
Curcuminoid Chalcones: Synthesis, Stability, and New Neuroprotective and Sonosensitising Activities

 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
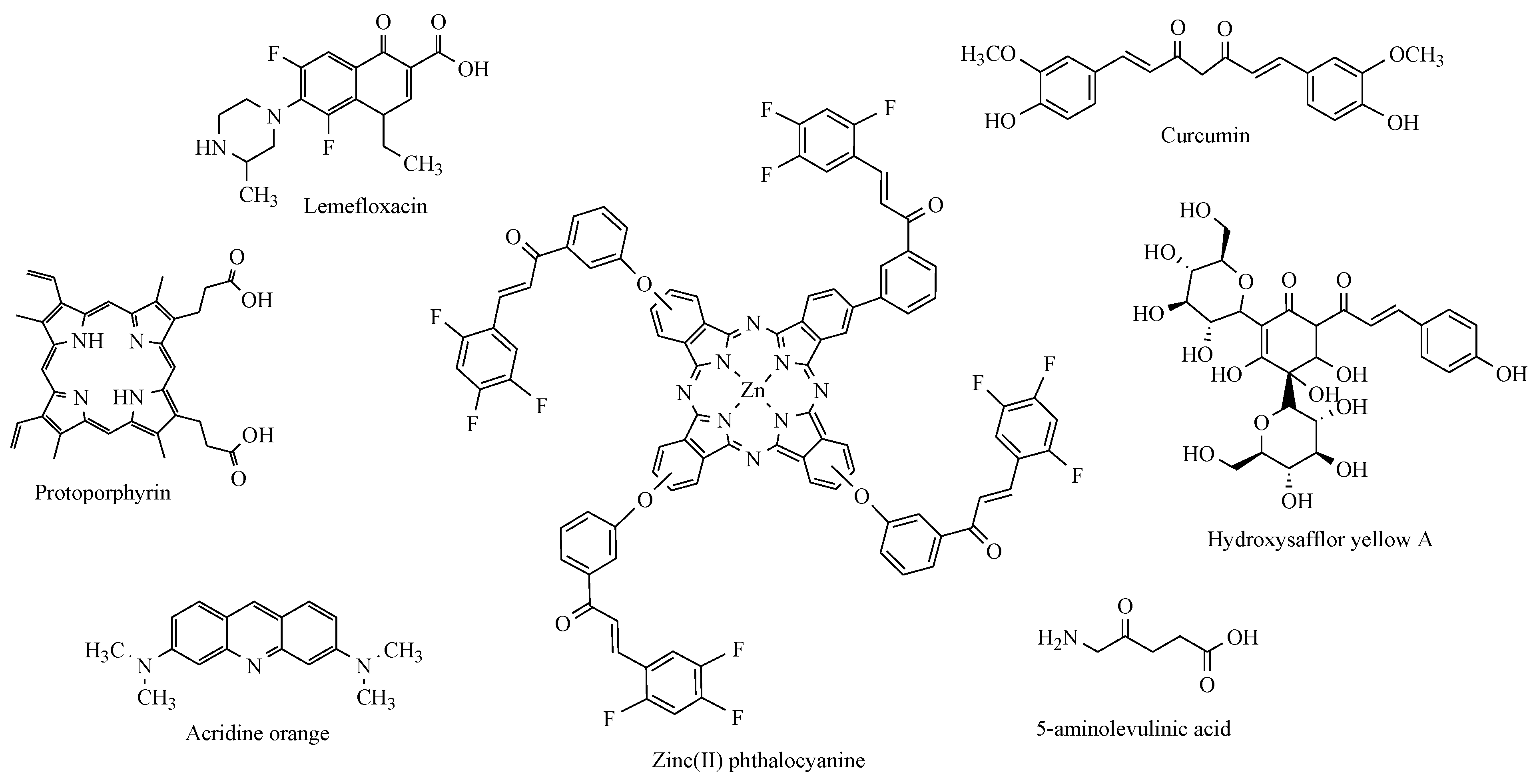
2. Results and Discussion
2.1. Chemical Parts
Synthesis of Curcuminoid Chalcone–NSAID Hybrid Derivatives
2.2. Biological Activity
2.2.1. Sonodynamic Studies
2.2.2. Anti-Inflammatory Activity
Inhibition of Cyclooxygenase-2 (COX-2)
Effect on Antioxidant Enzyme Activity
Hydroxyl Radical Antioxidant Capacity (HORAC)
2.2.3. Anticholinergic Activity
2.2.4. Estimation of Stability of Ester-Type Hybrid Compounds
Stress Test
Stability Test in Dissolution Solutions
3. Materials and Methods
3.1. Solvents and Chemicals
3.2. Instrumental Analysis
3.3. General Synthesis Procedure for Curcuminoid Chalcones 3a and 3b
3.4. General Synthesis Procedure for Curcuminoid Chalcone–NSAID Hybrid Derivatives (3ai–3biii) in the Reaction of Curcuminoid Chalcones with NSAIDs
3.5. General Procedure Synthesis of Aromatic Aldehyde–NSAID Intermediate Derivatives (2ai–2biii)
3.6. General Synthesis Procedure of Curcuminoid Chalcone–NSAID Hybrid Derivatives (3ai–3biii) in the Reaction of Aromatic Aldehyde–NSAID Intermediate Derivatives (2ai–2biii) with Acetophenone
3.7. Stability Tests of Hybrid Curcuminoid Chalcones
3.7.1. Apparatus and Chromatographic Conditions
3.7.2. Stress Test
Strong Acid, Strong Base, and Neutral Degradation
Oxidative Degradation
Photostability of Compound 2 and Compound 6 in the Solutions
Photostability of Compound 2 and Compound 6 in the Solid State
Stability Test in Release Solutions
3.8. Sonodynamic Method
Cell Culture and Viability Cells
3.9. Estimation of Antineurodegenerative, Anti-Inflammatory, and Antioxidant Activity
3.9.1. Sample Preparation
3.9.2. Effect on AChE and BChE Activity
3.9.3. Effect on COX-2 Activity
3.9.4. Effect on GPx and GR Activity
3.9.5. Effect on CAT Activity
3.9.6. HORAC
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pawełczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Molecular Consortia—Various Structural and Synthetic Concepts for More Effective Therapeutics Synthesis. Int. J. Mol. Sci. 2018, 19, 1104. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A.O.; et al. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef] [PubMed]
- Elkanzi, N.A.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Fatin, M.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega. 2022, 7, 27769–27786. [Google Scholar] [CrossRef] [PubMed]
- Tekale, S.; Mashele, S.; Pooe, O.; Thore, S.; Kendrekar, P.; Pawar, R. Biological Role of Chalcones in Medicinal Chemistry, from the Edited Volume Vector-Borne Diseases 2020; Claborn, D., Bhattacharya, S., Roy, S., Eds.; IntechOpen: London, UK, 2020; ISBN 978-1-83880-038-3. [Google Scholar] [CrossRef]
- Rammohan, A.; Reddy, J.S.; Sravya, G.; Rao, C.N.; Zyryanov, G.V. Synthesis and Pharmacological Study of Some Novel Pyrimidines Chalcone synthesis, properties and medicinal applications: A review. Environ. Chem. Lett. 2020, 18, 433–458. [Google Scholar] [CrossRef]
- Kamei, R.; Kadokura, M.; Kitagawa, Y.; Hazeki, O.; Oikawa, S. 2′-benzyloxychalcone derivatives stimulate glucose uptake in 3T3-L1 adipocytes. Life Sci. 2003, 73, 2091–2099. [Google Scholar] [CrossRef]
- Gaonkar, S.L.; Vignesh, U.N. Synthesis and Pharmacological Properties of Chalcones: A Review. Res. Chem. Intermed. 2017, 43, 6043–6077. [Google Scholar] [CrossRef]
- Thapa, P.; Upadhyay, S.P.; Suo, W.Z.; Singh, V.; Gurung, P.; Lee, E.S.; Sharma, R.; Sharma, M. Chalcone and its analogs: Therapeutic and diagnostic applications in Alzheimer’s disease. Bioorg. Chem. 2021, 108, 104681. [Google Scholar] [CrossRef]
- Marquina, S.; Maldonado-Santiago, M.; Sánchez-Carranza, J.N.; Antúnez-Mojica, M.; González-Maya, L.; Razo-Hernández, R.S.; Alvarez, L. Design, synthesis and QSAR study of 2′-hydroxy-4′-alkoxy chalcone derivatives that exert cytotoxic activity by the mitochondrial apoptotic pathway. Bioorg. Med. Chem. 2019, 27, 43–54. [Google Scholar] [CrossRef]
- Go, M.L.; Wu, X.; Liu, X.L. Chalcones: An update on cytotoxic and chemoprotective properties. Curr. Med. Chem. 2005, 12, 483–499. [Google Scholar] [CrossRef]
- Constantinescu, T.; Lungu, C.N. Anticancer Activity of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2021, 22, 11306. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Burmaoğlu, S. Fluoro-substituted chalcones as the compounds having anticancer activity. Karaelmas Fen Ve Müh. Derg. 2017, 7, 658–664. [Google Scholar]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, H.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73, e557s. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-J.; Li, J.-P.; Fan, C.-H.; Liu, H.-L.; Yeh, C.-K. Ultrasound in tumor immunotherapy: Current status and future developments. J. Control. Release 2020, 323, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Toccaceli, G.; Barbagallo, G.; Peschillo, S. Low-intensity focused ultrasound for the treatment of brain diseases: Safety and feasibility. Theranostics 2019, 9, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Bonosi, L.; Marino, S.; Benigno, U.E.; Musso, S.; Buscemi, F.; Giardina, K.; Gerardi, R.; Brunasso, L.; Costanzo, R.; Iacopino, D.G.; et al. Sonodynamic therapy and magnetic resonance-guided focused ultrasound: New therapeutic strategy in glioblastoma. J. Neurooncol. 2023, 163, 219–238. [Google Scholar] [CrossRef]
- Wan, G.Y.; Liu, Y.; Chen, B.W.; Liu, Y.Y.; Wang, Y.S.; Zhang, N. Recent advances of sonodynamic therapy in cancer treatment. Cancer Biol. Med. 2016, 13, 325–338. [Google Scholar] [CrossRef]
- Costley, D.; Mc Ewan, C.; Fowley, C.; McHale, A.P.; Atchison, J.; Nomikou, N.; Callan, J.F. Treating cancer with sonodynamic therapy: A review. Int. J. Hyperth. 2015, 31, 107–117. [Google Scholar] [CrossRef]
- Suzuki, N.; Okada, K.; Chida, S.; Komori, C.; Shimada, Y.; Suzuki, T. Antitumor effect of acridine orange under ultrasonic irradiation in vitro. Anticancer Res. 2007, 27, 4179–4184. [Google Scholar] [PubMed]
- Yumita, N.; Nishigaki, R.; Sakata, I.; Nakajima, S.; Umemura, S. Sonodynamically Induced Antitumor Effect of 4-Formyloximethylidene-3-hydroxy-2-vinyl-deuterio-porphynyl(IX)-6,7-diaspartic Acid (ATX-S10). Jpn. J. Cancer Res. 2000, 91, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Kantekin, H.; Yalazan, H.; Kahriman, N.; Ertem, B.; Serdaroğlu, V.; Pişkin, M.; Durmuş, M. New peripherally and non-peripherally tetra-substituted metal-free, magnesium(II) and zinc(II) phthalocyanine derivatives fused chalcone units: Design, synthesis, spectroscopic characterization, photochemistry and photophysics. J. Photochem. Photobiol. 2018, 361, 1–11. [Google Scholar] [CrossRef]
- Kamiloglu, A. Photochemical properties of fluoro-chalcone substituted peripherally tetra Zn(II)Pc and Mg(II)Pc. J. Incl. Phenom. Macrocycl. Chem. 2021, 99, 185–196. [Google Scholar] [CrossRef]
- Gong, Z.; Dai, Z. Design and Challenges of Sonodynamic Therapy System for Cancer Theranostics: From Equipment to Sensitizers. Adv. Sci. 2021, 12, 2002178. [Google Scholar] [CrossRef] [PubMed]
- Umemura, K.; Yumita, N.; Nishigaki, R.; Umemura, S.I. Sonodynamically induced antitumor effect of pheophorbide a. Cancer lett. 1996, 102, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Rengeng, L.; Qianyu, Z.; Yuehong, L.; Zhongzhong, P.; Libo, L. Sonodynamic therapy, a treatment developing from photodynamic therapy. Photodiagnosis Photodyn. Ther. 2017, 19, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yumita, N.; Nishigaki, R.; Umemura, S. Sonodynamically induced antitumor effect of Photofrin II on colon 26 carcinoma. J. Cancer Res. Clin. Oncol. 2000, 126, 601–606. [Google Scholar] [CrossRef]
- Yumita, N.; Iwase, Y.; Umemura, S.I.; Chen, F.S.; Momose, Y. Sonodynamically-induced anticancer effects of polyethylene glycol-modified carbon nano tubes. Anticancer Res. 2020, 40, 2549–2557. [Google Scholar] [CrossRef]
- Li, D.; Yang, Y.; Li, D.; Pan, J.; Chu, C.; Liu, G. Organic sonosensitizers for sonodynamic therapy: From small molecules and nanoparticles toward clinical development. Small 2021, 17, 2101976. [Google Scholar] [CrossRef]
- Sakusabe, N.; Okada, K.; Sato, K.; Kamada, S.; Yoshida, Y.; Suzuki, T. Enhanced sonodynamic antitumor effect of ultrasound in the presence of nonsteroidal anti-inflammatory drugs. Jpn. J. Cancer Res. 1999, 90, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Sowa-Kasprzak, K.; Józkowiak, M.; Olender, D.; Pawełczyk, A.; Piotrowska-Kempisty, H.; Zaprutko, L. Curcumin–Triterpene Type Hybrid as Effective Sonosensitizers for Sonodynamic Therapy in Oral Squamous Cell Carcinoma. Pharmaceutics 2023, 15, 2008. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Itoi, E.; Miyakoshi, N.; Nakajima, M.; Suzuki, T.; Nishida, J. Enhanced antitumor effect of ultrasound in the presence of piroxicam in a mouse air pouch model. Jpn. J. Cancer Res. 2002, 93, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Olender, D.; Sowa-Kasprzak, K.; Pawełczyk, A.; Skóra, B.; Zaprutko, L.; Szychowski, K.A. Curcuminoid Chalcones: Synthesis and Biological Activity against the Human Colon Carcinoma (Caco-2) Cell Line. Curr. Med. Chem. 2023; submitted and accepted to press. [Google Scholar]
- Teimuri-Mdfrad, R.; Rahimpour, K.; Gholizadeh, M. Design, synthesis, characterization and fluorescence property evaluation of dehydroacetic acid-based chalcones. J. Iran. Chem. Soc. 2020, 17, 1103–1109. [Google Scholar] [CrossRef]
- Ganapati, D.; Yadav, D.; Wagh, P. Claisen-Schmidt Condensation using Green Catalytic Processes: A Critical Review. ChemistrySelect 2020, 5, 9059–9085. [Google Scholar] [CrossRef]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-2 inhibitors. Lancet 1999, 353, 307–314. [Google Scholar] [CrossRef]
- Gasparini, L.; Ongini, E.; Wenk, G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: Old and new mechanisms of action. J. Neurochem. 2004, 91, 521–536. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Navarrete, L.P.; González, A.; González-Canacer, A.; Guzmán-Martínez, L.; Cortés, N. Inflammation: A Major Target for Compounds to Control Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1199–1213. [Google Scholar] [CrossRef]
- Studzińska-Sroka, E.; Majchrzak-Celińska, A.; Zalewski, P.; Szwajgier, D.; Baranowska-Wójcik, E.; Kaproń, B.; Plech, T.; Żarowski, T.; Cielecka-Piontek, J. Lichen-derived compounds and extracts as biologically active substances with anticancer and neuroprotective properties. Pharmaceuticals 2021, 14, 1293. [Google Scholar] [CrossRef]
- Rehmann, A.U.; Ijaz, A.S.; Murtaza, G.; Hussain, I. Prediction of chemical stability of ibuprofen in solution: An accelerated aging study. Asian. J. Chem. 2012, 24, 4939–4943. [Google Scholar]
- Watanabe, M.; de Moura Neiva, L.B.; da Costa Santos, C.X.; Martins Laurindo, F.R.; de Fátima Fernandes Vattimo, M. Isoflavone and the heme oxygenase system in ischemic acute kidney injury in rats. Food Chem. Toxicol. 2007, 45, 2366–2371. [Google Scholar] [CrossRef]
- Szwajgier, D.; Baranowska-Wójcik, E.; Kukula-Koch, V.; Kowalik, K.; Polak-Berecka, M.; Waśko, A. Evolution of the anticholinesterase, antioxidant, and anti-inflammatory activity of Epilobium angustifolium L. infusion during in vitro digestion. J. Funct. Foods 2021, 85, 104645. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | COX-2 Inhibition (%) | Equivalent Concentration of Acetylsalicylic Acid (mg/mL) |
|---|---|---|
| 1 | 6.7 ± 0.1 | 6.1 ± 0.1 |
| 2 | - | - |
| 3 | 15.2 ± 0.1 | 7.1 ± 0.1 |
| 4 | 22.4 ± 0.1 | 8.0 ± 0.1 |
| 5 | - | - |
| 6 | - | - |
| 7 | 20.2 ± 0.2 | 7.7 ± 0.1 |
| Compound | GPx | GR | CAT | |||
|---|---|---|---|---|---|---|
| Inhibition (%) | Inhibition (nmol Depleted NADPH/min) | Inhibition (%) | Inhibition (nmol Depleted NADPH/min) | Inhibition (%) | Inhibition of the H2O2 Depletion (mm/dm3/min) | |
| 1 | 30.3 ± 3.6 | 60.3 ± 7.1 | 7.6 ± 2.4 | 286 ± 90 | 78.6 ± 2.8 | 0.26 ± 0.01 |
| 2 | 17.0 ± 3.4 | 33.8 ± 6.8 | 27.1 ± 4.0 | 1018 ± 150 | - | - |
| 3 | 31.9 ± 4.0 | 63.0 ± 7.9 | - | - | 75.2 ± 5.8 | 0.24 ±0.02 |
| 4 | - | - | 19.9 ± 1.1 | 747 ± 41 | - | - |
| 5 | - | - | 29.1 ± 4.4 | 1093 ± 165 | 12.5 ±2.4 | 0.03 ± 0.01 |
| 6 | 17.4 ± 1.7 | 34.6 ± 3.3 | - | - | 41.9 ± 1.2 | 0.15 ± 0.01 |
| 7 | 32.4 ± 2.8 | 64.4 ± 5.5 | 41.8 ± 3.1 | 1570 ± 116 | 88.1 ± 4.0 | 0.22 ± 0.01 |
| Compound | Gallic Acid Equivalents (μg/mL) |
|---|---|
| 1 | 18.7 ± 3.8 |
| 2 | 24.2 ± 2.3 |
| 3 | 21.0 ± 4.2 |
| 4 | 35.7 ± 4.2 |
| 5 | 6.1 ± 2.5 |
| 6 | 20.4 ± 3.0 |
| 7 | 11.5 ± 1.3 |
| Compound | AChE % | AChE (µg/mL) (Equivalent Concentration of Reference Compound) | BChE % | BChE (µg/mL) (Equivalent Concentration of Reference Compound | ||
|---|---|---|---|---|---|---|
| Rivastigmine | Magniflorine | Rivastigmine | Magniflorine | |||
| 1 | 80.5 ± 8.2 | 0.46 ± 0.05 | 0.31 ± 0.03 | 78.0 ± 4.6 | 0.23 ± 0.01 | 0.30 ± 0.02 |
| 2 | 90.5 ± 4.2 | 0.52 ± 0.02 | 0.34 ± 0.02 | 89.7 ± 4.8 | 0.26 ± 0.01 | 0.35 ± 0.02 |
| 3 | 83.4 ± 6.0 | 0.48 ± 0.03 | 0.32 ± 0.02 | 90.8 ± 3.0 | 0.26 ± 0.01 | 0.35 ± 0.01 |
| 4 | 87.3 ± 4.9 | 0.50 ± 0.03 | 0.33 ± 0.02 | 64.2 ± 4.9 | 0.19 ± 0.01 | 0.25 ± 0.02 |
| 5 | 65.7 ± 4.7 | 0.37 ± 0.03 | 0.25 ± 0.02 | 58.1 ± 3.4 | 0.17 ± 0.01 | 0.23 ± 0.01 |
| 6 | 73.7 ± 5.1 | 0.42 ± 0.03 | 0.28 ± 0.02 | 77.3 ± 5.2 | 0.22 ± 0.02 | 0.30 ± 0.02 |
| 7 | 89.8 ± 5.8 | 0.51 ± 0.03 | 0.34 ± 0.01 | 94.4 ± 4.1 | 0.27 ± 0.01 | 0.37 ± 0.02 |
| Environment | Conditions | C0, μg/mL | Ct, μg/mL | % Decomposition | ICH Classification |
|---|---|---|---|---|---|
| 2 | |||||
| HCl | 0.1 M, 90 °C, 16 h | 61.3 | 5.0 | 91.8 | |
| HCl | 0.01 M, 40 °C, 8 h | 60.0 | 33.7 | 43.8 | very unstable |
| NaOH | 0.1 M, 90 °C, 16 h | 61.3 | 0 | 100 | |
| NaOH | 0.01 M, 25 °C, 2 h | 60.0 | 0 | 100 | extremely unstable |
| H2O | 90 °C, 24 h | 61.3 | 46.3 | 24.4 | unstable |
| H2O2 | 1%, 30 min | 30.0 | 29.9 | <0.5 | |
| H2O2 | 3%, 24 h | 30.0 | 29.8 | <0.5 | |
| H2O2 | 10%, 24 h | 30.0 | 30.1 | <0.5 | practically stable |
| 6 | |||||
| HCl | 0.1 M, 90 °C, 16 h | 66.9 | 1.5 | 97.8 | |
| HCl | 0.01 M, 40 °C, 8 h | 60.0 | 49.3 | 17.8 | very unstable |
| NaOH | 0.1 M, 90 °C, 16 h | 66.9 | 0 | 100 | |
| NaOH | 0.01 M, 25 °C, 2 h | 60.0 | 0 | 100 | extremely unstable |
| H2O | 90 °C, 24 h | 66.9 | 50.9 | 23.9 | unstable |
| H2O2 | 1%, 30 min | 30.0 | 29.7 | <0.5 | |
| H2O2 | 3%, 24 h | 30.0 | 29.9 | <0.5 | |
| H2O2 | 10%, 24 h | 30.0 | 30.0 | <0.5 | practically stable |
| Environment | Sample | C0 [μg/mL] | Ct [μg/mL] | %Decomposition | ICH Classification |
| 2 | |||||
| MeOH | Protected | 15.0 | 4.8 | 68.2 | |
| Not Protected | 15.0 | 0 | 100 | Photolabile | |
| 6 | |||||
| MeOH | Protected | 15.0 | 13.8 | 7.9 | |
| Not Protected | 15.0 | 0 | 100 | Photolabile | |
| Environment | Sample | Dose [lux·h] | Compatibility of the IR spectrum with the spectrum before irradiation | ICH classification | |
| 2 | |||||
| Solid phase | Protected | 1.2 × 106 | 0.9995 | ||
| Not Protected | 1.2 × 106 | 0.9404 | |||
| Protected | 6.0 × 106 | 0.9994 | |||
| Not Protected | 6.0 × 106 | 0.9017 | Photolabile | ||
| 6 | |||||
| Solid phase | Protected | 1.2 × 106 | 0.9997 | ||
| Not Protected | 1.2 × 106 | 0.9962 | |||
| Protected | 6.0 × 106 | 0.9993 | |||
| Not Protected | 6.0 × 106 | 0.9752 | Photolabile | ||
| Time (Days) | 2 | 6 | ||
|---|---|---|---|---|
| pH 1.2 | pH 6.8 | pH 1.2 | pH 6.8 | |
| C, % | C, % | C, % | C, % | |
| 0 | 100 | 100 | 100 | 100 |
| 1 | 88.9 | 94.9 | 87.7 | 90.5 |
| 2 | 88.9 | 92.7 | 85.1 | 88.2 |
| 5 | 83.1 | 88.6 | 81.0 | 84.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olender, D.; Józkowiak, M.; Piotrowska-Kempisty, H.; Sowa-Kasprzak, K.; Zaprutko, L.; Muszalska-Kolos, I.; Baranowska-Wójcik, E.; Szwajgier, D. Curcuminoid Chalcones: Synthesis, Stability, and New Neuroprotective and Sonosensitising Activities. Pharmaceuticals 2023, 16, 1331. https://doi.org/10.3390/ph16091331
Olender D, Józkowiak M, Piotrowska-Kempisty H, Sowa-Kasprzak K, Zaprutko L, Muszalska-Kolos I, Baranowska-Wójcik E, Szwajgier D. Curcuminoid Chalcones: Synthesis, Stability, and New Neuroprotective and Sonosensitising Activities. Pharmaceuticals. 2023; 16(9):1331. https://doi.org/10.3390/ph16091331
Chicago/Turabian StyleOlender, Dorota, Małgorzata Józkowiak, Hanna Piotrowska-Kempisty, Katarzyna Sowa-Kasprzak, Lucjusz Zaprutko, Izabela Muszalska-Kolos, Ewa Baranowska-Wójcik, and Dominik Szwajgier. 2023. "Curcuminoid Chalcones: Synthesis, Stability, and New Neuroprotective and Sonosensitising Activities" Pharmaceuticals 16, no. 9: 1331. https://doi.org/10.3390/ph16091331
APA StyleOlender, D., Józkowiak, M., Piotrowska-Kempisty, H., Sowa-Kasprzak, K., Zaprutko, L., Muszalska-Kolos, I., Baranowska-Wójcik, E., & Szwajgier, D. (2023). Curcuminoid Chalcones: Synthesis, Stability, and New Neuroprotective and Sonosensitising Activities. Pharmaceuticals, 16(9), 1331. https://doi.org/10.3390/ph16091331