



Cellular Efficacy of Fattigated Nanoparticles and Real-Time ROS Occurrence Using Microfluidic Hepatocarcinoma Chip System: Effect of Anticancer Drug Solubility and Shear Stress

 , , , and
, , , and
Abstract
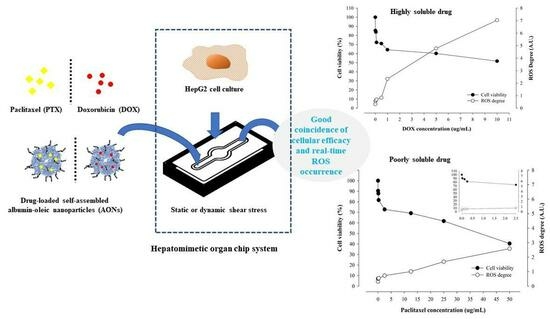
:
1. Introduction
2. Results and Discussion
2.1. Identification of AOC and Physicochemical Properties of AONs
2.1.1. Identification of AOCs
2.1.2. Physicochemical Properties of AONs
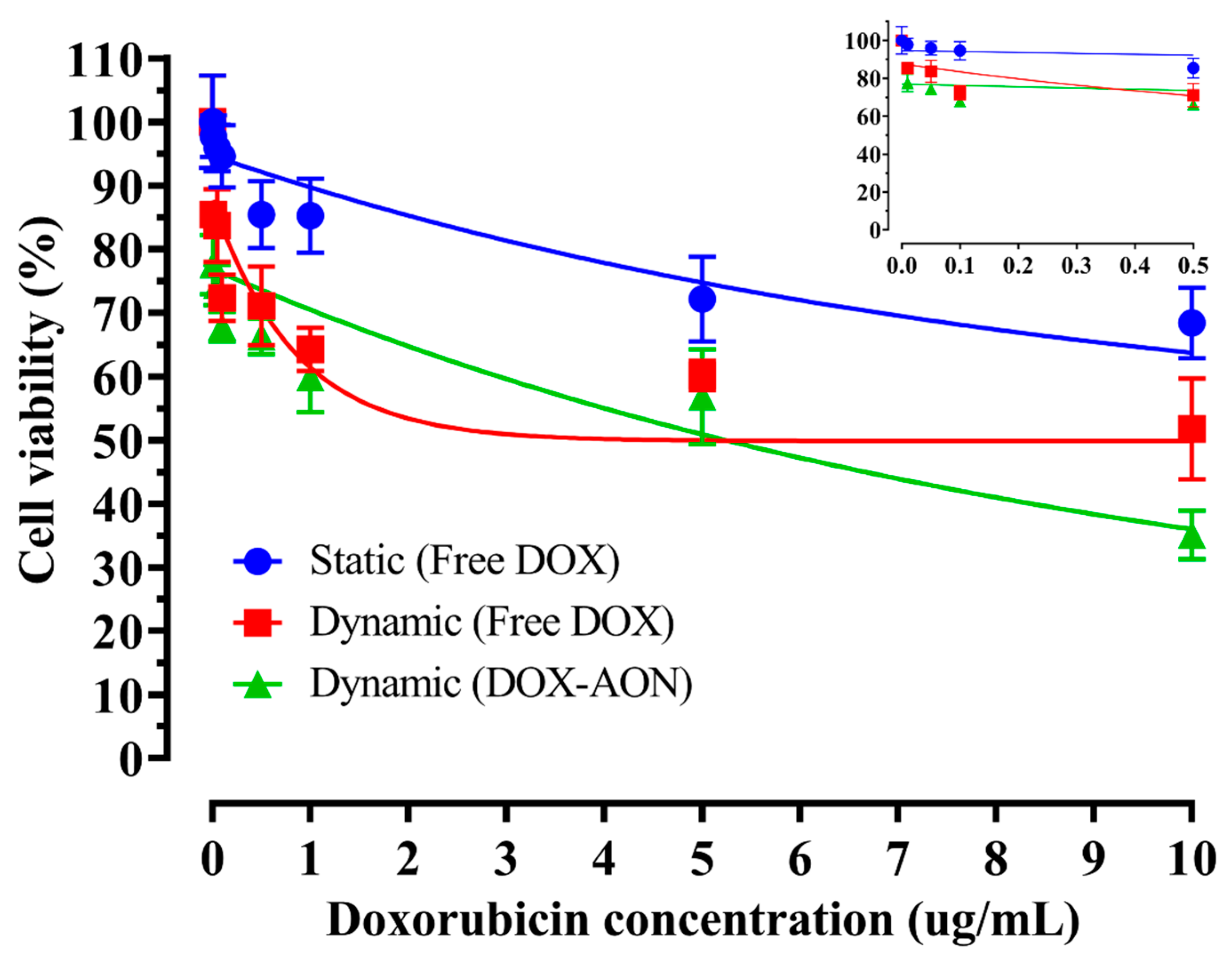
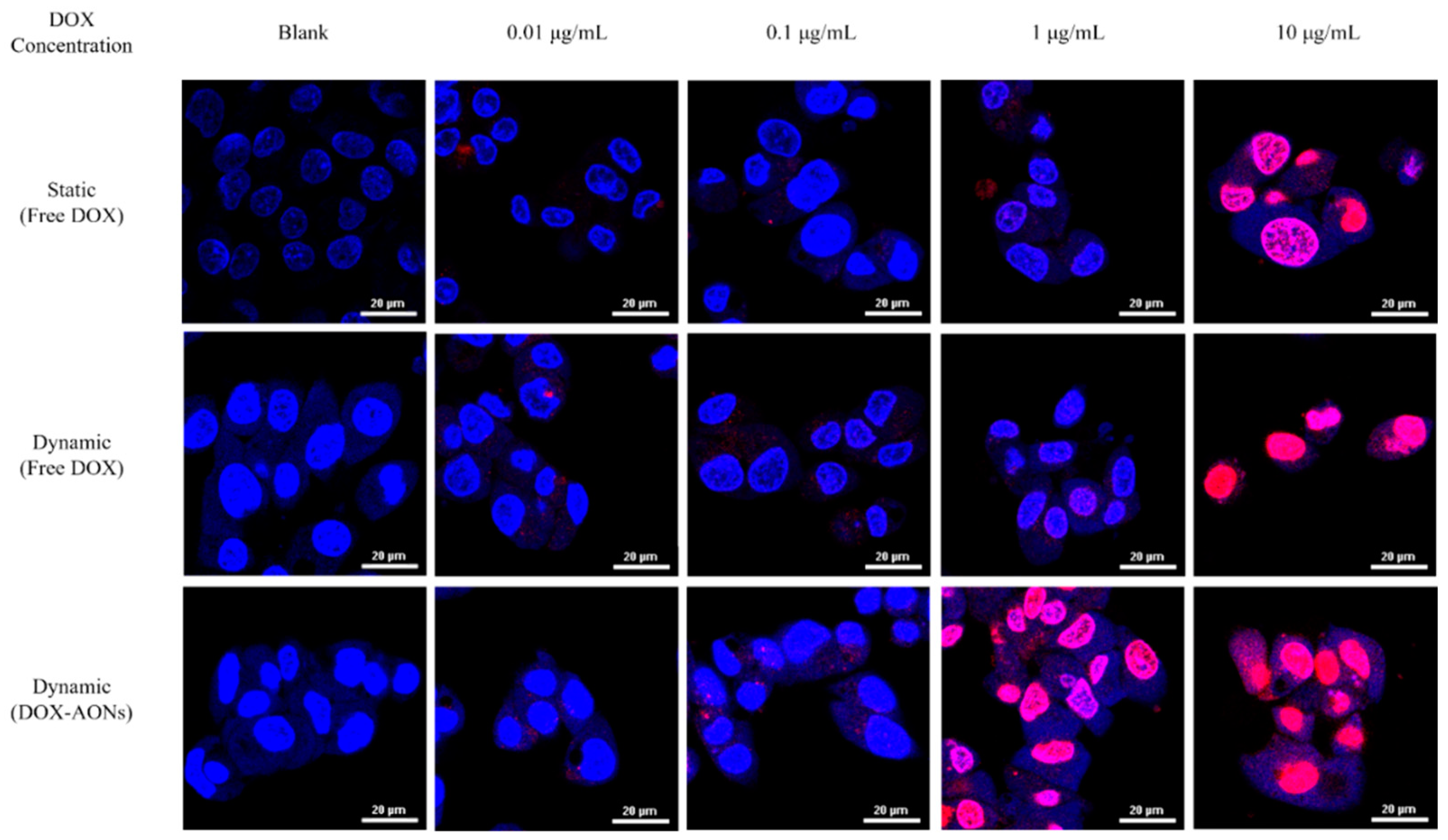
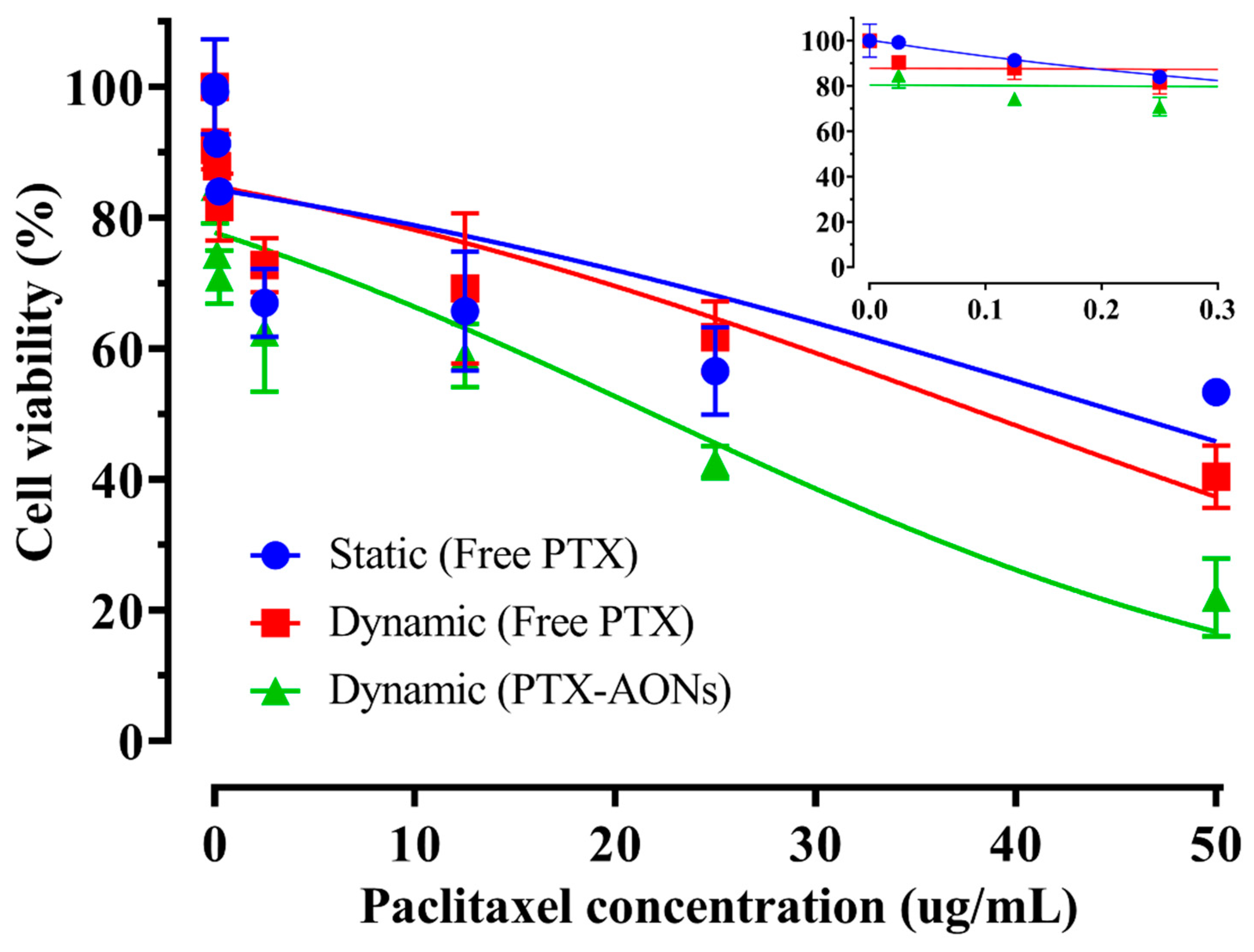
2.2. Cellular Efficacy According to Shear Stress and Drug Type
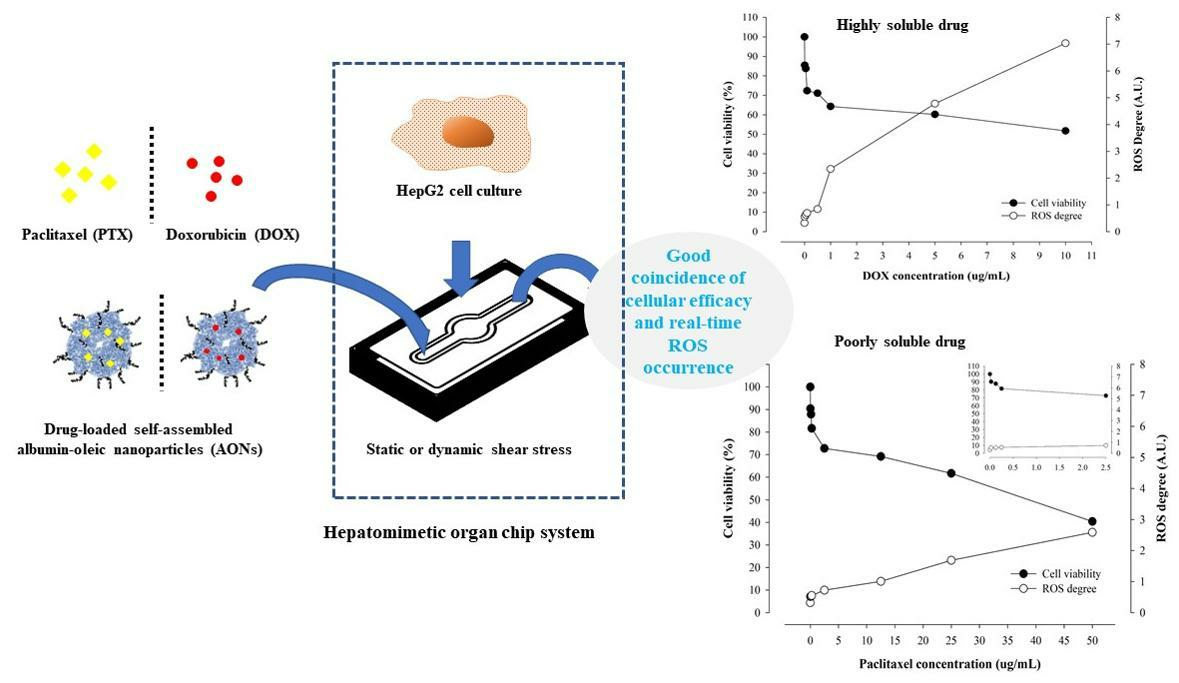
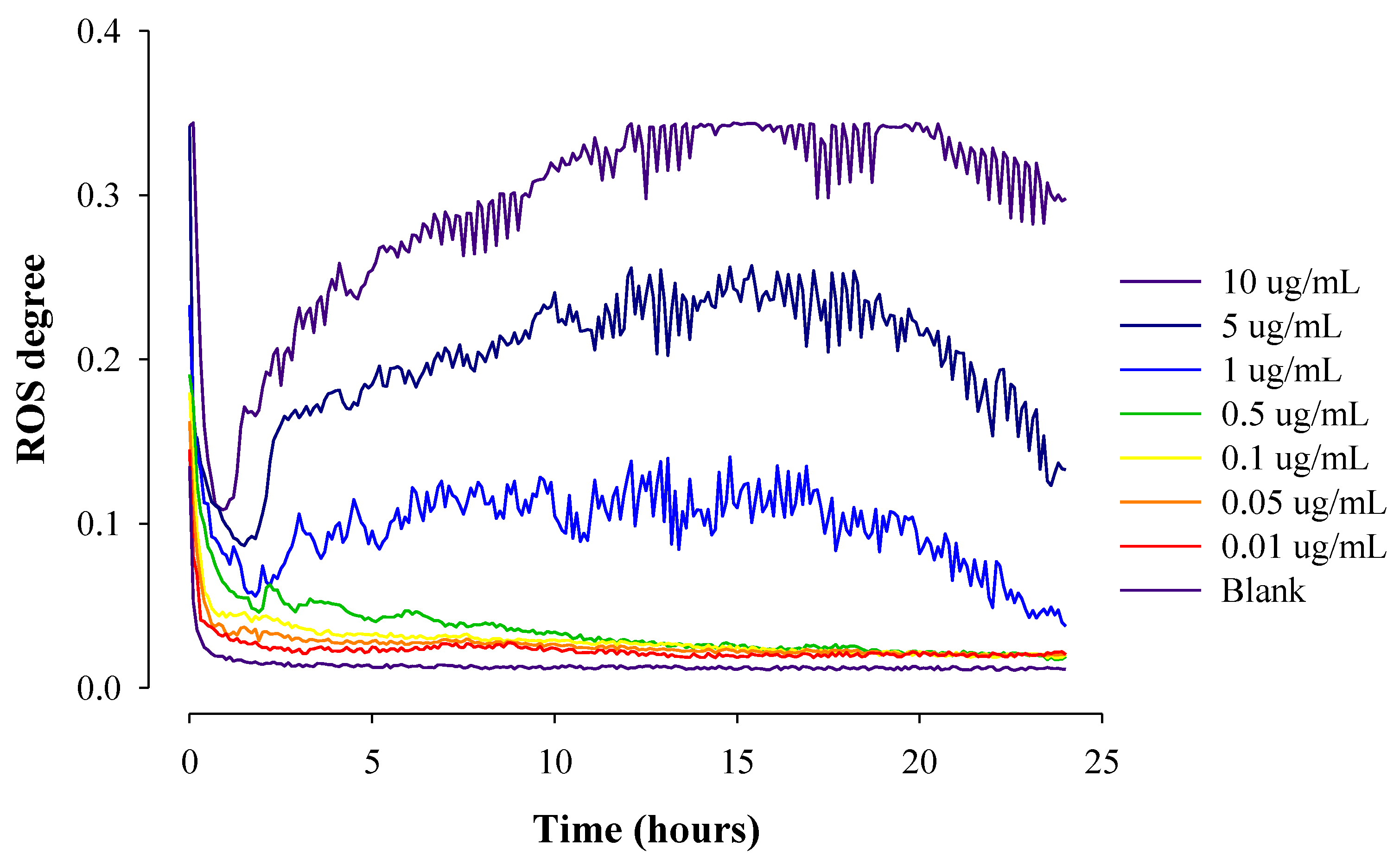
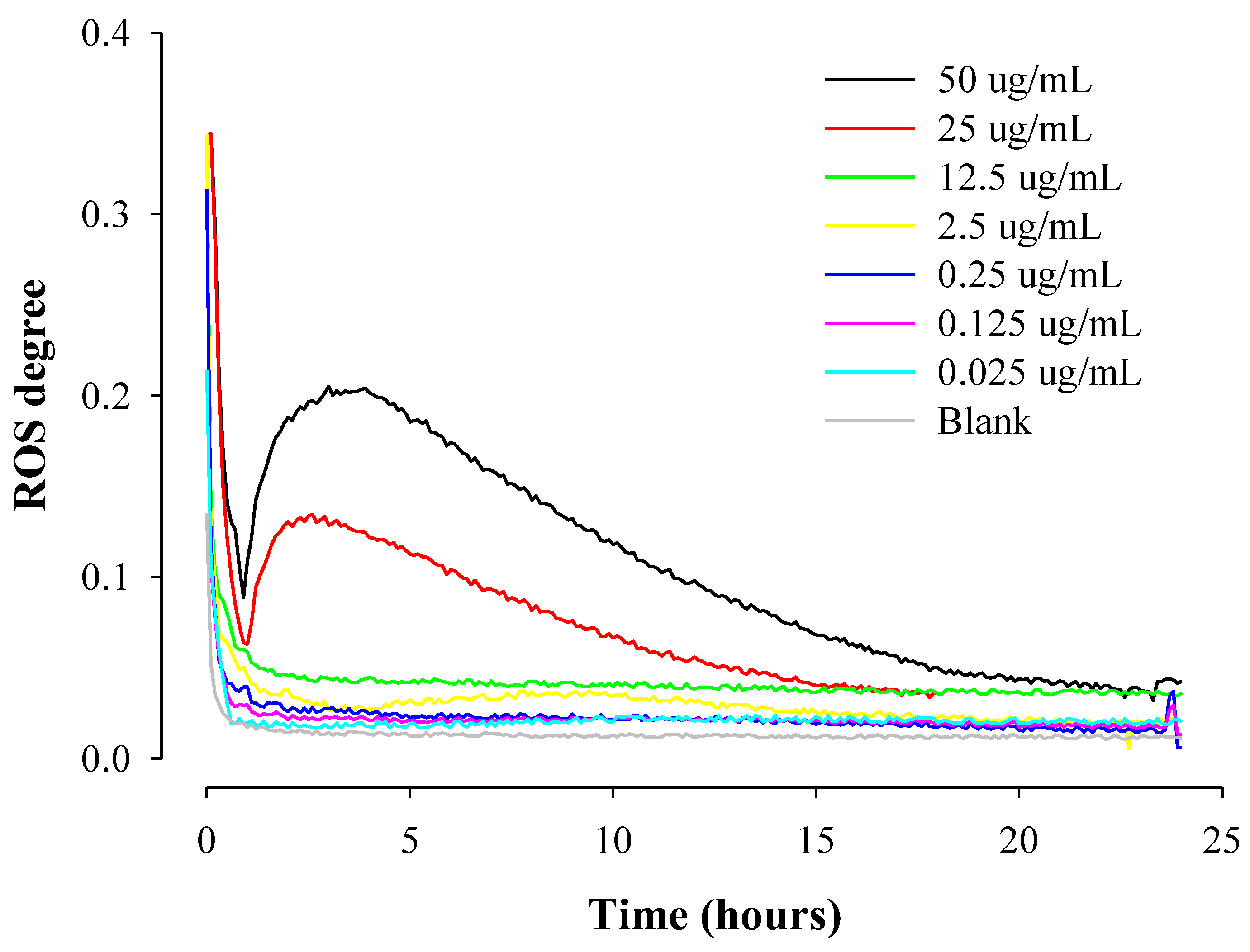
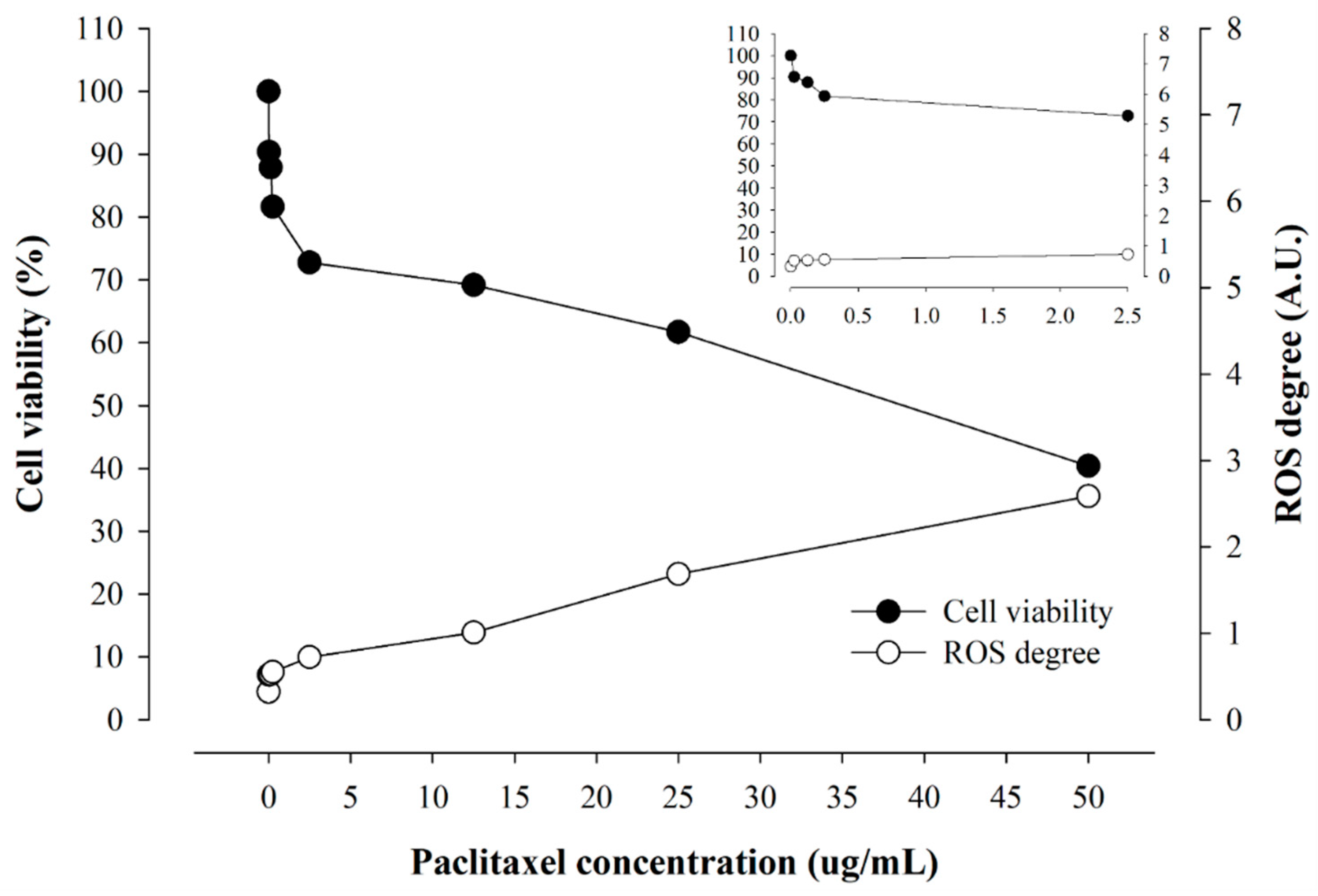
2.3. Real-Time ROS Sensing of Anticancer Drugs with Different Solubility
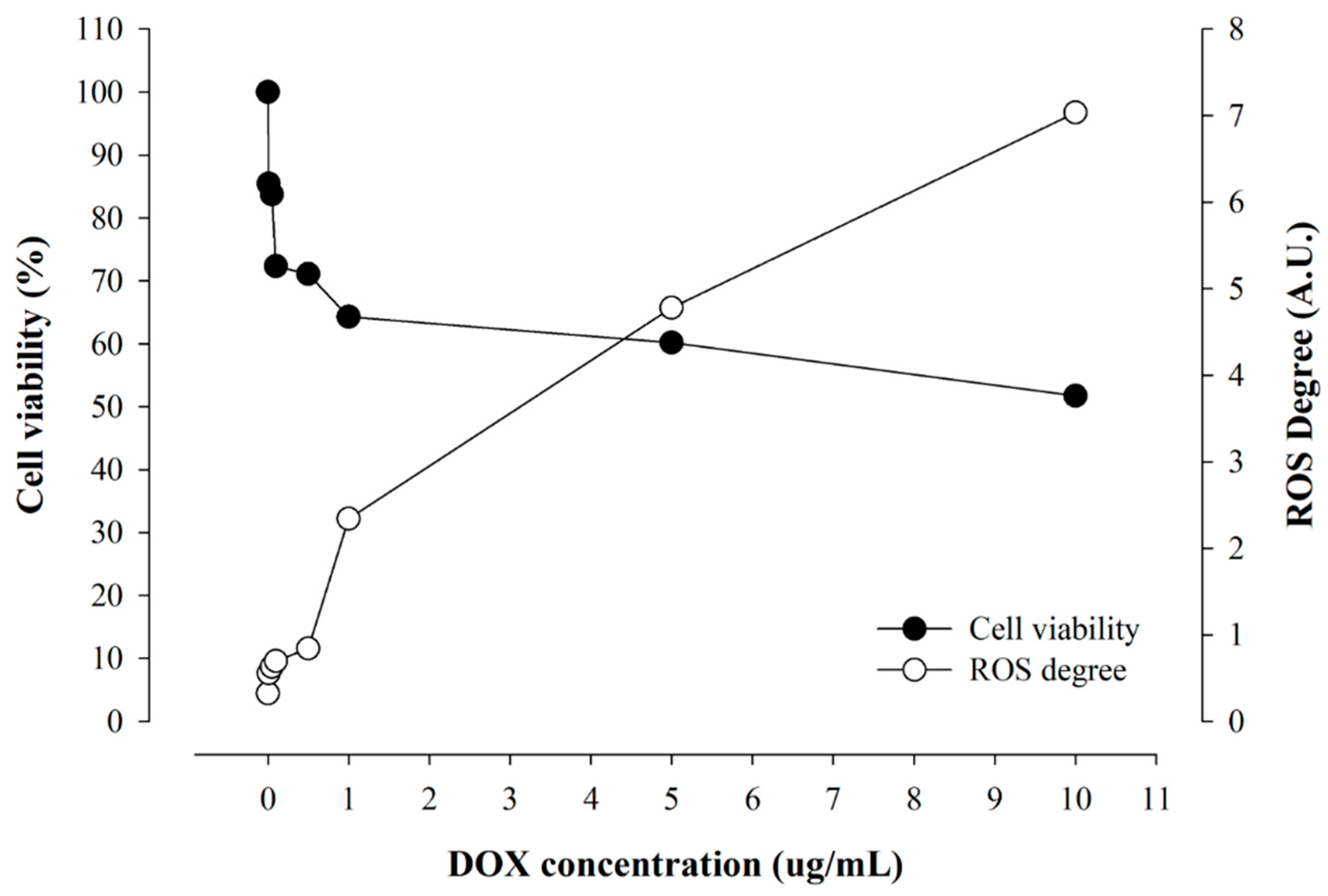
2.4. Correlation of Cellular Viability and Real-Time ROS Occurrence
3. Materials and Methods
3.1. Materials
3.2. Preparation of AONs, DOX-AONs, and PTX-AONs
3.2.1. Synthesis of AOC
3.2.2. Conversion of AOCs to AONs Using a Desolvation Method
3.2.3. Preparation of Drug-Loaded AONs
3.3. Physicochemical Properties of NPs
3.3.1. Fourier Transform-Infrared (FT-IR) Spectrometer
3.3.2. Particle Size and Zeta Potential Measurements
3.3.3. Loading Content (LC) and Encapsulation Efficiency (EE)
3.3.4. Morphologies of NPs using FE-TEM and FE-SEM
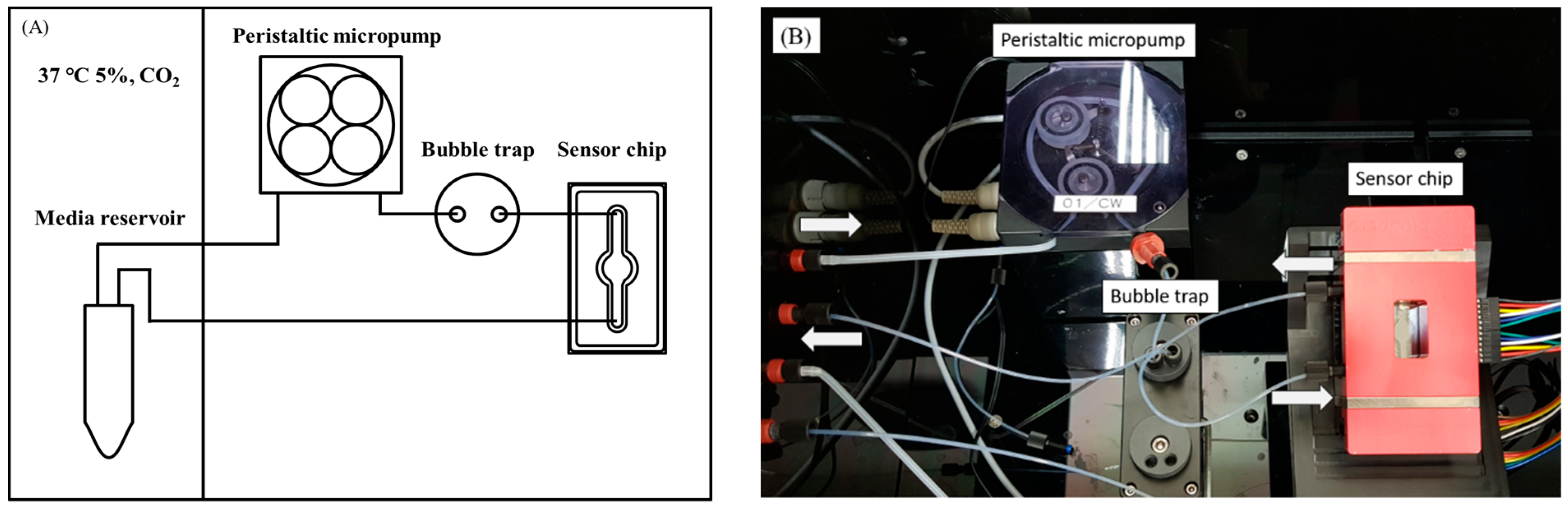
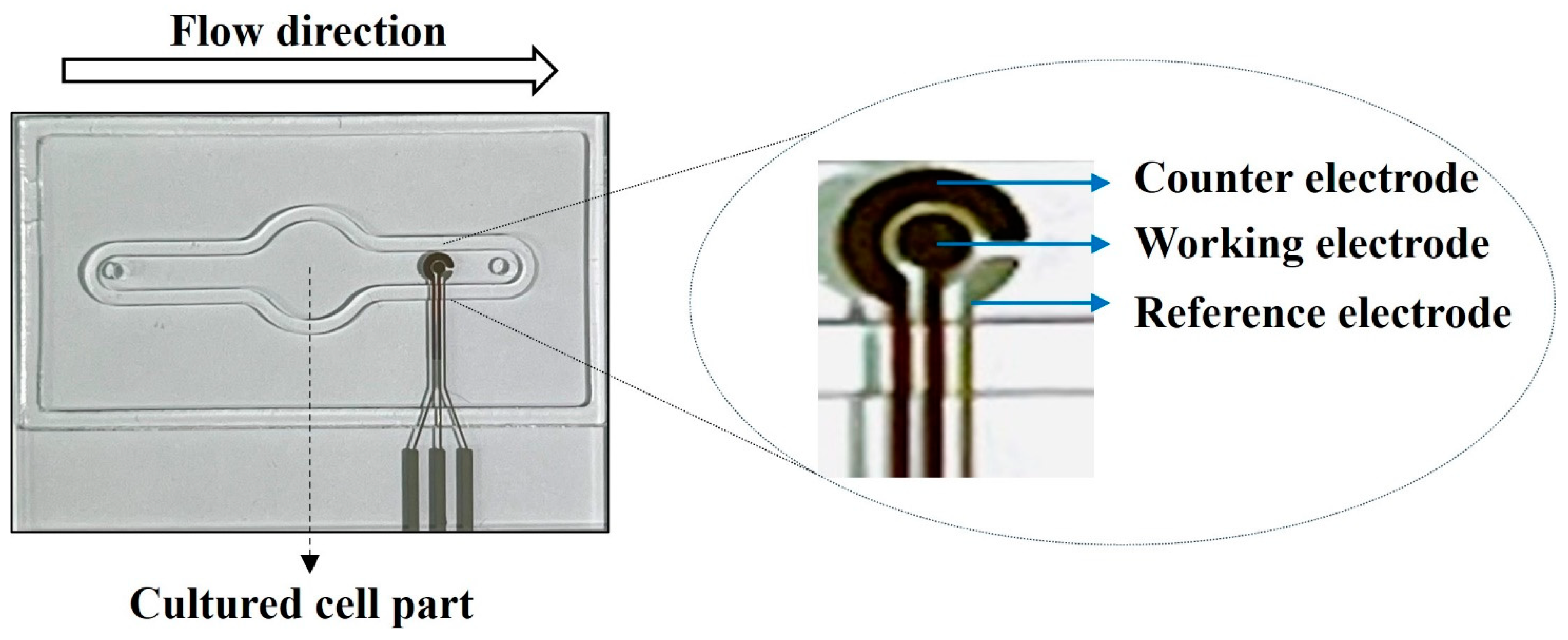
3.4. Establishment of Biomimetic Microfluidic System with Real-Time ROS Sensor Chip
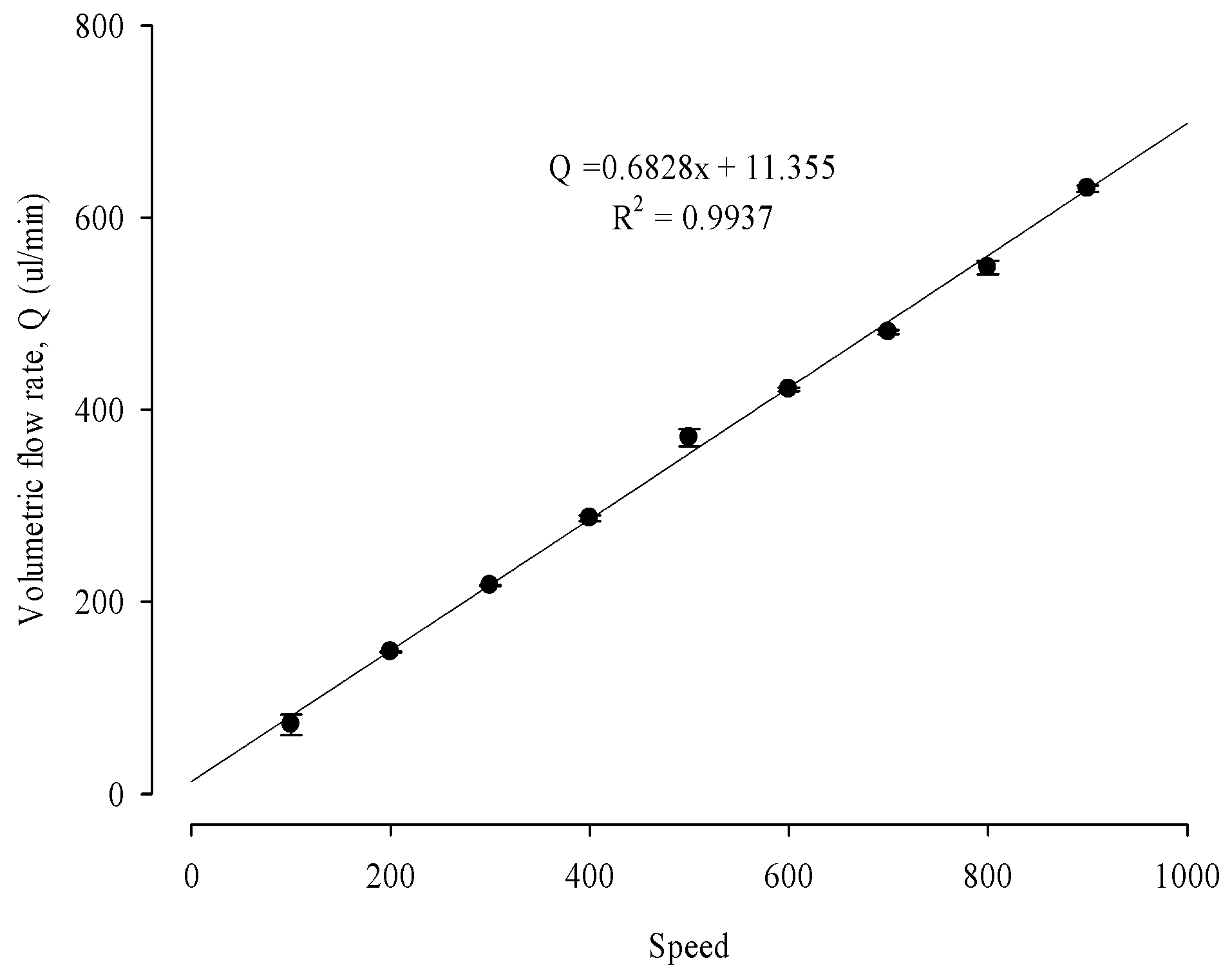
3.4.1. Biomimetic Microfluidic System Calibration
3.4.2. Cell Culture in a Real-Time ROS Sensor Chip
3.4.3. Cellular Viability Assay
3.4.4. Real-Time Sensing of the Generated ROS
3.5. Cellular Images Using Confocal Laser Scanning Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McDonald, T.A.; Zepeda, M.L.; Tomlinson, M.J.; Bee, W.H.; Ivens, I.A. Subcutaneous administration of biotherapeutics: Current experience in animal models. Curr. Opin. Mol. Ther. 2010, 12, 461–470. [Google Scholar]
- Makalowski, W.; Zhang, J.; Boguski, M.S. Comparative analysis of 1196 orthologous mouse and human full-length mRNA and protein sequences. Genome Res. 1996, 6, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.; Coto, E.; Setien, F.; Gonzalez, S.; Gonzalez-Roces, S.; Lopez-Larrea, C. Characterization of interleukin-8 receptors in non-human primates. Immunogenetics 1996, 43, 261–267. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. C Embryo Today 2015, 105, 140–156. [Google Scholar] [CrossRef]
- Pallocca, G.; Leist, M. Animal Experimentation: Working Towards a Paradigm Change. ALTEX 2019, 36, 148–150. [Google Scholar]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Pisapia, F.; Balachandran, W.; Rasekh, M. Organ-on-a-Chip: Design and simulation of various microfluidic channel geometries for the influence of fluid dynamic parameters. Appl. Sci. 2022, 12, 3829. [Google Scholar] [CrossRef]
- Haddrick, M.; Simpson, P.B. Organ-on-a-chip technology: Turning its potential for clinical benefit into reality. Drug Discov. Today 2019, 24, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Wang, S. Application of microfluidic chip technology in pharmaceutical analysis: A review. J. Pharm. Anal. 2019, 9, 238–247. [Google Scholar] [CrossRef]
- Kilic, T.; Navaee, F.; Stradolini, F.; Renaud, P.; Carrara, S. Organs-on-chip monitoring: Sensors and other strategies. Microphysiol. Syst. 2018, 2, 1–32. [Google Scholar] [CrossRef]
- Thompson, D.M.; King, K.R.; Wieder, K.J.; Toner, M.; Yarmush, M.L.; Jayaraman, A. Dynamic gene expression profiling using a microfabricated living cell array. Anal. Chem. 2004, 76, 4098–4103. [Google Scholar] [CrossRef]
- Cong, Y.; Han, X.; Wang, Y.; Chen, Z.; Lu, Y.; Liu, T.; Wu, Z.; Jin, Y.; Luo, Y.; Zhang, X. Drug Toxicity Evaluation Based on Organ-on-a-chip Technology: A Review. Micromachines 2020, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.-H.; Wu, Y.-Y.; Liu, Y.-S.; Rimando, M.; Ho, J.H.-C.; Lee, O.K.-S. Efficient generation of hepatic cells from mesenchymal stromal cells by an innovative bio-microfluidic cell culture device. Stem Cell Res. Ther. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, A.M.; Hancock, M.J.; Harrington, H.; Kaji, H.; Khademhosseini, A. Biomimetic tissues on a chip for drug discovery. Drug Discov. Today 2012, 17, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Razu, M.E.; Wang, X.; Lacerda, C.; Kim, J.J. Biomimetic cardiac microsystems for pathophysiological studies and drug screens. J. Lab. Autom. 2015, 20, 96–106. [Google Scholar] [CrossRef]
- Gough, A.; Soto-Gutierrez, A.; Vernetti, L.; Ebrahimkhani, M.R.; Stern, A.M.; Taylor, D.L. Human biomimetic liver microphysiology systems in drug development and precision medicine. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 252–268. [Google Scholar] [CrossRef]
- Li, Y.; Sella, C.; Lemaitre, F.; Guille-Collignon, M.; Amatore, C.; Thouin, L. Downstream Simultaneous Electrochemical Detection of Primary Reactive Oxygen and Nitrogen Species Released by Cell Populations in an Integrated Microfluidic Device. Anal. Chem. 2018, 90, 9386–9394. [Google Scholar] [CrossRef]
- Weltin, A.; Slotwinski, K.; Kieninger, J.; Moser, I.; Jobst, G.; Wego, M.; Ehret, R.; Urban, G.A. Cell culture monitoring for drug screening and cancer research: A transparent, microfluidic, multi-sensor microsystem. Lab Chip 2014, 14, 138–146. [Google Scholar] [CrossRef]
- Dervisevic, E.; Tuck, K.L.; Voelcker, N.H.; Cadarso, V.J. Recent Progress in Lab-On-a-Chip Systems for the Monitoring of Metabolites for Mammalian and Microbial Cell Research. Sensors 2019, 19, 5027. [Google Scholar] [CrossRef]
- Park, C.; Meghani, N.; Amin, H.; Tran, P.H.; Tran, T.T.; Nguyen, V.H.; Lee, B.J. The roles of short and long chain fatty acids on physicochemical properties and improved cancer targeting of albumin-based fattigation-platform nanoparticles containing doxorubicin. Int. J. Pharm. 2019, 564, 124–135. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.J.; Simpson, A.M.; Swan, M.A.; Tao, C.; Tuch, B.E.; Crawford, R.M.; Jovanovic, A.; Martin, D.K. ATP-sensitive potassium channels induced in liver cells after transfection with insulin cDNA and the GLUT2 transporter regulate glucose-stimulated insulin secretion. FASEB J. 2003, 17, 1682. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.H.; Rather, M.A. Effect of calcination temperature on the crystallite size, particle size and zeta potential of TiO2 nanoparticles synthesized via polyol-mediated method. Mater. Today-Proc. 2021, 44, 482–488. [Google Scholar] [CrossRef]
- Borzova, V.A.; Markossian, K.A.; Chebotareva, N.A.; Kleymenov, S.Y.; Poliansky, N.B.; Muranov, K.O.; Stein-Margolina, V.A.; Shubin, V.V.; Markov, D.I.; Kurganov, B.I. Kinetics of Thermal Denaturation and Aggregation of Bovine Serum Albumin. PLoS ONE 2016, 11, e0153495. [Google Scholar] [CrossRef] [PubMed]
- Munaweera, I.; Hong, J.; D’Souza, A.; Balkus, K.J. Novel wrinkled periodic mesoporous organosilica nanoparticles for hydrophobic anticancer drug delivery. J. Porous Mater. 2015, 22, 1–10. [Google Scholar] [CrossRef]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Susa, M.; Iyer, A.K.; Ryu, K.; Hornicek, F.J.; Mankin, H.; Amiji, M.M.; Duan, Z. Doxorubicin loaded Polymeric Nanoparticulate Delivery System to overcome drug resistance in osteosarcoma. BMC Cancer 2009, 9, 399. [Google Scholar] [CrossRef]
- Mobaraki, M.; Faraji, A.; Zare, M.; Dolati, P.; Ataei, M.; Manshadi, H.R.D. Molecular Mechanisms of Cardiotoxicity: A Review on the Major Side-effects of Doxorubicin. Indian J. Pharm. Sci. 2017, 79, 335–344. [Google Scholar] [CrossRef]
- Park, C.; Baek, N.; Loebenberg, R.; Lee, B.J. Importance of the fatty acid chain length on in vitro and in vivo anticancer activity of fattigation-platform albumin nanoparticles in human colorectal cancer xenograft mice model. J. Control. Release 2020, 324, 55–68. [Google Scholar] [CrossRef]
- Dowdy, S.C.; Jiang, S.; Zhou, X.C.; Hou, X.; Jin, F.; Podratz, K.C.; Jiang, S.W. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 2006, 5, 2767–2776. [Google Scholar] [CrossRef]
- Meshkini, A.; Yazdanparast, R. Involvement of oxidative stress in taxol-induced apoptosis in chronic myelogenous leukemia K562 cells. Exp. Toxicol. Pathol. 2012, 64, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Gonzalez-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H. Mechanisms of Anthracycline-Enhanced Reactive Oxygen Metabolism in Tumor Cells. Oxid. Med. Cell Longev. 2019, 2019, 9474823. [Google Scholar] [CrossRef]
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway. Front. Pharmacol. 2019, 10, 1531. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Meghani, N.; Loebenberg, R.; Cui, J.-H.; Cao, Q.-R.; Lee, B.-J. Fatty acid chain length impacts nanonizing capacity of albumin-fatty acid nanomicelles: Enhanced physicochemical property and cellular delivery of poorly water-soluble drug. Eur. J. Pharm. Biopharm. 2020, 152, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Park, C.; Meghani, N.; Tran, T.T.D.; Tran, P.H.L.; Lee, B.J. Shear Stress-Dependent Targeting Efficiency Using Self-Assembled Gelatin-Oleic Nanoparticles in a Biomimetic Microfluidic System. Pharmaceutics 2020, 12, 555. [Google Scholar] [CrossRef]
- Filipovic, N.; Nikolic, M.; Sustersic, T. Simulation of organ-on-a-chip systems. In Biomaterials for Organ and Tissue Regeneration; Elsevier: Amsterdam, The Netherlands, 2020; pp. 753–790. [Google Scholar]
- Choi, J.; Yip-Schneider, M.; Albertin, F.; Wiesenauer, C.; Wang, Y.; Schmidt, C.M. The effect of doxorubicin on MEK-ERK signaling predicts its efficacy in HCC. J. Surg. Res. 2008, 150, 219–226. [Google Scholar] [CrossRef]
- Prabha, G.; Raj, V. Sodium alginate-polyvinyl alcohol-bovin serum albumin coated Fe3O4 nanoparticles as anticancer drug delivery vehicle: Doxorubicin loading and in vitro release study and cytotoxicity to HepG2 and L02 cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 79, 410–422. [Google Scholar] [CrossRef]
- Choi, J.S.; Piao, Y.J.; Kang, K.W. Effects of quercetin on the bioavailability of doxorubicin in rats: Role of CYP3A4 and P-gp inhibition by quercetin. Arch. Pharm. Res. 2011, 34, 607–613. [Google Scholar] [CrossRef]
- Wen, S.H.; Su, S.C.; Liou, B.H.; Lin, C.H.; Lee, K.R. Sulbactam-enhanced cytotoxicity of doxorubicin in breast cancer cells. Cancer Cell Int. 2018, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Hanauske, A.; Faivre, S.; Izbicka, E.; Clark, G.; Rowinsky, E.K.; Von Hoff, D.D. Effects of prolonged versus short-term exposure paclitaxel (Taxol) on human tumor colony-forming units. Anticancer Drugs 1997, 8, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Shord, S.S.; Camp, J.R. Intravenous administration of paclitaxel in Sprague-Dawley rats: What is a safe dose? Biopharm. Drug Dispos. 2006, 27, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, H.M.U.; Kang, B.; Khalid, M.A.U.; Salih, A.R.C.; Hyun, K.; Park, S.H.; Huh, D.; Choi, K.H. Real-time monitoring of liver fibrosis through embedded sensors in a microphysiological system. Nano Converg. 2021, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.A.U.; Kim, K.H.; Chethikkattuveli Salih, A.R.; Hyun, K.; Park, S.H.; Kang, B.; Soomro, A.M.; Ali, M.; Jun, Y.; Huh, D.; et al. High performance inkjet printed embedded electrochemical sensors for monitoring hypoxia in a gut bilayer microfluidic chip. Lab Chip 2022, 22, 1764–1778. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Size (nm) | Polydispersity Index | Zeta Potential (mV) | Loading Content (%) | Encapsulation Efficiency (%) | |
|---|---|---|---|---|---|
| AONs | 181.20 ± 29.20 | 0.300 ± 0.005 | −40.57 ± 0.22 | - | - |
| DOX-AONs | 313.23 ± 3.97 | 0.180 ± 0.024 | −36.36 ± 0.10 | 6.97 ± 0.33 | 69.68 ± 3.26 |
| PTX-AONs | 438.90 ± 27.97 | 0.170 ± 0.081 | −21.04 ± 0.16 | 5.60 ± 0.14 | 59.34 ± 1.56 |
| Formulation | Shear Stress | IC50 Values (μg/mL) |
|---|---|---|
| Free DOX | Static | 13.4 ± 1.345 |
| Free DOX | Dynamic | 11.798 ± 1.721 |
| DOX-AONs | Dynamic | 5.613 ± 1.601 |
| Free PTX | Static | 45.44 ± 5.824 |
| Free PTX | Dynamic | 38.43 ± 3.123 |
| PTX-AONs | Dynamic | 21.86 ± 2.340 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Kim, E.-J.; Ngo, H.V.; Nguyen, H.D.; Park, C.; Choi, K.H.; Park, J.-B.; Lee, B.-J. Cellular Efficacy of Fattigated Nanoparticles and Real-Time ROS Occurrence Using Microfluidic Hepatocarcinoma Chip System: Effect of Anticancer Drug Solubility and Shear Stress. Pharmaceuticals 2023, 16, 1330. https://doi.org/10.3390/ph16091330
Kim H, Kim E-J, Ngo HV, Nguyen HD, Park C, Choi KH, Park J-B, Lee B-J. Cellular Efficacy of Fattigated Nanoparticles and Real-Time ROS Occurrence Using Microfluidic Hepatocarcinoma Chip System: Effect of Anticancer Drug Solubility and Shear Stress. Pharmaceuticals. 2023; 16(9):1330. https://doi.org/10.3390/ph16091330
Chicago/Turabian StyleKim, Hoyoung, Eun-Ji Kim, Hai V. Ngo, Hy D. Nguyen, Chulhun Park, Kyung Hyun Choi, Jun-Bom Park, and Beom-Jin Lee. 2023. "Cellular Efficacy of Fattigated Nanoparticles and Real-Time ROS Occurrence Using Microfluidic Hepatocarcinoma Chip System: Effect of Anticancer Drug Solubility and Shear Stress" Pharmaceuticals 16, no. 9: 1330. https://doi.org/10.3390/ph16091330
APA StyleKim, H., Kim, E.-J., Ngo, H. V., Nguyen, H. D., Park, C., Choi, K. H., Park, J.-B., & Lee, B.-J. (2023). Cellular Efficacy of Fattigated Nanoparticles and Real-Time ROS Occurrence Using Microfluidic Hepatocarcinoma Chip System: Effect of Anticancer Drug Solubility and Shear Stress. Pharmaceuticals, 16(9), 1330. https://doi.org/10.3390/ph16091330