
Small-Angle X-ray Scattering (SAXS) Used for the Identification of Nicomorphine Polymorphic Changes at the Early Stage to Avoid Varied Stability and Possible Side Effects

 , , , and
, , , and
Abstract
: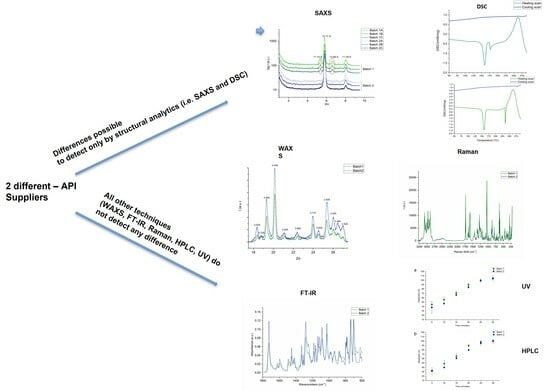
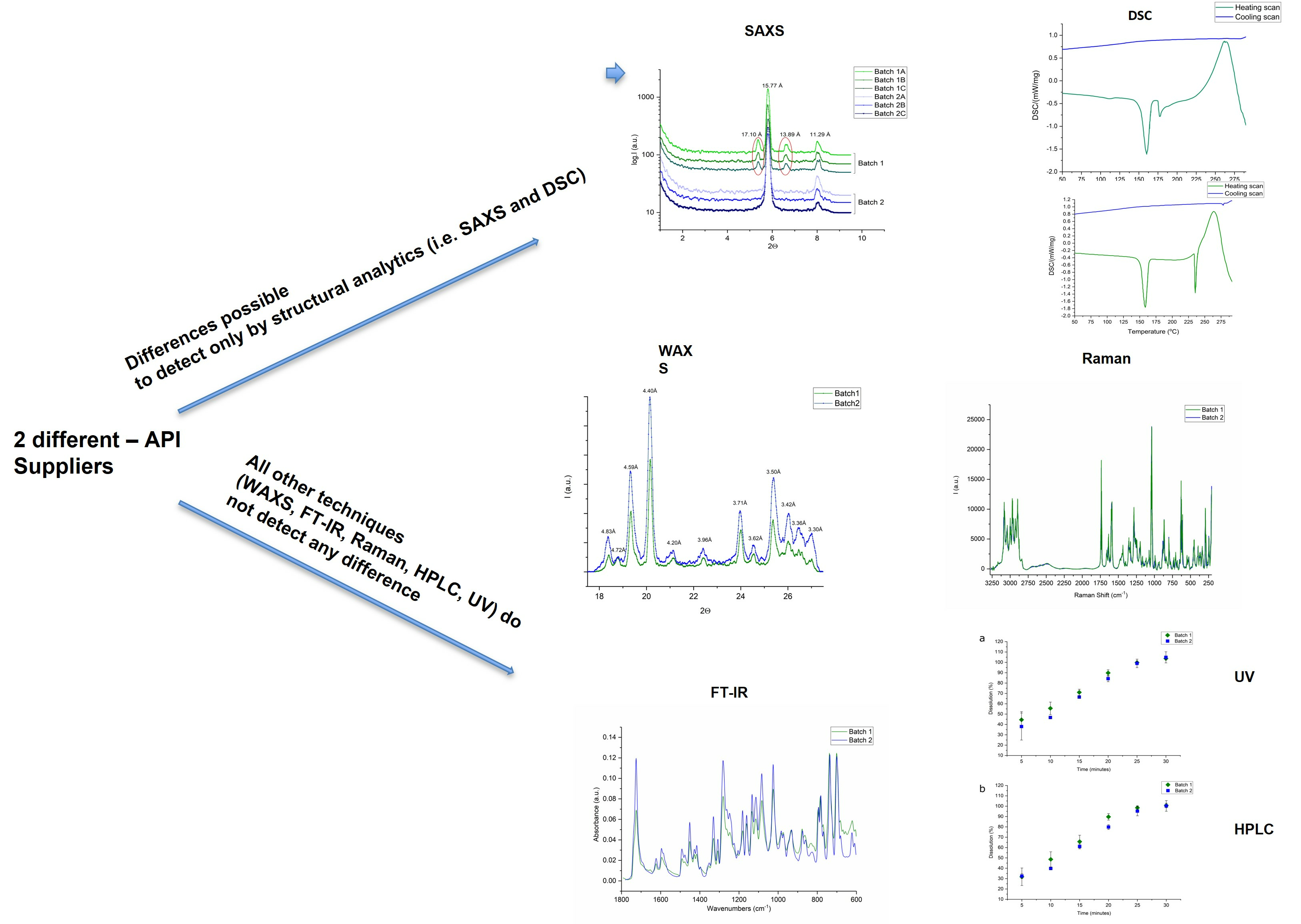{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. SWAXS
2.2. ATR-FTIR

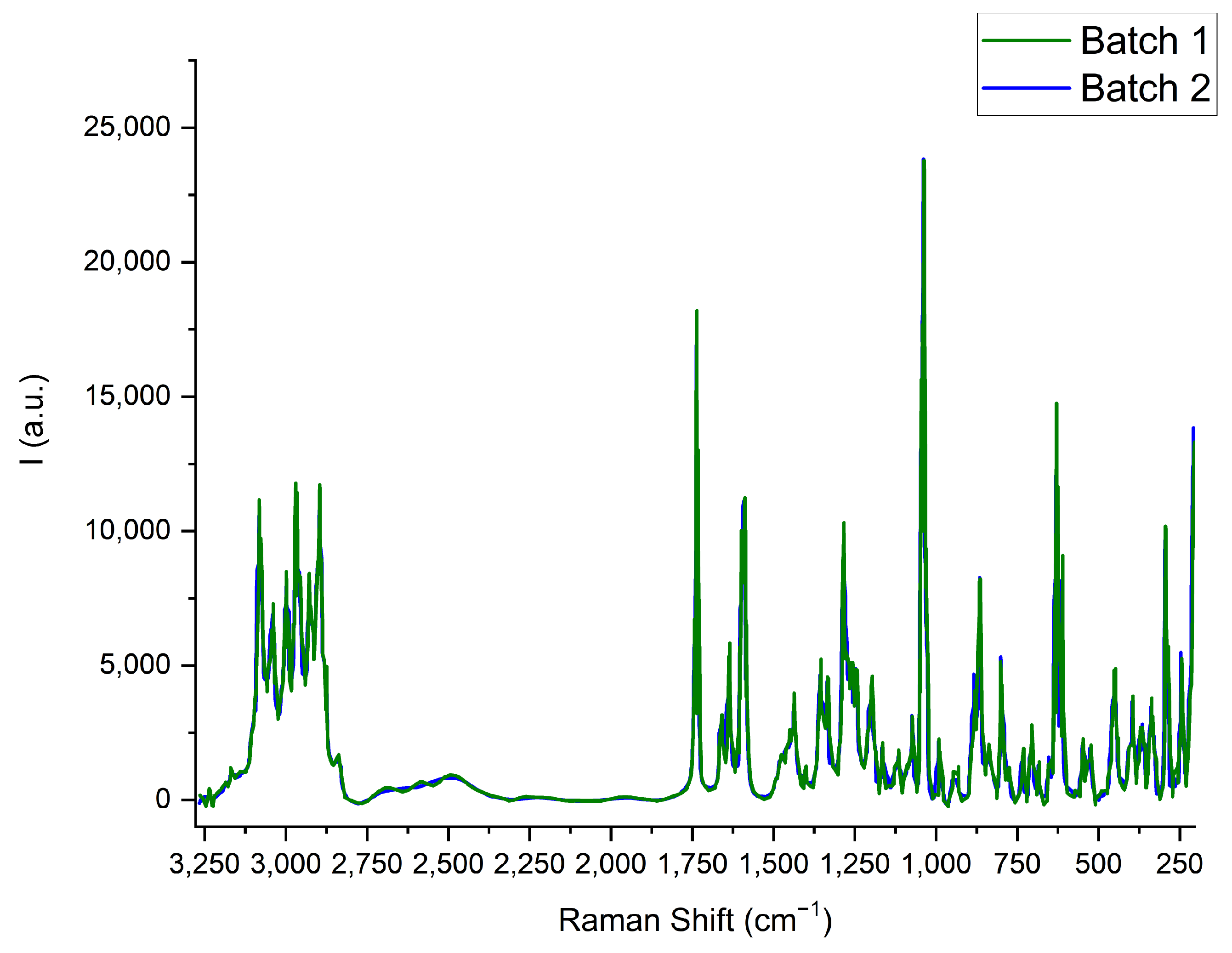
2.3. Raman
2.4. Dissolution
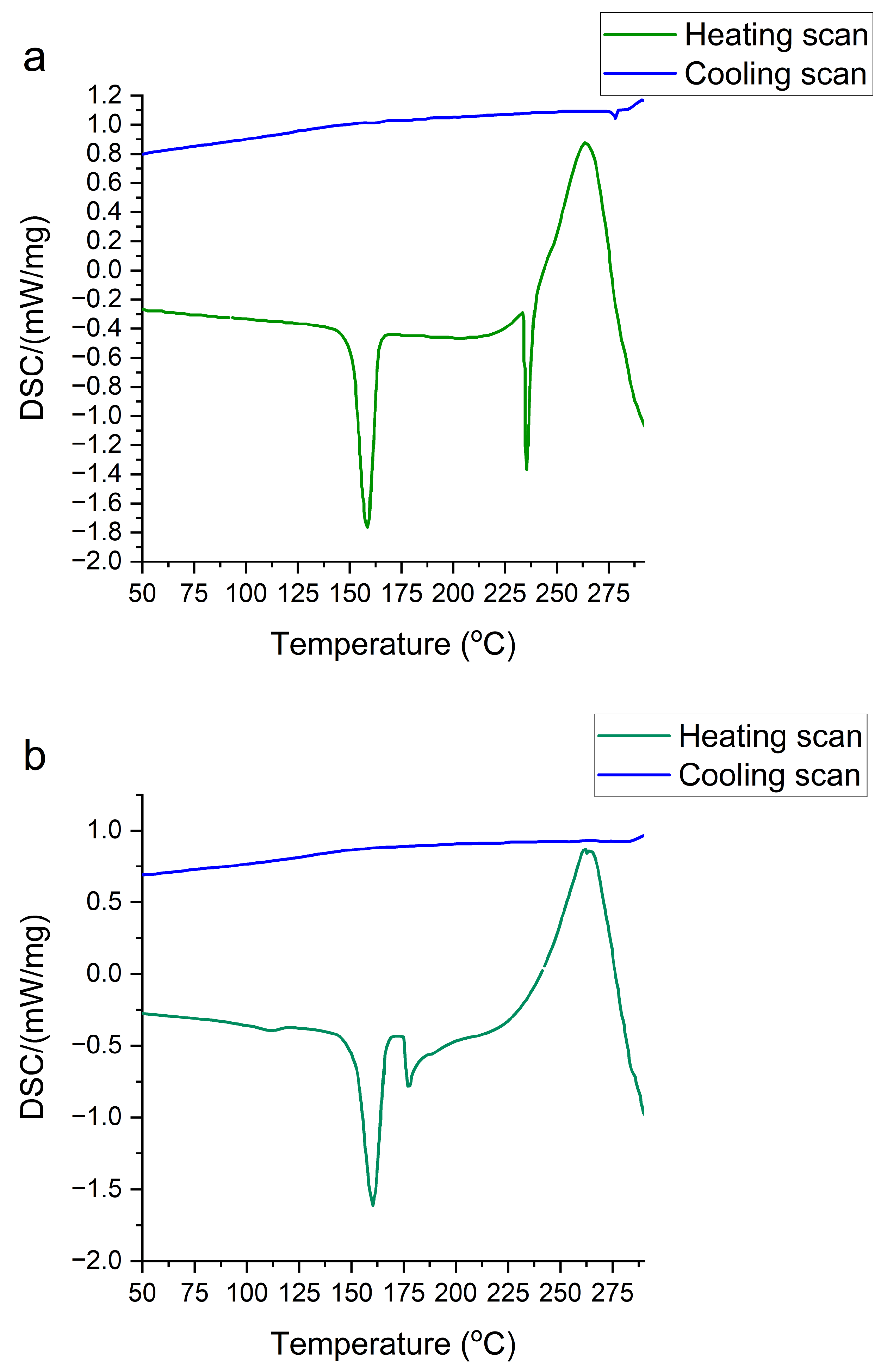
2.5. DSC
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. SWAXS
3.2.2. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR)
3.2.3. Raman
3.2.4. Dissolution
3.2.5. Differential Scanning Calorimetry (DSC)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hodzic, A.; Llusa, M.; Heigl, N.; Tritthart, W.; Fraser, S.D.; Laggner, P.; Khinnast, J.G. Effects of process variables on the Small and Wide Angle X-Ray Scattering (SWAXS) patterns of powders, granulates and pkarmaceutical tablets. Powder Technol. 2012, 221, 447–452. [Google Scholar] [CrossRef]
- Heigl, N.; Hodzic, A.; Llusa, M.; Tritthart, W.; Reiter, F.; Fraser, S.D.; Laggner, P.; Khinast, J.G. Potential of Raman Spectroscopy for Evaluating Crushing Strength of Tablets. J. Pharm Innov. 2012, 7, 76–86. [Google Scholar] [CrossRef]
- Hodzic, A.; Zoumpoulakis, P.; Pabst, G.; Mavromoustakos, T.; Rappolt, M. Losartan’s affinity to fluid bilayers modulates lipid–cholesterol interactions. Phys. Chem. Chem. Phys. 2012, 14, 4780–4788. [Google Scholar] [CrossRef]
- Brittain, H.G. Polymorphism in Pharmaceutical Solids; Marcel Dekker: New York, NY, USA, 1999; Volume 95. [Google Scholar]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An extraordinary example of conformational polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef]
- Merkle, H.P.; Jen, A. A crystal clear solution for insulin delivery. Nat. Biotechnol. 2002, 20, 789–790. [Google Scholar] [CrossRef]
- Tedesco, E.; Giron, D.; Pfeffer, S. Crystal structure elucidation and morphology study of pharmaceuticals in development. Cryst. Eng. Commun. 2002, 4, 393–400. [Google Scholar] [CrossRef]
- Hodzic, A.; Llusa, M.; Fraser, S.D.; Scheibelhofer, O.; Koller, D.M.; Reiter, F.; Laggner, P.; Khinast, J.G. Small- and wide-angle X-ray scattering (SWAXS) for quantification of aspirin content in a binary powder mixture. Inter. J. Pharm. 2012, 428, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Raw, A.S.; Furness, M.S.; Gill, D.S.; Adams, R.C.; Holcombe, F.O., Jr.; Yu, L.X. Regulatory considerations of pharmaceutical solid polymorphism in abbreviated new drug applications (ANDAs). Adv. Drug. Del. Rev. 2004, 56, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Rappolt, M.; Hodzic, A.; Sartori, B.; Ollivon, M.; Laggner, P. Conformational and hydrational properties during the Lβ- to Lα- and Lα- to HII-phase transition in phosphatidylethanolamine. Chem. Phys. Lipids 2008, 154, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Roblegg, E.; Jäger, E.; Hodzic, A.; Koscher, G.; Mohr, S.; Zimmer, A.; Khinast, J. Development of sustained-release lipophilic calcium stearate pellets via hot melt extrusion. Eur. J. Pharm. Biopharm. 2011, 79, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Jednacak, T.; Hodzic, A.; Scheibelhofer, O.; Marijan, M.; Khinast, J.G.; Novak, P. Fast real-time monitoring of entacapone crystallization and characterization of polymorphs via Raman spectroscopy, statistics and SWAXS. Acta Pharm. 2014, 64, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schrank, S.; Hodzic, A.; Zimmer, A.; Glasser, B.J.; Khinast, J.; Roblegg, E. Ibuprofen-Loaded Calcium Stearate Pellets: Drying-Induced Variations in Dosage Form Properties. AAPS PharmSciTech 2012, 13, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Besenhard, M.O.; Hohl, R.; Hodzic, A.; Eder, R.J.P.; Khinast, J.G. Modeling a seeded continuous crystallizer for the production of active pharmaceutical ingredients. Cryst. Res. Technol. 2014, 49, 92–108. [Google Scholar] [CrossRef]
- Hodzic, A.; Kriechbaum, M.; Schrank, S.; Reiter, F. Monitoring of Pentoxifylline Thermal Behavior by Novel Simultaneous Laboratory Small and Wide X-Ray Scattering (SWAXS) and Differential Scanning Calorimetry (DSC). PLoS ONE 2016, 11, e0159840. [Google Scholar] [CrossRef] [PubMed]
- Jednacak, T.; Novak, P.; Hodzic, A.; Scheibelhofer, O.; Khinast, J.G.; Plavec, J.; Sket, P.; Vukovic, J.P. Condensation reaction between carbohydrazide and salicylaldehyde: In-line vibrational spectroscopy monitoring and characterization of the reaction products in solution and solid state. Acta Chim Slov. 2014, 61, 161–169. [Google Scholar]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Discov. 2004, 3, 42–57. [Google Scholar] [CrossRef]
- Potamitis, C.; Chatzigeorgiou, P.; Siapi, E.; Viras, K.; Mavromoustakos, T.; Hodzic, A.; Pabst, G.; Cacho-Nerin, F.; Laggner, P.; Rappolt, M. Interactions of the AT1 antagonist valsartan with dipalmitoyl-phosphatidylcholine bilayers. Bioch. Et Biophys. Acta 2011, 1808, 1753–1763. [Google Scholar] [CrossRef]
- Suda, M.; Takayama, K.; Otsuka, M. An accurate quantitative analysis of polymorphic content by chemometric X-ray powder diffraction. Anal. Sci. 2008, 24, 451–457. [Google Scholar] [CrossRef]
- Hodzic, A.; Rappolt, M.; Amenitsch, H.; Laggner, P.; Pabst, G. Differential modulation of membrane structure and fluctuations by plant sterols and cholesterol. Biophys. J. 2008, 94, 3935–3944. [Google Scholar] [CrossRef]
- Hodzic, A.; Birarda, G.; Juraic, K.; Sket, P.; Eder, S.; Kriechbaum, M.; D’amico, F.; De Giacomo, O.; Roblegg, E. Revealing hidden molecular nanostructure details in the pellet formulation of ibuprofen by combining Synchrotron and laboratory sources. J. Drug Deliv. Sci. Technol. 2022, 68, 103114. [Google Scholar] [CrossRef]
- Glatter, O.; Kratky, O. Small Angle X-ray Scattering; Academic Press: London, UK; Tokyo, Japan, 1982. [Google Scholar]
- Liang, Y.; Noda, L.K.; Sala, O. Polarizability, and concentration effects on the Raman spectra of picolinic acid species in aqueous solution. J. Molec. Struct. 2000, 554, 271–277. [Google Scholar] [CrossRef]
- Rana, V.; Cañamares, M.V.; Kubic, T.; Leona, M.; Lombardi, J.R. Surface-enhanced Raman Spectroscopy for Trace Identification of Controlled Substances: Morphine, Codeine, and Hydrocodone. J. Forensic Sci. 2011, 56, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.; Angelov, B.; Mutafchieva, R.; Lesieur, S. Biocompatible Mesoporous and Soft Nanoarchitectures. J. Inorg. Organomet. Polym. 2015, 25, 214–232. [Google Scholar] [CrossRef]
- Santos, O.M.M.; Dias Reis, M.E.; Tavares Jacon, J.; Esselin de Sousa Lino, M.; Savioli Simões, J.; Carlos Doriguetto, A. Polymorphism: An evaluation of the potential risk to the quality of drug products from the Farmácia Popular Rede Própria. Braz. J. Pharmac. Sci. 2014, 50, 1–24. [Google Scholar] [CrossRef]
- Randall, C.S.; Rocco, W.L.; Ricou, P. XRD in pharmaceutical analysis: A versatile tool for problem-solving. Am. Pharm. Rev. 2010, 13, 52–59. [Google Scholar]
- Thakral, N.K.; Zanon, R.L.; Kelly, R.C.; Thakral, S. Applications of powder X-ray diffraction in small molecule pharmaceuticals: Achievements and aspirations. J. Pharm. Sci. 2018, 107, 2969–2982. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, I.; Gautam, R.; Tinoco, A.D. Using X-ray diffraction techniques for biomimetic drug development, formulation, and polymorphic characterization. Biomimetics 2021, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Thakral, S.; Terban, M.W.; Thakral, N.K.; Suryanarayanan, R. Recent advances in the characterization of amorphous pharmaceuticals by X-Ray diffractometry. Adv. Drug Deliv. Rev. 2016, 100, 183–193. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M.; Kariuki, B.M. Contemporary advances in the use of powder X-ray diffraction for structure determination. Angew. Chem. Int. Ed. 2001, 40, 1626–1651. [Google Scholar] [CrossRef]
- Tsue, H.; Horiguchi, M.; Tamura, R.; Fujii, K.; Uekusa, H. Crystal structure solution of organic compounds from X-ray powder diffraction data. J. Synth. Org. Chem. Jpn. 2007, 65, 1203–1212. [Google Scholar] [CrossRef]
- Altomare, A.; Ciriaco, F.; Cuocci, C.; Falcicchio, A.; Fanelli, F. Combined powder X-ray diffraction data and quantum-chemical calculations in EXPO2014. Powder Diffr. 2017, 32, S123–S128. [Google Scholar] [CrossRef]
- Kabova, E.A.; Cole, J.C.; Korb, O.; López-Ibáñez, M.; Williams, A.C.; Shankland, K. Improved performance of crystal structure solution from powder diffraction data through parameter tuning of a simulated annealing algorithm. J. Appl. Crystallogr. 2017, 50, 1411–1420. [Google Scholar] [CrossRef]
- Vioglio, P.C.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Kabova, E.A.; Blundell, C.D.; Shankland, K. Pushing the limits of molecular crystal structure determination from powder diffraction data in high-throughput chemical environments. J. Pharm. Sci. 2018, 107, 2042–2047. [Google Scholar] [CrossRef]
- Haleblian, J.; McCrone, W. Pharmaceutical applications of polymorphism. J. Pharm. Sci. 1969, 58, 911–929. [Google Scholar] [CrossRef]
- Yao, C.; Zhang, S.; Wang, L.; Tao, X. Recent advances in polymorph discovery methods of organic crystals. Cryst. Growth Des. 2023, 23, 637–654. [Google Scholar] [CrossRef]
- Brittain, H.G. Pharmaceutical cocrystals: The coming wave of new drug substances. J. Pharm. Sci. 2013, 102, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Munjal, B.; Suryanarayanan, R. Applications of synchrotron powder X-ray diffractometry in drug substance and drug product characterization. Trends Anal. Chem. 2021, 136, 116181. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Schlesinger, C.; Fitterer, A.; Buchsbaum, C.; Habermehl, S.; Chierotti, M.R.; Nervi, C.; Schmidt, M.U. Ambiguous structure determination from powder data: Four different structural models of 4,11-difluoroquinacridone with similar X-ray powder patterns, fit to the PDF, SSNMR and DFT-D. IUCrJ 2022, 9, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.D.M. NMR Crystallography as a vital tool in assisting crystal structure determination from powder XRD data. Crystals 2022, 12, 1277. [Google Scholar] [CrossRef]
- Chan, F.C.; Anwar, J.; Cernik, R.; Barnes, P.; Wilson, R.M. Ab initio structure determination of sulfathiazole polymorph V from synchrotron X-ray powder diffraction data. J. Appl. Crystallogr. 1999, 32, 436–441. [Google Scholar] [CrossRef]
- Grzesiak, A.L.; Matzger, A.J. New form discovery for the analgesis flurbiprofen and sulindac facilitated by polymer-induced heteronucleation. J. Pharm. Sci. 2007, 96, 2978–2986. [Google Scholar] [CrossRef]
- Chernyshev, V.V.; Machula, A.A.; Kukushkin, S.Y.; Velikodny, Y.A. Carvedilol dihydrogen phosphate hemihydrate: A powder study. Acta Crystallogr. Sect. E 2009, 65, o2020–o2021. [Google Scholar] [CrossRef]
- Vogt, F.G.; Copley, R.C.B.; Mueller, R.L.; Spoors, G.P.; Cacchio, T.N.; Carlton, R.A.; Katrincic, L.M.; Kennady, J.M.; Parsons, S.; Chetina, O.V. Isomorphism, disorder, and hydration in the crystal structures of racemic and single-enantiomer carvedilol phosphate. Cryst. Growth Des. 2010, 10, 2713–2733. [Google Scholar] [CrossRef]
- Shimpi, M.R.; Childs, S.L.; Bostrom, D.; Velaga, S.P. New cocrystals of ezetimibe with L-proline and imidazole. CrystEngComm 2014, 16, 8984–8993. [Google Scholar] [CrossRef]
- Bortolotti, M.; Lonardelli, I.; Pepponi, G. Determination of the crystal structure of nifedipine form C by synchrotron powder diffraction. Acta Crystallogr. Sect. B 2011, 67, 357–364. [Google Scholar] [CrossRef]
- Gunn, E.; Guzei, I.A.; Cai, T.; Yu, L. Polymorphism of nifedipine: Crystal structure and reversible transition of the metastable β polymorph. Cryst. Growth Des. 2012, 12, 2037–2043. [Google Scholar] [CrossRef]
- Martins, I.C.B.; Al-Sabbagh, D.; Meyer, K.; Maiwald, M.; Scholz, G.; Emmerling, F. Insight into the structure and properties of novel imidazole-based salts of salicylic acid. Molecules 2019, 24, 4144. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.W. A fully automatic program for finding the unit cell from powder data. J. Appl. Crystallogr. 1969, 2, 89–95. [Google Scholar] [CrossRef]
- Zhukov, S.G.; Babaev, E.V.; Chernyshev, V.V.; Rybakov, V.B.; Sonneveld, E.J.; Schenk, H. Crystal structure determination of 2-oxo-3-benzoyloxazolo[3,2-a]pyridine from X-ray powder data. Z. Krist.-Cryst. Mater. 2000, 215, 306–308. [Google Scholar] [CrossRef]
- Mirocki, A.; Lopresti, M.; Palin, L.; Conterosito, E.; Sikorski, A.; Milanesio, M. Exploring the molecular landscape of multicomponent crystals formed by naproxen drug and acridines. CrystEngComm 2022, 24, 6839–6853. [Google Scholar] [CrossRef]
- Allu, S.; Garai, A.; Chernyshev, V.V.; Nangia, A.K. Synthesis of ternary cocrystals, salts, and hydrates of acefylline with enhanced dissolution and high permeability. Cryst. Growth Des. 2022, 22, 4165–4181. [Google Scholar] [CrossRef]
- MacMillan, S.D.; Roberts, K.J.; Rossi, A.; Wells, M.A.; Polgreen, M.C.; Smith, I.H. In Situ Small Angle X-ray Scattering (SAXS) Studies of Polymorphism with the Associated Crystallization of Cocoa Butter Fat Using Shearing Conditions. Cryst. Growth Des. 2002, 2, 221–226. [Google Scholar] [CrossRef]
- Vella, J.; Hemar, Y.; Gu, Q.; Wu, Z.R.; Li, N.; Schoenel, T. In-situ SAXS investigation of high-pressure triglyceride polymorphism in milk cream and anhydrous milk fat. LWT 2021, 135, 110174. [Google Scholar] [CrossRef]
- Woodrow, I.L.; deMan, J.M. Polymorphism in Milk Fat Shown by X-ray Diffraction and Infrared Spectroscopy. J. Dairy Sci. 1968, 51, 996–1000. [Google Scholar] [CrossRef]
- Li, L.; Salamończyk, M.; Shadpour, S.; Zhu, C.; Jákli, A.; Hegmann, T. An unusual type of polymorphism in a liquid crystal. Nat Commun. 2018, 9, 714. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekhar, V.K. Lipid crystallization: From self-assembly to hierarchical and biological ordering. Nanoscale 2012, 4, 5779. [Google Scholar]
- Parada, M.L.; Sadeghpour, A.; Vieira, J.; Povey, M.; Rappolt, M. Global Small-Angle X-ray Scattering Data Analysis of Triacylglycerols in the α-Phase (Part II). J. Phys. Chem. B 2018, 122, 10330–10336. [Google Scholar] [CrossRef] [PubMed]
- Petoukhov, M.V.; Konarev, P.V.; Volkov, V.V.; Mozhaev, A.A.; Shtykova, E.V. The Ambiguity Issue in Solving Inverse Problems of Small-Angle Scattering: A Consistent Approach Using an Insulin Receptor-Related Receptor as an Example. Methods for Interpreting SAXS Data. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2021, 15, 270–283. [Google Scholar] [CrossRef]
- Otsuka, C.; Takahashi, S.; Isobe, A.; Saito, T.; Aizawa, T.; Tsuchida, R.; Yamashita, S.; Harano, K.; Hanayama, H.; Shimizu, N.; et al. Supramolecular Polymer Polymorphism: Spontaneous Helix–Helicoid Transition through Dislocation of Hydrogen-Bonded π-Rosettes. J. Am. Chem. Soc. 2023, 145, 22563–22576. [Google Scholar] [CrossRef]
- Leea, Y.L.; Ristica, R.I.; DeMatosa, L.L.; Martinb, C.M. Crystallisation Pathways of Polymorphic Triacylglycerols Induced by Mechanical Energy. J. Phys. Conf. Ser. 2010, 247, 012049. [Google Scholar] [CrossRef]
- Bertoni, S.; Simone, E.; Sangiorgi, S.; Albertini, B.; Passerini, N. The use of polymorphic state modifiers in solid lipid microparticles: The role of structural modifications on drug release performance. Eur. J. Pharm. Sci. 2024, 192, 106650. [Google Scholar] [CrossRef]
- Rodríguez-Negrette, A.C.; Rodríguez-Batiller, M.J.; García-Londoño, V.A.; Borroni, V.; Candal, R.J.; Herrera, M.L. Effect of sucrose esters on polymorphic behavior and crystallization kinetics of cupuassu fat and its fractions. J. Am. Oil Chem. Soc. 2022, 99, 27–41. [Google Scholar] [CrossRef]
- Cholakova, D.; Denkov, N. Polymorphic phase transitions in triglycerides and their mixtures studied by SAXS/WAXS techniques: In bulk and in emulsions. Adv. Colloid Interf. Sci. 2024, 323, 103071. [Google Scholar] [CrossRef]
- Shtykova, E.V. Shape determination of polydisperse and polymorphic nanoobjects from small-angle X-ray scattering data (Computer simulation). Nanotechnol. Russ. 2015, 10, 408–419. [Google Scholar] [CrossRef]
- Blázquez-Blázquez, E.; Barranco-García, R.; Cerrada, M.L.; Martínez, J.C.; Pérez, E. Synchrotron and Raman Study of the Rotator Phases and Polymorphism in Tricosane Paraffin. Polymers 2020, 12, 1341. [Google Scholar] [CrossRef] [PubMed]
- Sirota, E.B.; King, H.E.; Singer, D.M.; Shao, H.H. Rotator phases of the normal alkanes: An X-ray scattering study. J. Chem. Phys. 1993, 98, 5809–5824. [Google Scholar] [CrossRef]
- Doucet, J.; Denicolo, I.; Craievich, A. X-ray study of the “rotator” phase of the odd-numbered paraffins C17H36, C19H40, and C21H44. J. Chem. Phys. 1981, 75, 1523–1529. [Google Scholar] [CrossRef]
- Denicolo, I.; Doucet, J.; Craievich, A.F. X-ray study of the rotator phase of paraffins (III): Even-numbered paraffins C18H38, C20H42, C22H46, C24H50, and C26H54. J. Chem. Phys. 1983, 78, 1465–1469. [Google Scholar] [CrossRef]
- Doucet, J.; Denicolo, I.; Craievich, A.F.; Germain, C. X-ray study of the rotator phase of paraffins (IV): C27H56,C28H58, C29H60, C30H62, C32H66, and C34H70. J. Chem. Phys. 1984, 80, 1647–1651. [Google Scholar] [CrossRef]
- Nozaki, K.; Higashitani, N.; Yamamoto, T.; Hara, T. Solid-solid phase transitions in n-alkanes C23H48 andC25H52: X-ray power diffraction study on new layer stacking in phase-V. J. Chem. Phys. 1995, 103, 5762–5766. [Google Scholar] [CrossRef]
- Hu, W.G.; Srinivas, S.; Sirota, E.B. Crystalline structure and properties of EP and EB copolymers by solid-state NMR, DSC, and WAXS. Macromolecules 2002, 35, 5013–5024. [Google Scholar] [CrossRef]
- Liu, L.Z.; Hsiao, B.S.; Ran, S.F.; Fu, B.X.; Toki, S.; Zuo, F.; Tsou, A.H.; Chu, B. In situ WAXD study of structure changes during uniaxial deformation of ethylene-based semicrystalline ethylene-propylene copolymer. Polymer 2006, 47, 2884–2893. [Google Scholar] [CrossRef]
- Muller, A. An X-ray investigation of normal paraffins near their melting points. Proc. R. Soc. London Ser. A 1932, 138, 514–530. [Google Scholar]
- Strobl, G. From the melt via mesomorphic and granular crystalline layers to lamellar crystallites: A major route followed in polymer crystallization? Eur. Phys. J. E 2000, 3, 165–183. [Google Scholar] [CrossRef]
- Snyder, R.G.; Krause, S.J.; Scherer, J.R. Determination of distribution of straight-chain segment lengths in crystalline polyethylene from Raman LAM-1 band. J. Polym. Sci. Part B Polym. Phys. 1978, 16, 1593–1609. [Google Scholar] [CrossRef]
- Jin, Y.; Kotula, A.P.; Walker, A.R.H.; Migler, K.B.; Lee, Y.J. Phase-specific Raman analysis of n-alkane melting by moving-window two-dimensional correlation spectroscopy. J. Raman Spectrosc. 2016, 47, 1375–1384. [Google Scholar] [CrossRef]
- Rastogi, S.; Kurelec, L.; Lemstra, P.J. Chain mobility in polymer systems: On the borderline between solid and melt. 2. Crystal size influence in phase transition and sintering of ultrahigh molecular weight polyethylene via the mobile hexagonal phase. Macromolecules 1998, 31, 5022–5031. [Google Scholar] [CrossRef]
- Sirota, E.B. Polymer crystallization: Metastable mesophases and morphology. Macromolecules 2007, 40, 1043–1048. [Google Scholar] [CrossRef]
- Snyder, R.G.; Maroncelli, M.; Qi, S.P.; Strauss, H.L. Phase transitions and nonplanar conformers in crystallinen-alkanes. Science 1981, 214, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Schaufele, R.F.; Shimanouchi, T. Longitudinal acoustical vibrations of finite polymethylene chains. J. Chem. Phys. 1967, 47, 3605–3610. [Google Scholar] [CrossRef]
- Olf, H.G.; Fanconi, B. Low frequency Raman-active lattice vibrations of n-paraffins. J. Chem. Phys. 1973, 59, 534–544. [Google Scholar] [CrossRef]
- Androsch, R.; Blackwell, J.; Chvalun, S.N.; Wunderlich, B. Wide- and small-angle X-ray analysis of poly(ethylene-co-octene). Macromolecules 1999, 32, 3735–3740. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malanovic, N.; Birarda, G.; Eder, S.; Gruber-Woelfler, H.; Reiter, F.; Juraic, K.; Hodzic, A. Small-Angle X-ray Scattering (SAXS) Used for the Identification of Nicomorphine Polymorphic Changes at the Early Stage to Avoid Varied Stability and Possible Side Effects. Pharmaceuticals 2024, 17, 375. https://doi.org/10.3390/ph17030375
Malanovic N, Birarda G, Eder S, Gruber-Woelfler H, Reiter F, Juraic K, Hodzic A. Small-Angle X-ray Scattering (SAXS) Used for the Identification of Nicomorphine Polymorphic Changes at the Early Stage to Avoid Varied Stability and Possible Side Effects. Pharmaceuticals. 2024; 17(3):375. https://doi.org/10.3390/ph17030375
Chicago/Turabian StyleMalanovic, Nermina, Giovanni Birarda, Simone Eder, Heidrun Gruber-Woelfler, Franz Reiter, Krunoslav Juraic, and Aden Hodzic. 2024. "Small-Angle X-ray Scattering (SAXS) Used for the Identification of Nicomorphine Polymorphic Changes at the Early Stage to Avoid Varied Stability and Possible Side Effects" Pharmaceuticals 17, no. 3: 375. https://doi.org/10.3390/ph17030375
APA StyleMalanovic, N., Birarda, G., Eder, S., Gruber-Woelfler, H., Reiter, F., Juraic, K., & Hodzic, A. (2024). Small-Angle X-ray Scattering (SAXS) Used for the Identification of Nicomorphine Polymorphic Changes at the Early Stage to Avoid Varied Stability and Possible Side Effects. Pharmaceuticals, 17(3), 375. https://doi.org/10.3390/ph17030375