienzyme cascades assembled on a DNA origami triangle. ACS Nano 2019, 13, 13677–13689. [Google Scholar] [CrossRef]
- Girelli, A.M.; Astolfi, M.L.; Scuto, F.R. Agro-industrial wastes as potential carriers for enzyme immobilization: A review. Chemosphere 2020, 244, 125368. [Google Scholar] [CrossRef]
- Chandrasekhar, S.A.T.H.Y.; Satyanarayana, K.G.; Pramada, P.N.; Raghavan, P.; Gupta, T.N. Review processing, properties and applications of reactive silica from rice huskdan overview. J. Mater. Sci. 2003, 38, 3159–3168. [Google Scholar] [CrossRef]
- Girelli, A.M.; Scuto, F.R. Eggshell membrane as feedstock in enzyme immobilization. J. Biotechnol. 2021, 10, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Kessi, E.; Arias, J.L. Using natural waste material as a matrix for the immobilization of enzymes: Chicken eggshell membrane powder for β-galactosidase immobilization. Appl. Biochem. Biotechnol. 2019, 187, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Budžaki, S.; Velić, N.; Ostojčić, M.; Stjepanović, M.; Rajs, B.B.; Šereš, Z.; Maravić, N.; Stanojev, J.; Hessel, V.; Strelec, I. Waste management in the agri-food industry: The conversion of eggshells, spent coffee grounds, and brown onion skins into carriers for lipase immobilization. Foods 2022, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Brígida, A.I.; Pinheiro, A.D.; Ferreira, A.L.; Gonçalves, L.R. Immobilization of Candida antarctica lipase B by adsorption to green coconut fiber. Appl. Biochem. Biotechnol. 2008, 146, 173–187. [Google Scholar] [CrossRef]
- Costa-Silva, T.A.; Souza, C.R.F.; Said, S.; Oliveira, W.P. Drying of enzyme immobilized on eco-friendly supports. Afr. J. Biotechnol. 2015, 14, 3019–3026. [Google Scholar] [CrossRef]
- Nájera-Martínez, E.F.; Melchor-Martínez, E.M.; Sosa-Hernández, J.E.; Levin, L.N.; Parra-Saldívar, R.; Iqbal, H.M.N. Lignocellulosic residues as supports for enzyme immobilization, and biocatalysts with potential applications. Int. J. Biol. Macromol. 2022, 208, 748–759. [Google Scholar] [CrossRef]
- Bassan, J.C.; de Souza Bezerra, T.M.; Peixoto, G.; da Cruz, C.Z.P.; Galan, J.P.M.; Vaz, A.B.D.S.; Garrido, S.S.; Filice, M.; Monti, R. Immobilization of trypsin in lignocellulosic waste material to produce peptides with bioactive potential from whey protein. Materials 2016, 9, 357. [Google Scholar] [CrossRef]
- Nuraliyah, A.; Wijanarko, A.; Hermansyah, H. Immobilization of Candida rugosa lipase by adsorption-crosslinking onto corn husk. IOP Conf. Ser. Mater. Sci. Eng. 2018, 345, 012042. [Google Scholar] [CrossRef]
- Ittrat, P.; Chacho, T.; Pholprayoon, J.; Suttiwarayanon, N.; Charoenpanich, J. Application of agriculture waste as a support for lipase immobilization. Biocatal. Agric. Biotechnol. 2014, 3, 77–82. [Google Scholar] [CrossRef]
- Kumari, M.; Chattopadhyay, S. The evaluation of the performance of rice husk and rice straw as potential matrix to obtain the best lipase immobilized system: Creating wealth from wastes. Prep. Biochem. Biotechnol. 2022, 53, 763–772. [Google Scholar] [CrossRef]
- Chen, K.I.; Lo, Y.C.; Liu, C.W.; Yu, R.C.; Chou, C.C.; Cheng, K.C. Enrichment of two isoflavone aglycones in black soymilk by using spent coffee grounds as an immobiliser for β-glucosidase. Food Chem. 2013, 139, 79–85. [Google Scholar] [CrossRef]
- Buntic, A.V.; Pavlovic, M.D.; Antonovic, D.G.; Siler-Marinkovic, S.S.; Dimitrijevic-Brankovic, S.I. Utilization of spent coffee grounds for isolation and stabilization of Paenibacillus chitinolyticus CKS1 cellulase by immobilization. Heliyon 2016, 2, e00146. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.; Gonçalves, M.P.; Teixeira, J.A. Immobilization of trypsin on spent grains for whey protein hydrolysis. Process Biochem. 2011, 46, 505–511. [Google Scholar] [CrossRef]
- Ye, N.; Kou, X.; Shen, J.; Huang, S.; Chen, G.; Ouyang, G. Metal-organic frameworks: A new platform for enzyme immobilization. ChemBioChem 2020, 21, 2585–2590. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Anand, B.; Tsang, Y.F.; Kim, K.-H.; Khullar, S.; Wang, B. Regeneration, degradation, and toxicity effect of MOFs: Opportunities and challenges. Environ. Res. 2019, 176, 108488. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Zhao, Y.; Rasheed, T.; Iqbal, H.M. Magnetic nanoparticles as versatile carriers for enzymes immobilization: A review. Int. J. Biol. Macromol. 2018, 120, 2530–2544. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Cui, L.; Jia, S.; Su, Z.; Zhang, S. Hybrid cross-linked lipase aggregates with magnetic nanoparticles: A robust and recyclable biocatalysis for the epoxidation of oleic acid. J. Agric. Food Chem. 2016, 64, 7179–7187. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yu, H.; Zhou, L.; He, Y.; Ma, L.; Jiang, Y. Formation of cross-linked nitrile hydratase aggregates in the pores of tannic acid-templated magnetic mesoporous silica: Characterization and catalytic application. Biochem. Eng. J. 2017, 117, 92–101. [Google Scholar] [CrossRef]
- Cai, L.; Gao, Y.; Chu, Y.; Lin, Y.; Liu, L.; Zhang, G. Green synthesis of silica-coated magnetic nanocarriers for simultaneous purification-immobilization of β-1,3-xylanase. Int. J. Biol. Macromol. 2023, 233, 123223. [Google Scholar] [CrossRef]
- Hu, F.; Deng, C.; Zhang, X. Development of high performance liquid chromatography with immobilized enzyme onto magnetic nanospheres for screening enzyme inhibitor. J. Chromatogr. B 2008, 87, 67–71. [Google Scholar] [CrossRef]
- Liu, D.M.; Chen, J.; Shi, Y.P. Screening of enzyme inhibitors from traditional Chinese medicine by magnetic immobilized α-glucosidase coupled with capillary electrophoresis. Talanta 2017, 164, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Veale, C.G.V. Into the fray! A beginner’s guide to medicinal chemistry. ChemMedChem 2021, 16, 199–1225. [Google Scholar] [CrossRef]
- Das, B.; Baidya, A.T.K.; Mathew, A.T.; Kumar Yadav, A.; Kumar, R. Structural modification aimed for improving solubility of lead compounds in early phase drug discovery. Bioorg. Med. Chem. 2022, 56, 116614. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; He, M.; Wang, L.; He, Y.; Rao, Y. Chemistries of bifunctional PROTAC degraders. Chem. Soc. Rev. 2022, 51, 7066–7114. [Google Scholar] [CrossRef]
- Jayashree, B.S.; Nikhil, P.S.; Paul, S. Bioisosterism in drug discovery and development—An overview. Med. Chem. 2022, 18, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, J.K.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Adelusi, T.I.; Kehinde Oyedele, A.-Q.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Hasan, M.R.; Alsaiari, A.A.; Fakhurji, B.Z.; Molla, M.H.R.M.; Asseri, A.H.; Sumon, M.A.A.; Park, M.N.; Ahammad, F.; Kim, B. Application of mathematical modeling and computational tools in the modern drug design and development process. Molecules 2022, 27, 4169. [Google Scholar] [CrossRef] [PubMed]
- Belhassan, A.; Chtita, S.; Zaki, H.; Alaqarbeh, M.; Alsakhen, N.; Almohtaseb, F.; Lakhlifi, T.; Bouachrine, M. In silico detection of potential inhibitors from vitamins and their derivatives compounds against SARS-CoV-2 main protease by using molecular docking, molecular dynamic simulation and ADMET profiling. J. Mol. Struct. 2022, 1258, 132652. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Barale, S.S.; Parulekar, R.S.; Fandilolu, P.M.; Dhanavade, M.J.; Sonawane, K.D. Molecular insights into destabilization of Alzheimer’s Aβ protofibril by arginine containing short peptides: A molecular modeling approach. ACS Omega 2019, 4, 892–903. [Google Scholar] [CrossRef]
- Khan, A.N.; Khan, R.H. Protein misfolding and related human diseases: A comprehensive review of toxicity, proteins involved, and current therapeutic strategies. Int. J. Biol. Macromol. 2022, 223 Pt A, 143–160. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein–protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L. Small molecules: The missing link in the central dogma. Nat. Chem. Biol. 2005, 1, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, T.; Hashemian, S.M.R. Computer-aided design of amino acid-based therapeutics: A review. Drug Des. Dev. Ther. 2018, 12, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.-S.; Xie, N.-Z.; Huang, R.-B. Recent development of peptide drugs and advance on theory and methodology of peptide inhibitor design. Med. Chem. 2015, 11, 235–247. [Google Scholar] [CrossRef]
- Yao, T.; Xiao, H.; Wang, H.; Xu, X. Recent advances in PROTACs for drug targeted protein research. Int. J. Mol. Sci. 2022, 23, 10328. [Google Scholar] [CrossRef]
- Krüger, A.; Zimbres, F.M.; Kronenberger, T.; Wrenger, C. Molecular modeling applied to nucleic acid-based molecule development. Biomolecules 2018, 8, 83. [Google Scholar] [CrossRef]
- Alshaer, W.; Zureigat, H.; Al Karaki, A.; Al-Kadash, A.; Gharaibeh, L.; Hatmal, M.M.; Aljabali, A.A.A.; Awidi, A. siRNA: Mechanism of action, challenges, and therapeutic approaches. Eur. J. Pharmacol. 2021, 905, 174178. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zheng, L.; Chen, J. Molecular dynamics simulation in RNA interference. Curr. Med. Chem. 2014, 21, 1968–1975. [Google Scholar] [CrossRef]
- Pandya, A.K.; Patravale, V.B. Computational avenues in oral protein and peptide therapeutics. Drug Discov. Today 2021, 26, 1510–1520. [Google Scholar] [CrossRef]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. Machine intelligence in peptide therapeutics: A next-generation tool for rapid disease screening. Med. Res. Rev. 2020, 40, 1276–1314. [Google Scholar] [CrossRef]
- Guedeney, N.; Cornu, M.; Schwalen, F.; Kieffer, C.; Voisin-Chiret, A.S. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 2023, 28, 103395. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zheng, C.; Wang, H.; Zhang, L.; Liu, Z.; Xu, P. The state of the art of PROTAC technologies for drug discovery. Eur. J. Med. Chem. 2022, 235, 114290. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hu, M.; Yang, Y.; Du, C.G.; Zhou, H.; Liu, C.; Chen, Y.; Fan, L.; Ma, H.; Gong, Y.; et al. An overview of PROTACs: A promising drug discovery paradigm. Mol. Biomed. 2022, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. PROTAC-DB 2.0: An updated database of PROTACs. Nucleic Acids Res. 2023, 6, D1367–D1372. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef]
- Tunjic, T.M.; Weber, N.; Brunsteiner, M. Computer aided drug design in the development of proteolysis targeting chimeras. Comput. Struct. Biotechnol. J. 2023, 21, 2058–2067. [Google Scholar] [CrossRef]
- Li, D.; Yu, D.; Li, Y.; Yang, R. A bibliometric analysis of PROTAC from 2001 to 2021. Eur. J. Med. Chem. 2022, 244, 114838. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Aldrich, C.C.; Go, M.-L.; Dick, T. PROTAC antibiotics: The time is now. Expert Opin. Drug Discov. 2023, 18, 363–370. [Google Scholar] [CrossRef]
- Ahmad, H.; Zia, B.; Husain, H.; Husain, A. Recent advances in PROTAC-based antiviral strategies. Vaccines 2023, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Hassan, M.I. Targeted protein degraders march towards the clinic for neurodegenerative diseases. Ageing Res. Rev. 2022, 78, 101616. [Google Scholar] [CrossRef]
- Wang, F.; Zhan, Y.; Li, M.; Wang, L.; Zheng, A.; Liu, C.; Wang, H.; Wang, T. Cell-permeable PROTAC degraders against KEAP1 efficiently suppress hepatic stellate cell activation through the antioxidant and anti-inflammatory pathway. ACS Pharmacol. Transl. Sci. 2022, 6, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, G.; Ozpolat, B.; Coleman, R.L.; Sood, A.K.; Lopez-Berestein, G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 2015, 87, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Johnson, T.L.; Clugston, S.; Huang, H.; Butenhof, K.J.; Stanton, R.V. Molecular dynamics simulation and binding energy calculation for estimation of oligonucleotide duplex thermostability in RNA-based therapeutics. J. Chem. Inf. Model. 2011, 51, 1957–1965. [Google Scholar] [CrossRef]
- Bhandare, V.V.; Ramaswamy, A. Identification of possible siRNA molecules for TDP43 mutants causing amyotrophic lateral sclerosis: In silico design and molecular dynamics study. Comput. Biol. Chem. 2016, 61, 97–108. [Google Scholar] [CrossRef]
- Harikrishna, S.; Pradeepkumar, P.I. Probing the binding interactions between chemically modified siRNAs and human argonaute 2 using microsecond molecular dynamics simulations. J. Chem. Inf. Model. 2017, 57, 883–896. [Google Scholar] [CrossRef]
- Gao, H.; Kan, S.; Ye, Z.; Feng, Y.; Jin, L.; Zhang, X.; Deng, J.; Chan, G.; Hu, Y.; Wang, Y.; et al. Development of in silico methodology for siRNA lipid nanoparticle formulations. Chem. Eng. J. 2022, 442, 136310. [Google Scholar] [CrossRef]
- Rissanou, A.N.; Ouranidis, A.; Karatasos, K. Complexation of single stranded RNA with an ionizable lipid: An all-atom molecular dynamics simulation study. Soft Matter 2020, 16, 6993–7005. [Google Scholar] [CrossRef]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Shehroz, M.; Zaheer, T.; Hussain, T. Computer-aided drug design against spike glycoprotein of SARS-CoV-2 to aid COVID-19 treatment. Heliyon 2020, 6, e05278. [Google Scholar] [CrossRef] [PubMed]
- Gaudêncio, S.P.; Pereira, F. A computer-aided drug design approach to predict marine drug-like leads for SARS-CoV-2 main protease inhibition. Mar. Drugs 2020, 18, 633. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gan, J.; Wang, R.; Yang, X.; Xiao, Z.; Cao, Y. DrugDevCovid19: An atlas of anti-COVID-19 compounds derived by computer-aided drug design. Molecules 2022, 27, 683. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, J.; Wei, F. Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan, China. J. Med. Virol. 2020, 92, 441–447. [Google Scholar] [CrossRef]
- Chowdhury, U.F.; Shohan, M.U.S.; Hoque, K.I.; Beg, M.A.; Siam, M.K.S.; Moni, M.A. A computational approach to design potential siRNA molecules as a prospective tool for silencing nucleocapsid phosphoprotein and surface glycoprotein gene of SARS-CoV-2. Genomics 2021, 113, 331–343. [Google Scholar] [CrossRef]
- Mahfuz, M.; Shawan, A.K.; Sharma, A.R.; Bhattacharya, M.; Mallik, B.; Akhter, F.; Shakil, M.S.; Hossain, M.M.; Banik, S.; Lee, S.-S.; et al. Designing an effective therapeutic siRNA to silence RdRp gene of SARS-CoV-2. Infect. Genet. Evol. 2021, 93, 104951. [Google Scholar] [CrossRef]
- Sohrab, S.S.; El-Kafrawy, S.A.; Azhar, E.I. Effect of in silico predicted and designed potential siRNAs on inhibition of SARS-CoV-2 in HEK-293 cells. J. King Saud. Univ. Sci. 2022, 34, 101965. [Google Scholar] [CrossRef]
- Qureshi, A.; Tantray, V.G.; Kirmani, A.R.; Ahangar, A.G. A review on current status of antiviral siRNA. Rev. Med. Virol. 2018, 28, e1976. [Google Scholar] [CrossRef]
- Sajid, M.I.; Moazzam, M.; Cho, Y.; Kato, S.; Xu, A.; Way, J.J.; Lohan, S.; Tiwari, R.K. siRNA therapeutics for the therapy of COVID-19 and other coronaviruses. Mol. Pharm. 2021, 18, 2105–2121. [Google Scholar] [CrossRef]
- Mirza, M.U.; Vanmeert, M.; Ali, A.; Iman, K.; Froeyen, M.; Idrees, M. Perspectives towards antiviral drug discovery against Ebola virus. J. Med. Virol. 2019, 91, 2029–2048. [Google Scholar] [CrossRef]
- Mishra, N.; Ashique, S.; Garg, A.; Kumar Rai, V.; Dua, K.; Goyal, A.; Bhatt, S. Role of siRNA-based nanocarriers for the treatment of neurodegenerative diseases. Drug Discov. Today 2022, 27, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Patisiran: First global approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Scott, L.J. Givosiran: First approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Inclisiran: First approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hattab, D.; Mohd Gazzali, A.; Bakhtiar, A. Clinical advances of siRNA-based nanotherapeutics for cancer treatment. Pharmaceutics 2021, 13, 1009. [Google Scholar] [CrossRef]
- Sargazi, S.; Arshad, R.; Ghamari, R.; Rahdar, A.; Bakhshi, A.; Fathi Karkan, S.; Ajalli, N.; Bilal, M.; Díez-Pascual, A.M. siRNA-based nanotherapeutics as emerging modalities for immune-mediated diseases: A preliminary review. Cell Biol. Int. 2022, 46, 1320–1344. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-like property concepts in pharmaceutical design. Curr. Pharm. Des. 2009, 15, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Kumar Singh, P.; Negi, A.; Kumar Gupta, P.; Chauhan, M.; Kumar, R. Toxicophore exploration as a screening technology for drug design and discovery: Techniques, scope and limitations. Arch. Toxicol. 2016, 90, 1785–1802. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Ni, W.; Xu, X.; Wang, H.; Wang, J.; Ji, M.; Li, J. Chemical structure-related drug-like criteria of global approved drugs. Molecules 2016, 21, 75. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Doak, B.C.; Zheng, J.; Dobritzsch, D.; Kihlberg, J. How beyond Rule of 5 drugs and clinical candidates bind to their targets. J. Med. Chem. 2016, 59, 2312–2327. [Google Scholar] [CrossRef]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Wang, X.; Lyu, Y.; Chen, X.; Liu, K.; et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.; Lombardo, F.; Dominy, B.; Feeney, P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Cirillo, D.; Livi, C.M.; Ponti, R.D.; Tartaglia, G.G. cc SOL omics: A webserver for solubility prediction of endogenous and heterologous expression in Escherichia coli. Bioinformatics 2014, 30, 2975–2977. [Google Scholar] [CrossRef]
- Wang, C.K.; Craik, D.J. Cyclic peptide oral bioavailability: Lessons from the past. Biopolymers 2016, 106, 901–909. [Google Scholar] [CrossRef]
- Santos, G.B.; Ganesan, A.; Emery, F.S. Oral administration of peptide-based drugs: Beyond Lipinski’s rule. ChemMedChem 2016, 11, 2245–2251. [Google Scholar] [CrossRef]
- Zheng, S.; Tan, Y.; Wang, Z.; Li, C.; Zhang, Z.; Sang, X.; Chen, H. Accelerated rational PROTAC design via deep learning and molecular simulations. Nat. Mach. Intell. 2022, 4, 739–748. [Google Scholar] [CrossRef]
- DeGoey, D.A.; Chen, H.-J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 2018, 61, 2636–2651. [Google Scholar] [CrossRef]
- Freitas, R.A., Jr. What is nanomedicine? Nanomedicine 2005, 1, 2–9. [Google Scholar] [CrossRef]
- Mascheroni, P.; Schrefler, B.A. In silico models for nanomedicine: Recent developments. Curr. Med. Chem. 2018, 5, 4192–4207. [Google Scholar] [CrossRef]
- Hassanzadeh, P.; Atyabi, F.; Dinarvand, R. Ignoring the modeling approaches: Towards the shadowy paths in nanomedicine. J. Control. Release 2018, 280, 58–75. [Google Scholar] [CrossRef]
- Sultana, A.; Zare, M.; Thomas, V.; Sampath Kumar, T.S.; Ramakrishna, S. Nano-based drug delivery systems: Conventional drug delivery routes, recent developments and future prospects. Med. Drug Discov. 2022, 15, 100134. [Google Scholar] [CrossRef]
- Kumar, P.; Khan, R.A.; Choonara, Y.E.; Pillay, V. A prospective overview of the essential requirements in molecular modeling for nanomedicine design. Future Med. Chem. 2013, 5, 929–946. [Google Scholar] [CrossRef] [PubMed]
- Serov, N.; Vinogradov, V. Artificial intelligence to bring nanomedicine to life. Adv Drug. Deliv. Rev. 2022, 184, 14194. [Google Scholar] [CrossRef]
- Effinger, A.; O’Driscoll, C.M.; McAllister, M.; Fotaki, N. In vitro and in silico ADME prediction. In ADME Processes in Pharmaceutical Sciences: Dosage, Design, and Pharmacotherapy Success, 1st ed.; Talevi, A., Quiroga, P., Eds.; Springer: Cham, Switzerland, 2018; Volume 1, pp. 301–330. [Google Scholar] [CrossRef]
- Rostami-Hodjegan, A.; Tucker, G.T. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat. Rev. Drug Discov. 2007, 6, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.F.; Jurima-Romet, M.; Reimer, M.L.J.; Mortimer, E.; Rolfe, B.; Cayen, M.N. Making better drugs: Decision gates in non-clinical drug development. Nat. Rev. Drug Discov. 2003, 2, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Choudhary, M.I.; Thomson, W.J. Bioassay Techniques for Drug Development, 1st ed.; Harwood Academic Publishers: Amsterdam, The Netherlands, 2005; 240p. [Google Scholar]
- Barry, C.E., III. The death of the ‘three Ms’. ACS Infect. Dis. 2015, 1, 578–579. [Google Scholar] [CrossRef]
- Larkins-Ford, J.; Aldridge, B.B. Advances in the design of combination therapies for the treatment of tuberculosis. Expert Opin. Drug Discov. 2023, 18, 83–97. [Google Scholar] [CrossRef]
- Cavaleri, M.; Manolis, E. Hollow fiber system model for tuberculosis: The European medicines agency experience. Clin. Infect. Dis. 2015, 61 (Suppl. S1), S1–S4. [Google Scholar] [CrossRef]
- Dartois, V.; Barry, C.E., III. A medicinal chemists’ guide to the unique difficulties of lead optimization for tuberculosis. Bioorg. Med. Chem. Lett. 2013, 23, 4741–4750. [Google Scholar] [CrossRef]
- Brancato, V.; Oliveira, J.M.; Correlo, V.M.; Reis, R.L.; Kundu, S.C. Could 3D models of cancer enhance drug screening? Biomaterials 2020, 232, 119744. [Google Scholar] [CrossRef]
- Booij, T.H.; Price, L.S.; Danen, E.H.J. 3D Cell-based assays for drug screens: Challenges in imaging, image analysis, and high-content analysis. SLAS Discov. Adv. Life Sci. RD 2019, 24, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. In vitro assays and techniques utilized in anticancer drug discovery. J. Appl. Toxicol. 2019, 39, 38–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rhoads, D.D.; Appleby, B.S. Human prion diseases. Curr. Opin. Infect. Dis. 2019, 32, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Nakamura, T. Human prion disease. Clin. Exp. Neuroimmunol. 2022, 13, 24–33. [Google Scholar] [CrossRef]
- Moda, F.; Bolognesi, M.L.; Legname, G. Novel screening approaches for human prion diseases drug discovery. Expert Opin. Drug Discov. 2019, 14, 983–993. [Google Scholar] [CrossRef]
- Patrick, G.L. An Introduction to Medicinal Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Schollenberger, A. Zasada 3R w ochronie zwierząt wykorzystywanych do badań naukowych. Życie Weter. 2017, 92, 424–426. [Google Scholar]
- Maestri, E. The 3Rs principle in animal experimentation: A legal review of the state of the art in Europe and the case in Italy. BioTech 2021, 10, 9. [Google Scholar] [CrossRef]
- Lubeńczuk, G. Admissibility of the use of animals for scientific purposes in the light of international public law and EU law. Stud. Iurid. Lublinensia 2021, 30, 133–146. [Google Scholar] [CrossRef]
- Potempska, A. Zasada zastąpienia w badaniach eksperymentalnych modelujących procesy biochemiczne u ludzi (głos w dyskusji). Przegląd Filoz. Nowa Ser. 2015, 2, 1230–1493. [Google Scholar]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Universities Federation for Animal Welfare (UFAW): London, UK, 1992. [Google Scholar]
- Blattner, C.E. Rethinking the 3Rs: From whitewashing to rights. In Animal Experimentation: Working Towards a Paradigm Change; Brill: Leiden, The Netherlands, 2019; pp. 168–193. [Google Scholar]
- Herrmann, K.; Pistollato, F.; Stephens, M.L. Beyond the 3Rs: Expanding the use of human-relevant replacement methods in biomedical research. ALTEX 2019, 36, 343–352. [Google Scholar] [CrossRef]
- Kedzierska, E.; Dabkowska, L.; Krzanowski, T.; Gibula, E.; Orzelska, J.; Wujec, M. New drugs—From necessity to delivery. Curr. Issues Pharm. Med. Sci. 2018, 31, 69–75. [Google Scholar] [CrossRef]
- Clark, J.M. The 3Rs in research: A contemporary approach to replacement, reduction and refinement. Br. J. Nutr. 2018, 120, S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Sornat, R.; Gruszka, K.; Polińska, A. The 3R rules in practice. Improvement of welfare, reduction of the number of animals and improvement of test methods in acute toxicity studies. Med. Weter. 2022, 78, 548–555. [Google Scholar] [CrossRef]
- Rahman Khan, F.; Sulaiman Alhewairini, S. Zebrafish (Danio rerio) as a model organism. In Current Trends in Cancer Management; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Teame, T.; Zhang, Z.; Ran, C.; Zhang, H.; Yang, Y.; Ding, Q.; Xie, M.; Gao, C.; Ye, Y.; Duan, M.; et al. The use of zebrafish (Danio rerio) as biomedical models. Anim. Front. 2019, 9, 68–77. [Google Scholar] [CrossRef]
- Spence, R.; Gerlach, G.; Lawrence, C.; Smith, C. The behaviour and ecology of the zebrafish, Danio Rerio. Biol. Rev. 2008, 83, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Korzeniowski, P.J.; Wiweger, M. Danio pręgowany jako zwierzę laboratoryjne. Podstawowe zagadnienia z zakresu pielęgnacji i opieki lekarsko-weterynaryjnej nad rybami w warunkach hodowli laboratoryjnej. Życie Weter. 2014, 89, 750–756. [Google Scholar]
- Lawrence, C.; Mason, T. Zebrafish housing systems: A review of basic operating principles and considerations for design and functionality. ILAR J. 2012, 53, 179–191. [Google Scholar] [CrossRef]
- Shen, C.; Zuo, Z. Zebrafish (Danio rerio) as an excellent vertebrate model for the development, reproductive, cardiovascular, and neural and ocular development toxicity Study of hazardous chemicals. Environ. Sci. Pollut. Res. 2020, 27, 43599–43614. [Google Scholar] [CrossRef]
- Rosa, J.G.S.; Lima, C.; Lopes-Ferreira, M. Zebrafish larvae behavior models as a tool for drug screenings and pre-clinical trials: A review. Int. J. Mol. Sci. 2022, 23, 6647. [Google Scholar] [CrossRef]
- Gawel, K.; Banono, N.S.; Michalak, A.; Esguerra, C.V. A critical review of zebrafish schizophrenia models: Time for validation? Neurosci. Biobehav. Rev. 2019, 107, 6–22. [Google Scholar] [CrossRef] [PubMed]
- Gemberling, M.; Bailey, T.J.; Hyde, D.R.; Poss, K.D. The zebrafish as a model for complex tissue regeneration. Trends Genet. 2013, 29, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Dooley, K.; Zon, L.I. Zebrafish: A model system for the study of human disease. Curr. Opin. Genet. Dev. 2000, 10, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Zon, L.I.; Langenau, D.M. Zebrafish disease models in drug discovery: From preclinical modelling to clinical trials. Nat. Rev. Drug Discov. 2021, 20, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of zebrafish in drug discovery toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef]
- MacRae, C.A.; Peterson, R.T. Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef]
- Guerra, E.; Garcia-Sanchez, Y.; Jornet-Gibert, M.; Nuñez, J.; Balaguer-Castro, M.; Madden, K. Clinical practice guidelines: The good, the bad, and the ugly. Injury 2022, 54 (Suppl. S3), S26–S29. [Google Scholar] [CrossRef]
- Brodniewicz, T. Badania Kliniczne; CEDEWU: Warszawa, Poland, 2015. [Google Scholar]
- Mahan, V. Clinical trial phases. Int. J. Clin. Med. 2014, 5, 1374–1383. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/ (accessed on 12 June 2023).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biala, G.; Kedzierska, E.; Kruk-Slomka, M.; Orzelska-Gorka, J.; Hmaidan, S.; Skrok, A.; Kaminski, J.; Havrankova, E.; Nadaska, D.; Malik, I. Research in the Field of Drug Design and Development. Pharmaceuticals 2023, 16, 1283. https://doi.org/10.3390/ph16091283
Biala G, Kedzierska E, Kruk-Slomka M, Orzelska-Gorka J, Hmaidan S, Skrok A, Kaminski J, Havrankova E, Nadaska D, Malik I. Research in the Field of Drug Design and Development. Pharmaceuticals. 2023; 16(9):1283. https://doi.org/10.3390/ph16091283
Chicago/Turabian StyleBiala, Grazyna, Ewa Kedzierska, Marta Kruk-Slomka, Jolanta Orzelska-Gorka, Sara Hmaidan, Aleksandra Skrok, Jakub Kaminski, Eva Havrankova, Dominika Nadaska, and Ivan Malik. 2023. "Research in the Field of Drug Design and Development" Pharmaceuticals 16, no. 9: 1283. https://doi.org/10.3390/ph16091283
APA StyleBiala, G., Kedzierska, E., Kruk-Slomka, M., Orzelska-Gorka, J., Hmaidan, S., Skrok, A., Kaminski, J., Havrankova, E., Nadaska, D., & Malik, I. (2023). Research in the Field of Drug Design and Development. Pharmaceuticals, 16(9), 1283. https://doi.org/10.3390/ph16091283