



Antiviral Potential of Azathioprine and Its Derivative 6- Mercaptopurine: A Narrative Literature Review




Abstract
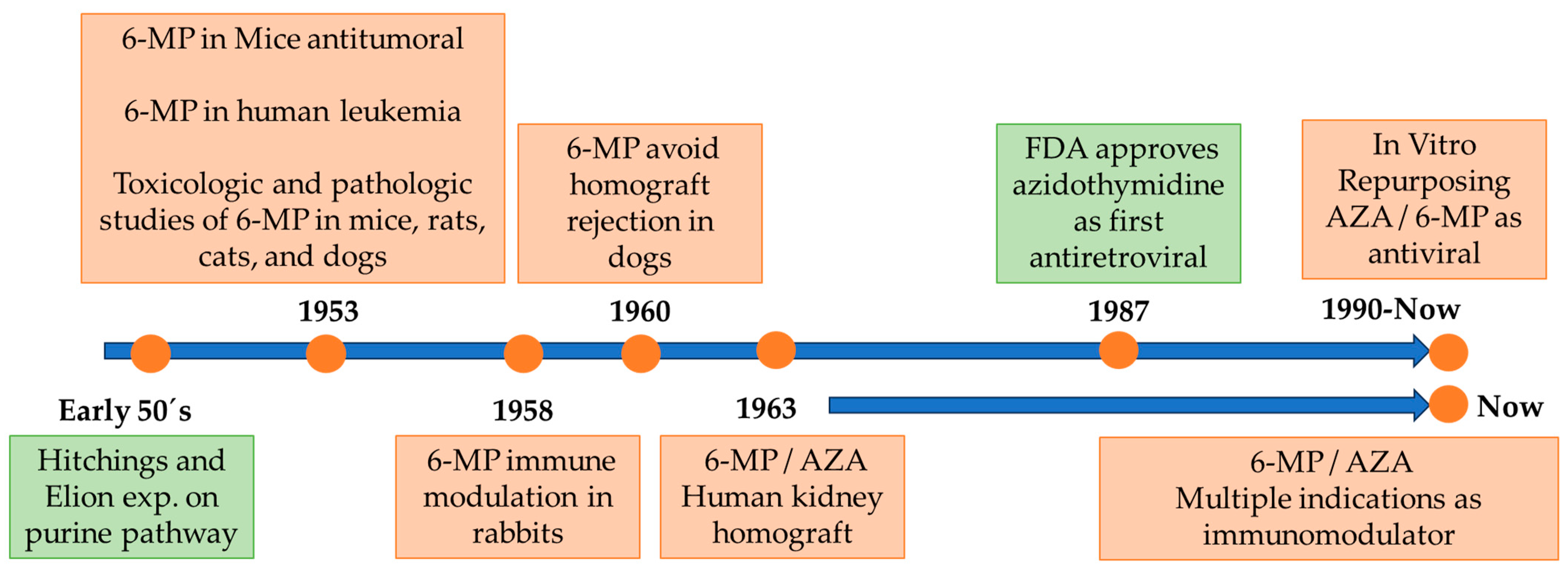
:1. Introduction: Brief History and Development of Azathioprine
2. Azathioprine Metabolism
3. AZA and Its Derivatives as Antiviral Agents
3.1. Herpesviruses
3.2. Flaviviruses
3.3. Morbilliviruses
3.4. Pneumoviruses
3.5. Coronaviruses
3.6. Influenzaviruses
4. Concluding Remarks and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Hitchings, G.H.; Elion, G.B. The chemistry and biochemistry of purine analogs. Ann. N. Y. Acad. Sci. 1954, 60, 195–199. [Google Scholar] [CrossRef]
- Elion, G.B.; Hitchings, G.H. Azathioprine. In Antineoplastic and Immunosuppressive Agents: Part II; Springer: Cham, Switzerland, 1975; pp. 404–425. [Google Scholar]
- Broen, J.C.A.; van Laar, J.M. Mycophenolate mofetil, azathioprine and tacrolimus: Mechanisms in rheumatology. Nat. Rev. Rheumatol. 2020, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.A.; Philips, F.; Sternberg, S.; Stock, C.; Elion, G. 6-Mercaptopurine: An inhibitor of mouse sarcoma 180. Proc. Am. Assoc. Cancer Res. 1953, 1, 9. [Google Scholar]
- Clarke, D.A.; Philips, F.S.; Sternberg, S.S.; Stock, C.C.; Elion, G.B.; Hitchings, G.H. 6-Mercaptopurine: Effects in mouse sarcoma 180 and in normal animals. Cancer Res. 1953, 13, 593–604. [Google Scholar] [PubMed]
- Sugiura, K. The effect of 6-thiopurine and of 1, 9-di (methane sulfonoxy) nonane on the growth of a variety of mouse and rat tumors. Proc. Am. Assoc. Cancer Res. 1953, 1, 55. [Google Scholar]
- Philips, F.; Sternberg, S.; Clarke, D.; Hitchings, G. Effects of 6-mercaptopurine in mammals. Proc. Am. Assoc. Cancer Res. 1953, 1, 42–43. [Google Scholar]
- Burchenal, J.H.; Murphy, M.L.; Ellison, R.R.; Sykes, M.P.; Tan, T.C.; Leone, L.A.; Karnofsky, D.A.; Craver, L.F.; Dargeon, H.W.; Rhoads, C.P. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases. Blood 1953, 8, 965–999. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.; Stack, J.; Dameshek, W. Effect of 6-mercaptopurine on antibody production. Proc. Soc. Exp. Biol. Med. 1958, 99, 164–167. [Google Scholar] [CrossRef]
- Calne, R.Y. The rejection of renal homografts. Inhibition in dogs by 6-mercaptopurine. Lancet 1960, 1, 417–418. [Google Scholar] [CrossRef]
- Murray, J.E.; Merrill, J.P.; Harrison, J.H.; Wilson, R.E.; Dammin, G.J. Prolonged survival of human-kidney homografts by immunosuppressive drug therapy. N. Engl. J. Med. 1963, 268, 1315–1323. [Google Scholar] [CrossRef]
- Hirsch, M.S.; Kaplan, J.C. Treatment of human immunodeficiency virus infections. Antimicrob. Agents Chemother. 1987, 31, 839–843. [Google Scholar] [CrossRef]
- Sariri, R.; Khalili, G. Synthesis of purine antiviral agents, hypoxanthine and 6-mercaptopurine. Russ. J. Org. Chem. 2002, 38, 1053–1055. [Google Scholar] [CrossRef]
- Dubinsky, M.C. Azathioprine, 6-mercaptopurine in inflammatory bowel disease: Pharmacology, efficacy, and safety. Clin. Gastroenterol. Hepatol. 2004, 2, 731–743. [Google Scholar] [CrossRef]
- Cara, C.J.; Pena, A.S.; Sans, M.; Rodrigo, L.; Guerrero-Esteo, M.; Hinojosa, J.; Garcia-Paredes, J.; Guijarro, L.G. Reviewing the mechanism of action of thiopurine drugs: Towards a new paradigm in clinical practice. Med. Sci. Monit. 2004, 10, RA247–RA254. [Google Scholar]
- Diehl, R.; Ferrara, F.; Muller, C.; Dreyer, A.Y.; McLeod, D.D.; Fricke, S.; Boltze, J. Immunosuppression for in vivo research: State-of-the-art protocols and experimental approaches. Cell Mol. Immunol. 2017, 14, 146–179. [Google Scholar] [CrossRef]
- Karran, P.; Attard, N. Thiopurines in current medical practice: Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8, 24–36. [Google Scholar] [CrossRef]
- McWilliam, M.; Khan, U. Azathioprine and the neurologist. Pract. Neurol. 2020, 20, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Orts-Arroyo, M.; Castro, I.; Martínez-Lillo, J. Detection of Hypoxanthine from Inosine and Unusual Hydrolysis of Immunosuppressive Drug Azathioprine through the Formation of a Diruthenium(III) System. Biosensors 2021, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- de Boer, N.K.; van Bodegraven, A.A.; Jharap, B.; de Graaf, P.; Mulder, C.J. Drug Insight: Pharmacology and toxicity of thiopurine therapy in patients with IBD. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 686–694. [Google Scholar] [CrossRef] [PubMed]
- van Gelder, T.; van Schaik, R.H.; Hesselink, D.A. Pharmacogenetics and immunosuppressive drugs in solid organ transplantation. Nat. Rev. Nephrol. 2014, 10, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Becker, C.; Kiesslich, R.; Neurath, M.F. Drug insight: Novel small molecules and drugs for immunosuppression. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Investig. 2003, 111, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Adam, L.; Phulukdaree, A.; Soma, P. Effective long-term solution to therapeutic remission in Inflammatory Bowel Disease: Role of Azathioprine. Biomed. Pharmacother. 2018, 100, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Chocair, P.R.; Neves, P.; Mohrbacher, S.; Neto, M.P.; Sato, V.A.H.; Oliveira, E.S.; Barbosa, L.V.; Bales, A.M.; da Silva, F.P.; Cuvello-Neto, A.L.; et al. Case Report: Azathioprine: An Old and Wronged Immunosuppressant. Front. Immunol. 2022, 13, 903012. [Google Scholar] [CrossRef] [PubMed]
- Nevins, T.E.; Thomas, W. Quantitative patterns of azathioprine adherence after renal transplantation. Transplantation 2009, 87, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L.; Van Loon, J.A.; Weinshilboum, R.M. Pharmacogenetics of acute azathioprine toxicity: Relationship to thiopurine methyltransferase genetic polymorphism. Clin. Pharmacol. Ther. 1989, 46, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L. TPMT in the treatment of Crohn’s disease with azathioprine. Gut 2002, 51, 143–146. [Google Scholar] [CrossRef]
- Stocco, G.; Pelin, M.; Franca, R.; De Iudicibus, S.; Cuzzoni, E.; Favretto, D.; Martelossi, S.; Ventura, A.; Decorti, G. Pharmacogenetics of azathioprine in inflammatory bowel disease: A role for glutathione-S-transferase? World J. Gastroenterol. 2014, 20, 3534–3541. [Google Scholar] [CrossRef]
- Schmiegelow, K.; Nielsen, S.N.; Frandsen, T.L.; Nersting, J. Mercaptopurine/Methotrexate maintenance therapy of childhood acute lymphoblastic leukemia: Clinical facts and fiction. J. Pediatr. Hematol. Oncol. 2014, 36, 503–517. [Google Scholar] [CrossRef]
- Wallisch, K.; Trepanier, L.A. Incidence, timing, and risk factors of azathioprine hepatotoxicosis in dogs. J. Vet. Intern. Med. 2015, 29, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Kidd, L.B.; Salavaggione, O.E.; Szumlanski, C.L.; Miller, J.L.; Weinshilboum, R.M.; Trepanier, L. Thiopurine methyltransferase activity in red blood cells of dogs. J. Vet. Intern. Med. 2004, 18, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Silvariño, R.; Tafuri, J.; Mérola, V.; Romero, C.; Alonso, J. Herpes zóster cutáneo diseminado en paciente con artritis reumatoide. Arch. Med. Interna 2010, 32, 22–24. [Google Scholar]
- Bermejo, A.; Uranga, A.I.; D’Atri, G.; Deane, L.; Olivares, L.; Pizzariello, G.; Forero, O.; Coringrato, M.; Maronna, E. Herpes simplex en pacientes inmunocomprometidos por HIV y terapia esteroidea en dermatosis ampollares. Dermatol. Argent. 2011, 17, 52–56. [Google Scholar]
- Borba, E.F.; Ribeiro, A.C.; Martin, P.; Costa, L.P.; Guedes, L.K.; Bonfa, E. Incidence, risk factors, and outcome of Herpes zoster in systemic lupus erythematosus. J. Clin. Rheumatol. 2010, 16, 119–122. [Google Scholar] [CrossRef]
- Ingelfinger, F.; Sparano, C.; Bamert, D.; Reyes-Leiva, D.; Sethi, A.; Rindlisbacher, L.; Zwicky, P.; Kreutmair, S.; Widmer, C.C.; Mundt, S.; et al. Azathioprine therapy induces selective NK cell depletion and IFN-gamma deficiency predisposing to herpesvirus reactivation. J. Allergy Clin. Immunol. 2023, 151, 280–286.e2. [Google Scholar] [CrossRef] [PubMed]
- Beale, K.M.; Altman, D.; Clemmons, R.R.; Bolon, B. Systemic toxicosis associated with azathioprine administration in domestic cats. Am. J. Vet. Res. 1992, 53, 1236–1240. [Google Scholar] [CrossRef]
- Willis, R.C.; Carson, D.A.; Seegmiller, J.E. Adenosine kinase initiates the major route of ribavirin activation in a cultured human cell line. Proc. Natl. Acad. Sci. USA 1978, 75, 3042–3044. [Google Scholar] [CrossRef]
- Haglund, S.; Taipalensuu, J.; Peterson, C.; Almer, S. IMPDH activity in thiopurine-treated patients with inflammatory bowel disease–relation to TPMT activity and metabolite concentrations. Br. J. Clin. Pharmacol. 2008, 65, 69–77. [Google Scholar] [CrossRef]
- Shiraki, K.; Ishibashi, M.; Okuno, T.; Kokado, Y.; Takahara, S.; Yamanishi, K. Effects of cyclosporine, azathioprine, mozoribine, and prednisolone on replication of human cytomegalovirus. Transplant. Proc. 1990, 22, 1682–1685. [Google Scholar]
- Shiraki, K.; Ishibashi, M.; Okuno, T.; Namazue, J.; Yamanishi, K.; Sonoda, T.; Takahashi, M. Immunosuppressive dose of azathioprine inhibits replication of human cytomegalovirus in vitro. Arch. Virol. 1991, 117, 165–171. [Google Scholar] [CrossRef]
- Stangl, J.R.; Carroll, K.L.; Illichmann, M.; Striker, R. Effect of antimetabolite immunosuppressants on Flaviviridae, including hepatitis C virus. Transplantation 2004, 77, 562–567. [Google Scholar] [CrossRef]
- Hoover, S.; Striker, R. Thiopurines inhibit bovine viral diarrhea virus production in a thiopurine methyltransferase-dependent manner. J. Gen. Virol. 2008, 89, 1000–1009. [Google Scholar] [CrossRef]
- Chou, C.Y.; Chien, C.H.; Han, Y.S.; Prebanda, M.T.; Hsieh, H.P.; Turk, B.; Chang, G.G.; Chen, X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008, 75, 1601–1609. [Google Scholar] [CrossRef]
- Lim, P.Y.; Keating, J.A.; Hoover, S.; Striker, R.; Bernard, K.A. A thiopurine drug inhibits West Nile virus production in cell culture, but not in mice. PLoS ONE 2011, 6, e26697. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.W.; Cheng, S.C.; Chen, W.Y.; Lin, M.H.; Chuang, S.J.; Cheng, I.H.; Sun, C.Y.; Chou, C.Y. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antivir. Res. 2015, 115, 9–16. [Google Scholar] [CrossRef]
- de Carvalho, O.V.; Felix, D.M.; de Mendonca, L.R.; de Araujo, C.; de Oliveira Franca, R.F.; Cordeiro, M.T.; Silva Junior, A.; Pena, L.J. The thiopurine nucleoside analogue 6-methylmercaptopurine riboside (6MMPr) effectively blocks Zika virus replication. Int. J. Antimicrob. Agents 2017, 50, 718–725. [Google Scholar] [CrossRef]
- de Carvalho, O.V.; Felix, D.M.; de Camargo Tozato, C.; Fietto, J.L.R.; de Almeida, M.R.; Bressan, G.C.; Pena, L.J.; Silva-Junior, A. 6-methylmercaptopurine riboside, a thiopurine nucleoside with antiviral activity against canine distemper virus in vitro. Virol. J. 2017, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Xu, Z.X.; Jiang, N.; Zheng, Y.P.; Rameix-Welti, M.A.; Jiao, Y.Y.; Peng, X.L.; Wang, Y.; Eleouet, J.F.; Cen, S.; et al. High-throughput screening of active compounds against human respiratory syncytial virus. Virology 2019, 535, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Slaine, P.D.; Kleer, M.; Duguay, B.A.; Pringle, E.S.; Kadijk, E.; Ying, S.; Balgi, A.; Roberge, M.; McCormick, C.; Khaperskyy, D.A. Thiopurines activate an antiviral unfolded protein response that blocks influenza A virus glycoprotein accumulation. J. Virol. 2021, 95, e00453-21. [Google Scholar] [CrossRef]
- Heller, T.; Saito, S.; Auerbach, J.; Williams, T.; Moreen, T.R.; Jazwinski, A.; Cruz, B.; Jeurkar, N.; Sapp, R.; Luo, G.; et al. An in vitro model of hepatitis C virion production. Proc. Natl. Acad. Sci. USA 2005, 102, 2579–2583. [Google Scholar] [CrossRef]
- Zekry, A.; Gleeson, M.; Guney, S.; McCaughan, G.W. A prospective cross-over study comparing the effect of mycophenolate versus azathioprine on allograft function and viral load in liver transplant recipients with recurrent chronic HCV infection. Liver Transpl. 2004, 10, 52–57. [Google Scholar] [CrossRef]
- Carrillo-Hernández, M.Y.; Ruiz-Saenz, J.; Martínez-Gutiérrez, M. Coinfection of Zika with Dengue and Chikungunya virus. In Zika Virus Biology, Transmission, and Pathology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 117–127. [Google Scholar]
- Chen, X.; Chou, C.Y.; Chang, G.G. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir. Chem. Chemother. 2009, 19, 151–156. [Google Scholar] [CrossRef]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef]
- Mercorelli, B.; Palu, G.; Loregian, A. Drug Repurposing for Viral Infectious Diseases: How Far Are We? Trends Microbiol. 2018, 26, 865–876. [Google Scholar] [CrossRef]
- Sepúlveda, C.S.; García, C.C.; Damonte, E.B. Inhibitors of Nucleotide Biosynthesis as Candidates for a Wide Spectrum of Antiviral Chemotherapy. Microorganisms 2022, 10, 1631. [Google Scholar] [CrossRef]
- Dos Santos Nascimento, I.J.; de Aquino, T.M.; da Silva-Junior, E.F. Drug Repurposing: A Strategy for Discovering Inhibitors against Emerging Viral Infections. Curr. Med. Chem. 2021, 28, 2887–2942. [Google Scholar] [CrossRef]
- O’Connor, K.A.; Roth, B.L. Finding new tricks for old drugs: An efficient route for public-sector drug discovery. Nat. Rev. Drug Discov. 2005, 4, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, J.; Mohan, M.; Byrareddy, S.N. Drug Repurposing Approaches to Combating Viral Infections. J. Clin. Med. 2020, 9, 3777. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Sahasranaman, S.; Howard, D.; Roy, S. Clinical pharmacology and pharmacogenetics of thiopurines. Eur. J. Clin. Pharmacol. 2008, 64, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Leigh, I.M.; Proby, C.M.; Inman, G.J.; Harwood, C.A. Azathioprine: Friend or foe? Br. J. Dermatol. 2019, 180, 961–963. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Family or Gender | Composite | Genome | Year | Ref |
|---|---|---|---|---|---|
| VZV | HERPESVIRIDAE | AZA | DNA | 1990,1991 | [41,42] |
| HSV | HERPESVIRIDAE | AZA | DNA | 1990,1991 | [41,42] |
| HCMV | HERPESVIRIDAE | AZA | DNA | 1990,1991 | [41,42] |
| BVDV | FLAVIVIRUS | AZA | Positive single-stranded RNA | 2004 | [43] |
| BVDV | FLAVIVIRUS | 6-MP, 6-TI, 6-TGr, 6-MMPr | Positive single-stranded RNA | 2008 | [44] |
| SARS | CORONAVIRIDAE | 6-MP, 6-TGN | Negative single-stranded RNA | 2008 | [45] |
| WNV | FLAVIVIRUS | 6-MMPr | Positive single-stranded RNA | 2011 | [46] |
| DENV-2 | FLAVIVIRUS | 6-MMPr | Positive single-stranded RNA | 2011 | [46] |
| YFV | FLAVIVIRUS | 6-MMPr | Positive single-stranded RNA | 2011 | [46] |
| MERS | CORONAVIRIDAE | 6-MP, 6-TGN | Negative single-stranded RNA | 2015 | [47] |
| ZIKV | FLAVIVIRUS | 6-MMPr | Positive single-stranded RNA | 2017 | [48] |
| CDV | MORBILLIVIRUS | 6-MMPr | Negative single-stranded RNA | 2017 | [49] |
| RSV | PNEUMOVIRUS | AZA, 6-MP | Negative single-stranded RNA | 2019 | [50] |
| IAV | ORTHOMYXOVIRIDAE | 6-TGN, 6-Tgo | Negative segmented RNA | 2021 | [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rios-Usuga, C.; Martinez-Gutierrez, M.; Ruiz-Saenz, J. Antiviral Potential of Azathioprine and Its Derivative 6- Mercaptopurine: A Narrative Literature Review. Pharmaceuticals 2024, 17, 174. https://doi.org/10.3390/ph17020174
Rios-Usuga C, Martinez-Gutierrez M, Ruiz-Saenz J. Antiviral Potential of Azathioprine and Its Derivative 6- Mercaptopurine: A Narrative Literature Review. Pharmaceuticals. 2024; 17(2):174. https://doi.org/10.3390/ph17020174
Chicago/Turabian StyleRios-Usuga, Carolina, Marlen Martinez-Gutierrez, and Julian Ruiz-Saenz. 2024. "Antiviral Potential of Azathioprine and Its Derivative 6- Mercaptopurine: A Narrative Literature Review" Pharmaceuticals 17, no. 2: 174. https://doi.org/10.3390/ph17020174
APA StyleRios-Usuga, C., Martinez-Gutierrez, M., & Ruiz-Saenz, J. (2024). Antiviral Potential of Azathioprine and Its Derivative 6- Mercaptopurine: A Narrative Literature Review. Pharmaceuticals, 17(2), 174. https://doi.org/10.3390/ph17020174