
Absorption Distribution Metabolism Excretion and Toxicity Property Prediction Utilizing a Pre-Trained Natural Language Processing Model and Its Applications in Early-Stage Drug Development

 , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. MoleculeNet Dataset
2.2. DrugBank Dataset
2.3. External Dataset
2.4. Applicability Domain
3. Discussion
4. Materials and Methods
4.1. Data Collection and Preprocessing
4.1.1. MoleculeNet Dataset
4.1.2. DrugBank Dataset
4.1.3. External Dataset
4.2. Deep Learning Models
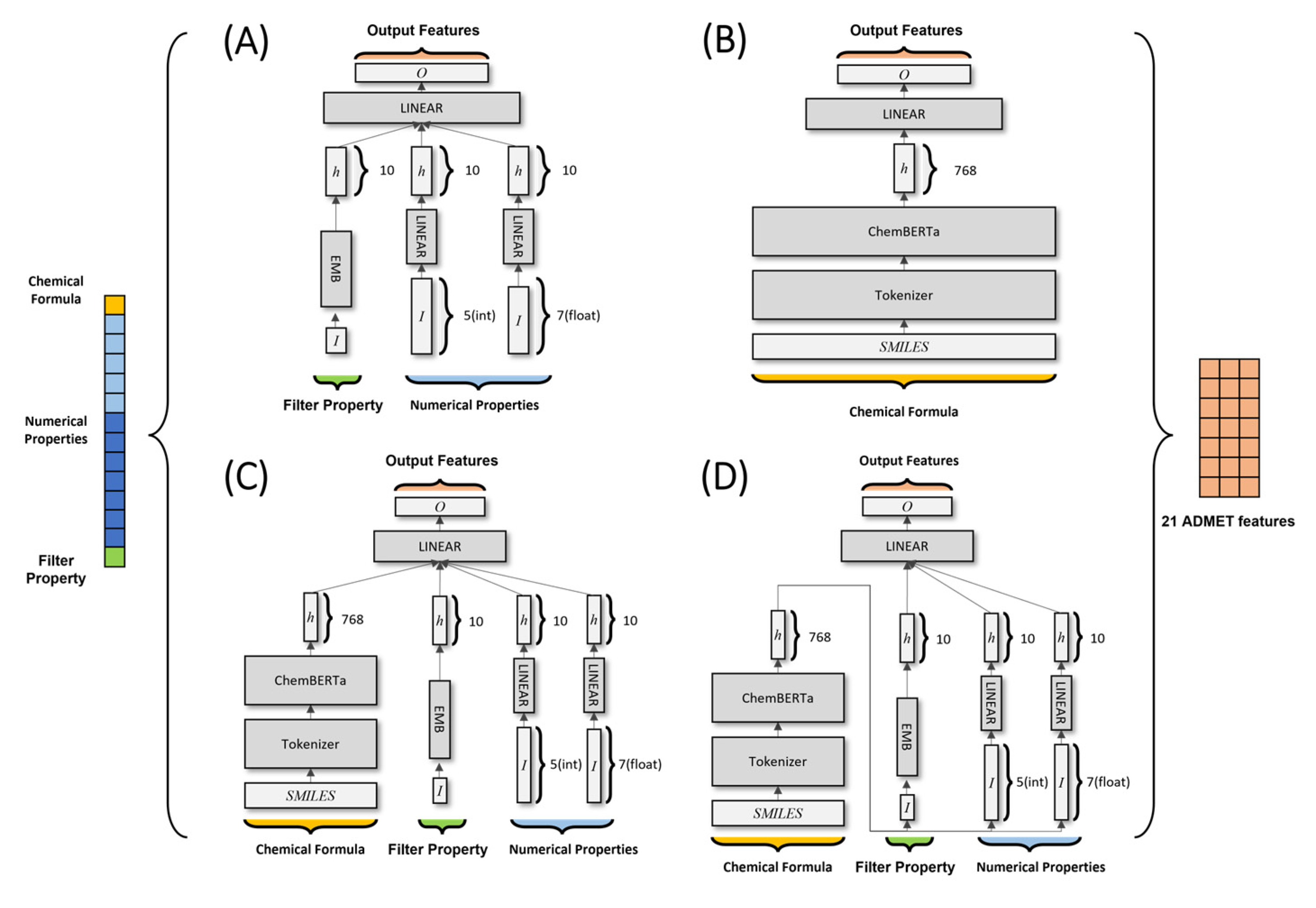
- A deep neural network (DNN) model consists of fully connected and embedded layers. The model uses 18 input physicochemical values as input properties. One Boolean feature was transformed into a 10-dimensional vector via embedding layers. Five integer-based and seven float features were transformed into a 10-dimensional vector via fully connected layers. Vectors were concatenated into a 30-dimensional vector, which passed through fully connected layers to return a vector with 21 dimensions (the predicted ADMET features).
- An encoder model includes a pre-trained ChemBERTa model. This model treats SMILES data as “natural sentences” and learns via MLM, which is RoBERTa. The SMILES data used for the pre-trained model are in the form of a 768-dimensional hidden vector, which is transformed into an 18-dimensional vector via fully connected layers.
- A concat model combines the DNN and encoder models described above. The 30-dimensional vector from the DNN model and the 768-dimensional hidden vector from the encoder model are concatenated and passed to the hidden layer of the concat model. This 798-dimensional hidden vector is then transformed into 21 dimensions.
- A pipe model, which subsumed a pre-trained ChemBERTa model, used a 768-dimensional hidden vector based on SMILES data to predict 21 physicochemical properties. Those physicochemical properties were then used as input for a DNN model to predict ADMET features.
- A modified version of the DNN model is DNN A (where A stands for attention). We incorporated dot-product self-attention into the model, which uses hidden vectors from the DNN as the query, key, and value. By implementing dot-product self-attention, it was possible to identify which input most affected ADMET feature predictions.
- A modified version of the pipe model is pipe A, into which dot-product self-attention can be incorporated.
4.3. Settings
4.4. Evaluation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Supplementary Results

{kind=link}
{kind=link}
{kind=link}
| Feature | Model | AUROC | F1 | Accuracy | Precision | Recall |
|---|---|---|---|---|---|---|
| Ames test | Concat | 0.679 | 0.730 | 0.901 | 0.790 | 0.679 |
| DNN | 0.504 | 0.583 | 0.878 | 0.689 | 0.504 | |
| DNN_A | 0.500 | 0.467 | 0.878 | 0.439 | 0.500 | |
| Encoder | 0.655 | 0.700 | 0.891 | 0.751 | 0.655 | |
| Pipe | 0.510 | 0.614 | 0.879 | 0.773 | 0.510 | |
| Pipe_A | 0.500 | 0.467 | 0.878 | 0.439 | 0.500 | |
| Biodegradation | Concat | 0.840 | 0.853 | 0.917 | 0.867 | 0.840 |
| DNN | 0.749 | 0.771 | 0.875 | 0.795 | 0.749 | |
| DNN_A | 0.757 | 0.779 | 0.879 | 0.802 | 0.757 | |
| Encoder | 0.854 | 0.857 | 0.917 | 0.860 | 0.854 | |
| Pipe | 0.806 | 0.821 | 0.899 | 0.835 | 0.806 | |
| Pipe_A | 0.500 | 0.452 | 0.823 | 0.412 | 0.500 | |
| Blood–Brain Barrier | Concat | 0.845 | 0.832 | 0.868 | 0.820 | 0.845 |
| DNN | 0.624 | 0.697 | 0.798 | 0.791 | 0.624 | |
| DNN_A | 0.677 | 0.715 | 0.805 | 0.757 | 0.677 | |
| Encoder | 0.835 | 0.844 | 0.885 | 0.854 | 0.835 | |
| Pipe | 0.757 | 0.804 | 0.861 | 0.857 | 0.757 | |
| Pipe_A | 0.669 | 0.733 | 0.818 | 0.811 | 0.669 | |
| Caco-2 permeable | Concat | 0.792 | 0.828 | 0.868 | 0.867 | 0.792 |
| DNN | 0.772 | 0.783 | 0.831 | 0.794 | 0.772 | |
| DNN_A | 0.776 | 0.790 | 0.837 | 0.804 | 0.776 | |
| Encoder | 0.859 | 0.865 | 0.893 | 0.872 | 0.859 | |
| Pipe | 0.768 | 0.796 | 0.845 | 0.827 | 0.768 | |
| Pipe_A | 0.624 | 0.644 | 0.739 | 0.665 | 0.624 | |
| Carcinogenicity | Concat | 0.607 | 0.690 | 0.973 | 0.801 | 0.607 |
| DNN | 0.500 | 0.493 | 0.971 | 0.485 | 0.500 | |
| DNN_A | 0.500 | 0.493 | 0.971 | 0.485 | 0.500 | |
| Encoder | 0.627 | 0.688 | 0.972 | 0.762 | 0.627 | |
| Pipe | 0.520 | 0.579 | 0.969 | 0.653 | 0.520 | |
| Pipe_A | 0.500 | 0.493 | 0.971 | 0.485 | 0.500 | |
| CYP450 1A2 substrate | Concat | 0.810 | 0.823 | 0.872 | 0.836 | 0.810 |
| DNN | 0.722 | 0.747 | 0.823 | 0.773 | 0.722 | |
| DNN_A | 0.758 | 0.777 | 0.841 | 0.796 | 0.758 | |
| Encoder | 0.782 | 0.811 | 0.866 | 0.843 | 0.782 | |
| Pipe | 0.791 | 0.821 | 0.873 | 0.853 | 0.791 | |
| Pipe_A | 0.782 | 0.804 | 0.860 | 0.826 | 0.782 | |
| CYP450 2C19 inhibitor | Concat | 0.762 | 0.788 | 0.884 | 0.816 | 0.762 |
| DNN | 0.575 | 0.637 | 0.831 | 0.713 | 0.575 | |
| DNN_A | 0.577 | 0.632 | 0.828 | 0.699 | 0.577 | |
| Encoder | 0.780 | 0.802 | 0.891 | 0.825 | 0.780 | |
| Pipe | 0.722 | 0.770 | 0.879 | 0.824 | 0.722 | |
| Pipe_A | 0.664 | 0.716 | 0.856 | 0.777 | 0.664 | |
| CYP450 2C9 inhibitor | Concat | 0.759 | 0.764 | 0.903 | 0.769 | 0.759 |
| DNN | 0.528 | 0.570 | 0.874 | 0.619 | 0.528 | |
| DNN_A | 0.515 | 0.608 | 0.883 | 0.742 | 0.515 | |
| Encoder | 0.751 | 0.754 | 0.898 | 0.756 | 0.751 | |
| Pipe | 0.673 | 0.737 | 0.907 | 0.814 | 0.673 | |
| Pipe_A | 0.514 | 0.547 | 0.874 | 0.585 | 0.514 | |
| CYP450 2C9 substrate | Concat | 0.500 | 0.800 | 0.997 | 0.999 | 0.667 |
| DNN | 0.500 | 0.499 | 0.996 | 0.498 | 0.500 | |
| DNN_A | 0.500 | 0.499 | 0.996 | 0.498 | 0.500 | |
| Encoder | 0.666 | 0.705 | 0.996 | 0.749 | 0.666 | |
| Pipe | 0.500 | 0.499 | 0.996 | 0.498 | 0.500 | |
| Pipe_A | 0.500 | 0.499 | 0.996 | 0.498 | 0.500 | |
| CYP450 2D6 inhibitor | Concat | 0.616 | 0.673 | 0.949 | 0.743 | 0.616 |
| DNN | 0.584 | 0.698 | 0.954 | 0.867 | 0.584 | |
| DNN_A | 0.610 | 0.752 | 0.959 | 0.979 | 0.610 | |
| Encoder | 0.669 | 0.765 | 0.962 | 0.894 | 0.669 | |
| Pipe | 0.608 | 0.722 | 0.957 | 0.888 | 0.608 | |
| Pipe_A | 0.500 | 0.487 | 0.948 | 0.474 | 0.500 | |
| CYP450 2D6 substrate | Concat | 0.698 | 0.782 | 0.973 | 0.888 | 0.698 |
| DNN | 0.500 | 0.490 | 0.962 | 0.481 | 0.500 | |
| DNN_A | 0.533 | 0.644 | 0.963 | 0.815 | 0.533 | |
| Encoder | 0.664 | 0.743 | 0.969 | 0.844 | 0.664 | |
| Pipe | 0.500 | 0.490 | 0.962 | 0.481 | 0.500 | |
| Pipe_A | 0.500 | 0.490 | 0.962 | 0.481 | 0.500 | |
| CYP450 3A4 inhibitor | Concat | 0.738 | 0.750 | 0.874 | 0.761 | 0.738 |
| DNN | 0.535 | 0.590 | 0.842 | 0.656 | 0.535 | |
| DNN_A | 0.520 | 0.588 | 0.845 | 0.675 | 0.520 | |
| Encoder | 0.758 | 0.764 | 0.879 | 0.771 | 0.758 | |
| Pipe | 0.654 | 0.704 | 0.868 | 0.763 | 0.654 | |
| Pipe_A | 0.558 | 0.620 | 0.847 | 0.696 | 0.558 | |
| CYP450 3A4 substrate | Concat | 0.870 | 0.864 | 0.879 | 0.858 | 0.870 |
| DNN | 0.755 | 0.766 | 0.803 | 0.778 | 0.755 | |
| DNN_A | 0.766 | 0.769 | 0.802 | 0.772 | 0.766 | |
| Encoder | 0.838 | 0.850 | 0.873 | 0.863 | 0.838 | |
| Pipe | 0.826 | 0.827 | 0.850 | 0.828 | 0.826 | |
| Pipe_A | 0.786 | 0.786 | 0.814 | 0.786 | 0.786 | |
| CYP450 inhibitory promiscuity | Concat | 0.843 | 0.832 | 0.874 | 0.822 | 0.843 |
| DNN | 0.723 | 0.736 | 0.818 | 0.750 | 0.723 | |
| DNN_A | 0.727 | 0.729 | 0.807 | 0.732 | 0.727 | |
| Encoder | 0.854 | 0.848 | 0.888 | 0.842 | 0.854 | |
| Pipe | 0.821 | 0.826 | 0.877 | 0.832 | 0.821 | |
| Pipe_A | 0.786 | 0.799 | 0.860 | 0.813 | 0.786 | |
| hERG inhibition (precisiondictor I) | Concat | 0.557 | 0.610 | 0.966 | 0.673 | 0.557 |
| DNN | 0.500 | 0.492 | 0.968 | 0.484 | 0.500 | |
| DNN_A | 0.500 | 0.492 | 0.968 | 0.484 | 0.500 | |
| Encoder | 0.538 | 0.603 | 0.967 | 0.685 | 0.538 | |
| Pipe | 0.500 | 0.492 | 0.968 | 0.484 | 0.500 | |
| Pipe_A | 0.500 | 0.492 | 0.968 | 0.484 | 0.500 | |
| hERG inhibition (precisiondictor II) | Concat | 0.835 | 0.830 | 0.905 | 0.825 | 0.835 |
| DNN | 0.641 | 0.688 | 0.854 | 0.743 | 0.641 | |
| DNN_A | 0.690 | 0.724 | 0.864 | 0.762 | 0.690 | |
| Encoder | 0.821 | 0.828 | 0.907 | 0.836 | 0.821 | |
| Pipe | 0.746 | 0.775 | 0.885 | 0.806 | 0.746 | |
| Pipe_A | 0.743 | 0.767 | 0.880 | 0.793 | 0.743 | |
| Human intestinal absorption | Concat | 0.884 | 0.854 | 0.896 | 0.827 | 0.884 |
| DNN | 0.727 | 0.771 | 0.868 | 0.820 | 0.727 | |
| DNN_A | 0.767 | 0.795 | 0.878 | 0.824 | 0.767 | |
| Encoder | 0.883 | 0.878 | 0.921 | 0.873 | 0.883 | |
| Pipe | 0.848 | 0.867 | 0.919 | 0.887 | 0.848 | |
| Pipe_A | 0.615 | 0.693 | 0.835 | 0.795 | 0.615 | |
| P-glycoprotein inhibitor I | Concat | 0.780 | 0.816 | 0.916 | 0.856 | 0.780 |
| DNN | 0.634 | 0.703 | 0.878 | 0.789 | 0.634 | |
| DNN_A | 0.711 | 0.744 | 0.885 | 0.780 | 0.711 | |
| Encoder | 0.800 | 0.828 | 0.920 | 0.857 | 0.800 | |
| Pipe | 0.736 | 0.782 | 0.903 | 0.835 | 0.736 | |
| Pipe_A | 0.672 | 0.748 | 0.893 | 0.845 | 0.672 | |
| P-glycoprotein inhibitor II | Concat | 0.683 | 0.716 | 0.894 | 0.753 | 0.683 |
| DNN | 0.577 | 0.629 | 0.879 | 0.692 | 0.577 | |
| DNN_A | 0.552 | 0.601 | 0.875 | 0.660 | 0.552 | |
| Encoder | 0.657 | 0.683 | 0.882 | 0.712 | 0.657 | |
| Pipe | 0.608 | 0.659 | 0.884 | 0.720 | 0.608 | |
| Pipe_A | 0.556 | 0.602 | 0.874 | 0.656 | 0.556 | |
| P-glycoprotein substrate | Concat | 0.851 | 0.850 | 0.849 | 0.850 | 0.851 |
| DNN | 0.798 | 0.799 | 0.800 | 0.800 | 0.798 | |
| DNN_A | 0.804 | 0.804 | 0.805 | 0.805 | 0.804 | |
| Encoder | 0.866 | 0.867 | 0.868 | 0.868 | 0.866 | |
| Pipe | 0.840 | 0.840 | 0.841 | 0.841 | 0.840 | |
| Pipe_A | 0.824 | 0.824 | 0.824 | 0.824 | 0.824 | |
| Renal organic cation transporter | Concat | 0.774 | 0.828 | 0.967 | 0.890 | 0.774 |
| DNN | 0.649 | 0.710 | 0.949 | 0.784 | 0.649 | |
| DNN_A | 0.626 | 0.678 | 0.944 | 0.739 | 0.626 | |
| Encoder | 0.808 | 0.863 | 0.973 | 0.926 | 0.808 | |
| Pipe | 0.597 | 0.683 | 0.948 | 0.798 | 0.597 | |
| Pipe_A | 0.556 | 0.708 | 0.949 | 0.974 | 0.556 | |
| Total | Concat | 0.749 | 0.786 | 0.911 | 0.824 | 0.757 |
| DNN | 0.624 | 0.660 | 0.879 | 0.705 | 0.624 | |
| DNN_A | 0.636 | 0.670 | 0.882 | 0.717 | 0.636 | |
| Encoder | 0.760 | 0.788 | 0.915 | 0.821 | 0.760 | |
| Pipe | 0.682 | 0.719 | 0.903 | 0.767 | 0.682 | |
| Pipe_A | 0.612 | 0.637 | 0.880 | 0.672 | 0.612 |
| Input | Property Type |
|---|---|
| SMILES | String |
| Physiological charge | Int |
| Number of rings | Int |
| Rotatable bond count | Int |
| H bond acceptor count | Int |
| H bond donor count | Int |
| polarizability | Float |
| Molar Refractivity | Float |
| Monoisotopic weight | Float |
| Molecular weight | Float |
| Polar surface area | Float |
| LogP | Float |
| LogS | Float |
| Water Solubility | Float |
| Bioavailability | Boolean |
| Rule of five | Boolean |
| Veber’s rule | Boolean |
| MDDR-like rule | Boolean |
| Ghose filter | Boolean |
| ADMET Property | TRUE | FALSE |
|---|---|---|
| Human intestinal absorption | TRUE | FALSE |
| Blood–Brain Barrier | TRUE | FALSE |
| Caco-2 permeable | TRUE | FALSE |
| P-glycoprotein substrate | Substrate | Non-substrate |
| P-glycoprotein inhibitor I | Inhibitor | Non-inhibitor |
| P-glycoprotein inhibitor II | Inhibitor | Non-inhibitor |
| Renal organic cation transporter | Inhibitor | Non-inhibitor |
| CYP450 2C9 substrate | Substrate | Non-substrate |
| CYP450 2D6 substrate | Substrate | Non-substrate |
| CYP450 3A4 substrate | Substrate | Non-substrate |
| CYP450 1A2 inhibitor | Inhibitor | Non-inhibitor |
| CYP450 2C9 inhibitor | Inhibitor | Non-inhibitor |
| CYP450 2D6 inhibitor | Inhibitor | Non-inhibitor |
| CYP450 2C19 inhibitor | Inhibitor | Non-inhibitor |
| CYP450 3A4 inhibitor | Inhibitor | Non-inhibitor |
| CYP450 inhibitory promiscuity | High CYP Inhibitory Promiscuity | Low CYP Inhibitory Promiscuity |
| Ames test | AMES toxic | Non-AMES toxic |
| Carcinogenicity | Carcinogens | Non-carcinogens |
| Biodegradation | Readily biodegradable | Not readily biodegradable |
| hERG inhibition (predictor I) | Week inhibitor | Strong inhibitor |
| hERG inhibition (predictor II) | Inhibitor | Non-inhibitor |
References
- Lavecchia, A. Machine-Learning Approaches in Drug Discovery: Methods and Applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef]
- Winkler, D.A. Neural Networks as Robust Tools in Drug Lead Discovery and Development. Mol. Biotechnol. 2004, 27, 139–167. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Chuang, K.V.; Gunsalus, L.M.; Keiser, M.J. Learning Molecular Representations for Medicinal Chemistry: Miniperspective. J. Med. Chem. 2020, 63, 8705–8722. [Google Scholar] [CrossRef] [PubMed]
- Kearnes, S.; Goldman, B.; Pande, V. Modeling Industrial ADMET Data with Multitask Networks. arXiv 2016, arXiv:1606.08793. [Google Scholar]
- Xu, Y.; Ma, J.; Liaw, A.; Sheridan, R.P.; Svetnik, V. Demystifying Multitask Deep Neural Networks for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2017, 57, 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhu, M.; Kang, Y.; Leung, E.L.-H.; Lei, T.; Shen, C.; Jiang, D.; Wang, Z.; Cao, D.; Hou, T. Do We Need Different Machine Learning Algorithms for QSAR Modeling? A Comprehensive Assessment of 16 Machine Learning Algorithms on 14 QSAR Data Sets. Brief. Bioinform. 2021, 22, bbaa321. [Google Scholar] [CrossRef] [PubMed]
- Göller, A.H.; Kuhnke, L.; Montanari, F.; Bonin, A.; Schneckener, S.; Ter Laak, A.; Wichard, J.; Lobell, M.; Hillisch, A. Bayer’s in Silico ADMET Platform: A Journey of Machine Learning over the Past Two Decades. Drug Discov. Today 2020, 25, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S. The next Era: Deep Learning in Pharmaceutical Research. Pharm. Res. 2016, 33, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Montanari, F.; Kuhnke, L.; Ter Laak, A.; Clevert, D.-A. Modeling Physico-Chemical ADMET Endpoints with Multitask Graph Convolutional Networks. Molecules 2019, 25, 44. [Google Scholar] [CrossRef]
- Cáceres, E.L.; Tudor, M.; Cheng, A.C. Deep Learning Approaches in Predicting ADMET Properties. Future Med. Chem. 2020, 12, 1995–1999. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Joshi, E.; Pande, V.S.; Cheng, A.C. Improvement in ADMET Prediction with Multitask Deep Featurization. J. Med. Chem. 2020, 63, 8835–8848. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Cao, Z.; Barati Farimani, A. Molecular Contrastive Learning of Representations via Graph Neural Networks. Nat. Mach. Intell. 2022, 4, 279–287. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-Training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Lewis, M.; Liu, Y.; Goyal, N.; Ghazvininejad, M.; Mohamed, A.; Levy, O.; Stoyanov, V.; Zettlemoyer, L. Bart: Denoising Sequence-to-Sequence Pre-Training for Natural Language Generation, Translation, and Comprehension. arXiv 2019, arXiv:1910.13461. [Google Scholar]
- Clark, K.; Luong, M.-T.; Le, Q.V.; Manning, C.D. Electra: Pre-Training Text Encoders as Discriminators Rather than Generators. arXiv 2020, arXiv:2003.10555. [Google Scholar]
- Irwin, R.; Dimitriadis, S.; He, J.; Bjerrum, E.J. Chemformer: A Pre-Trained Transformer for Computational Chemistry. Mach. Learn. Sci. Technol. 2022, 3, 015022. [Google Scholar] [CrossRef]
- Martins, I.F.; Teixeira, A.L.; Pinheiro, L.; Falcao, A.O. A Bayesian Approach to in Silico Blood-Brain Barrier Penetration Modeling. J. Chem. Inf. Model. 2012, 52, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xia, M.; Nguyen, D.-T.; Zhao, T.; Sakamuru, S.; Zhao, J.; Shahane, S.A.; Rossoshek, A.; Simeonov, A. Tox21Challenge to Build Predictive Models of Nuclear Receptor and Stress Response Pathways as Mediated by Exposure to Environmental Chemicals and Drugs. Front. Environ. Sci. 2016, 3, 85. [Google Scholar] [CrossRef]
- Gayvert, K.M.; Madhukar, N.S.; Elemento, O. A Data-Driven Approach to Predicting Successes and Failures of Clinical Trials. Cell Chem. Biol. 2016, 23, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- AIDS Antiviral Screen Data—NCI DTP Data—NCI Wiki. Available online: https://wiki.nci.nih.gov/display/NCIDTPdata/AIDS+Antiviral+Screen+Data (accessed on 1 March 2024).
- Subramanian, G.; Ramsundar, B.; Pande, V.; Denny, R.A. Computational Modeling of β-Secretase 1 (BACE-1) Inhibitors Using Ligand Based Approaches. J. Chem. Inf. Model. 2016, 56, 1936–1949. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER Database of Drugs and Side Effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, S.G.; Baumann, K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Kipf, T.N.; Welling, M. Semi-Supervised Classification with Graph Convolutional Networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Xu, K.; Hu, W.; Leskovec, J.; Jegelka, S. How Powerful Are Graph Neural Networks? arXiv 2018, arXiv:1810.00826. [Google Scholar]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.-J.; Tkatchenko, A.; Müller, K.-R. Schnet—A Deep Learning Architecture for Molecules and Materials. J. Chem. Phys. 2018, 148, 241722. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Liu, Q.; Wang, C.; Huang, Z.; Lin, P.; He, L. Molecular Property Prediction: A Multilevel Quantum Interactions Modeling Perspective. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; Volume 33, pp. 1052–1060. [Google Scholar]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef]
- Hu, W.; Liu, B.; Gomes, J.; Zitnik, M.; Liang, P.; Pande, V.; Leskovec, J. Strategies for Pre-Training Graph Neural Networks. arXiv 2019, arXiv:1905.12265. [Google Scholar]
- Liu, S.; Demirel, M.F.; Liang, Y. N-Gram Graph: Simple Unsupervised Representation for Graphs, with Applications to Molecules. In Proceedings of the 33rd Conference on Neural Information Processing Systems (NeurIPS 2019), Vancouver, BC, Canada, 8–14 December 2019. [Google Scholar]
- Mobley, D.L.; Guthrie, J.P. FreeSolv: A Database of Experimental and Calculated Hydration Free Energies, with Input Files. J. Comput.-Aided Mol. Des. 2014, 28, 711–720. Available online: https://link.springer.com/article/10.1007/s10822-014-9747-x (accessed on 1 March 2024). [CrossRef]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Hersey, A. ChEMBL Deposited Data Set—AZ Dataset 2015. Available online: https://www.ebi.ac.uk/chembl/document_report_card/CHEMBL3301361/ (accessed on 20 February 2024).
- Rupp, M.; Tkatchenko, A.; Müller, K.R.; Von Lilienfeld, O.A. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys. Rev. Lett. 2012, 108, 058301. Available online: https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.108.058301 (accessed on 1 March 2024). [CrossRef]
- Blum, L.C.; Reymond, J.-L. 970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I. Property Distribution of Drug-Related Chemical Databases. J. Comput.-Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Sennrich, R.; Haddow, B.; Birch, A. Neural Machine Translation of Rare Words with Subword Units. arXiv 2015, arXiv:1508.07909. [Google Scholar]
- Wu, Y.; Schuster, M.; Chen, Z.; Le, Q.V.; Norouzi, M.; Macherey, W.; Krikun, M.; Cao, Y.; Gao, Q.; Macherey, K.; et al. Google’s Neural Machine Translation System: Bridging the Gap between Human and Machine Translation. arXiv 2016, arXiv:1609.08144. [Google Scholar]



| Dataset | BBBP [19] | Tox21 [20] | ClinTox [21] | HIV [22] | BACE [23] | SIDER [24] | MUV [25] |
|---|---|---|---|---|---|---|---|
| # Molecules | 2039 | 7831 | 1478 | 41127 | 1513 | 1427 | 93,087 |
| # Tasks | 1 | 12 | 2 | 1 | 1 | 27 | 17 |
| RF | 71.4 (0.0) | 76.9 (1.5) | 71.3 (5.6) | 78.1 (0.6) | 86.7 (0.8) | 68.4 (0.9) | 63.2 (2.3) |
| SVM | 72.9 (0.0) | 81.8 (1.0) | 66.9 (9.2) | 79.2 (0.0) | 86.2 (0.0) | 68.2 (1.3) | 67.3 (1.3) |
| GCN [26] | 71.8 (0.0) | 70.9 (2.6) | 62.5 (2.8) | 74 (3.0) | 71.6 (2.0) | 53.6 (3.2) | 71.6 (4.0) |
| GIN [27] | 65.8 (4.5) | 74 (0.8) | 58 (4.4) | 75.3 (1.9) | 70.1 (5.4) | 57.3 (1.6) | 71.8 (2.5) |
| SchNet [28] | 84.8 (2.2) | 77.2 (2.3) | 71.5 (3.7) | 70.2 (3.4) | 76.6 (1.1) | 53.9 (3.7) | 71.3 (3.0) |
| MGCN [29] | 85 (6.4) | 70.7 (1.6) | 63.4 (4.2) | 73.8 (1.6) | 73.4 (3.0) | 55.2 (1.8) | 70.2 (3.4) |
| D-MPNN [30] | 71.2 (3.8) | 68.9 (1.3) | 90.5 (5.3) | 75 (2.1) | 85.3 (5.3) | 63.2 (2.3) | 76.2 (2.8) |
| Hu et al. [31] | 70.8 (1.5) | 78.7 (0.4) | 78.9 (2.4) | 80.2 (0.9) | 85.9 (0.8) | 65.2 (0.9) | 81.4 (2.0) |
| N-Gram [32] | 91.2 (3.0) | 76.9 (2.7) | 85.5 (3.7) | 83 (1.3) | 87.6 (3.5) | 63.2 (0.5) | 81.6 (1.9) |
| MolCLR-GCN [13] | 73.8 (0.2) | 74.7 (0.8) | 86.7 (1.0) | 77.8 (0.5) | 78.8 (0.5) | 66.9 (1.2) | 84 (1.8) |
| MolCLR-GIN [13] | 73.6 (0.5) | 79.8 (0.7) | 93.2 (1.7) | 80.6 (1.1) | 89 (0.3) | 68 (1.1) | 88.6 (2.2) |
| ChemBERTa | 73.4 (1.4) | 82.3 (0.9) | 88.9 (3.6) | 74.5 (3.1) | 79.2 (2.0) | 60.4 (2.0) | 73.9 (3.4) |
| ChemELECTRA | 72.5 (2.0) | 80 (1.0) | 84.6 (3.3) | 73.7 (2.9) | 76.9 (2.5) | 56.9 (1.8) | 73.7 (2.8) |
| Dataset | FreeSolv [33] | ESOL [34] | Lipo [35] | QM7 [36] | QM8 [37] | QM9 [37] |
|---|---|---|---|---|---|---|
| # Molecules | 642 | 1128 | 4200 | 6830 | 21,786 | 130,829 |
| # Tasks | 1 | 1 | 1 | 1 | 12 | 8 |
| RF | 2.03 (0.22) | 1.07 (0.19) | 0.88 (0.04) | 122.7 (4.2) | 0.0423 (0.0021) | 16.061 (0.019) |
| SVM | 3.14 (0.0) | 1.5 (0.0) | 0.82 (0.0) | 156.9 (0.0) | 0.0543 (0.001) | 24.613 (0.144) |
| GCN | 2.87 (0.14) | 1.43 (0.05) | 0.85 (0.08) | 122.9 (2.2) | 0.0366 (0.0011) | 5.796 (1.969) |
| GIN | 2.76 (0.18) | 1.45 (0.02) | 0.85 (0.07) | 124.8 (0.7) | 0.0371 (0.0009) | 4.741 (0.912) |
| SchNet | 3.22 (0.76) | 1.05 (0.06) | 0.91 (0.1) | 74.2 (6) | 0.0204 (0.0021) | 0.081 (0.001) |
| MGCN | 3.35 (0.01) | 1.27 (0.15) | 1.11 (0.04) | 77.6 (4.7) | 0.0223 (0.0021) | 0.05 (0.002) |
| D-MPNN | 2.18 (0.91) | 0.98 (0.26) | 0.65 (0.05) | 105.8 (13.2) | 0.0143 (0.0022) | 3.241 (0.119) |
| Hu et al. [31] | 2.83 (0.12) | 1.22 (0.02) | 0.74 (0.0) | 110.2 (6.4) | 0.0191 (0.0003) | 4.349 (0.061) |
| N-Gram | 2.51 (0.19) | 1.1 (0.03) | 0.88 (0.12) | 125.6 (1.5) | 0.032 (0.0032) | 7.636 (0.027) |
| MolCLR-GCN | 2.39 (0.14) | 1.16 (0.0) | 0.78 (0.01) | 83.1 (4.0) | 0.0181 (0.0002) | 3.552 (0.041) |
| MolCLR-GIN | 2.2 (0.2) | 1.11 (0.01) | 0.65 (0.08) | 87.2 (2.0) | 0.0174 (0.0013) | 2.357 (0.118) |
| ChemBERTa | 5 (0.11) | 2.06 (0.02) | 1.2 (0.0) | 187.7 (2.7) | 0.0333 (0.0003) | 20.941 (0.199) |
| ChemELECTRA | 5.03 (0.13) | 2.05 (0.0) | 1.2 (0.0) | 179.1 (0.7) | 0.0359 (0.0002) | 24.228 (0.314) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, W.; Goo, S.; Hwang, T.; Lee, H.; Kim, Y.-K.; Chae, J.-w.; Yun, H.-y.; Jung, S. Absorption Distribution Metabolism Excretion and Toxicity Property Prediction Utilizing a Pre-Trained Natural Language Processing Model and Its Applications in Early-Stage Drug Development. Pharmaceuticals 2024, 17, 382. https://doi.org/10.3390/ph17030382
Jung W, Goo S, Hwang T, Lee H, Kim Y-K, Chae J-w, Yun H-y, Jung S. Absorption Distribution Metabolism Excretion and Toxicity Property Prediction Utilizing a Pre-Trained Natural Language Processing Model and Its Applications in Early-Stage Drug Development. Pharmaceuticals. 2024; 17(3):382. https://doi.org/10.3390/ph17030382
Chicago/Turabian StyleJung, Woojin, Sungwoo Goo, Taewook Hwang, Hyunjung Lee, Young-Kuk Kim, Jung-woo Chae, Hwi-yeol Yun, and Sangkeun Jung. 2024. "Absorption Distribution Metabolism Excretion and Toxicity Property Prediction Utilizing a Pre-Trained Natural Language Processing Model and Its Applications in Early-Stage Drug Development" Pharmaceuticals 17, no. 3: 382. https://doi.org/10.3390/ph17030382
APA StyleJung, W., Goo, S., Hwang, T., Lee, H., Kim, Y. -K., Chae, J. -w., Yun, H. -y., & Jung, S. (2024). Absorption Distribution Metabolism Excretion and Toxicity Property Prediction Utilizing a Pre-Trained Natural Language Processing Model and Its Applications in Early-Stage Drug Development. Pharmaceuticals, 17(3), 382. https://doi.org/10.3390/ph17030382