3. Pseudopeptides
Another approach to construct peptidomimetics involves the design of conformationally restricted analogs that mimic and/or stabilise characteristics of the receptor-bound conformation of the endogenous peptide, such as β-turns and other.
The ability to access to such structures modified by incorporation of heterocycles allows the study of the associated biological processes, with an opportunity for further drug design and development. 1,2,3-Triazoles, for example, offer an appealing structural motif in peptidomimetic research because their structural and electronic characteristics are similar to those of a peptide bond [
24]. It is also known that secondary structures (β-turns, α-helices, β-strands) are sites of recognition by the enzymes, such as proteases [
25].
This idea can be extended to scaffold peptidomimetics in which important pharmacophoric residues are held in the appropriate orientation by a rigid template. So in this field, much effort has been devoted to the design and synthesis of conformationally constrained compounds that mimic, or induce, specific secondary structural features of peptides and proteins.
Many scaffolds possessing the functionalities of peptides (
i.e., amine and carboxyl groups) together with well defined spatial properties, thus reproducing the desired orientation of a growing peptide chain, have hence been created [
26].
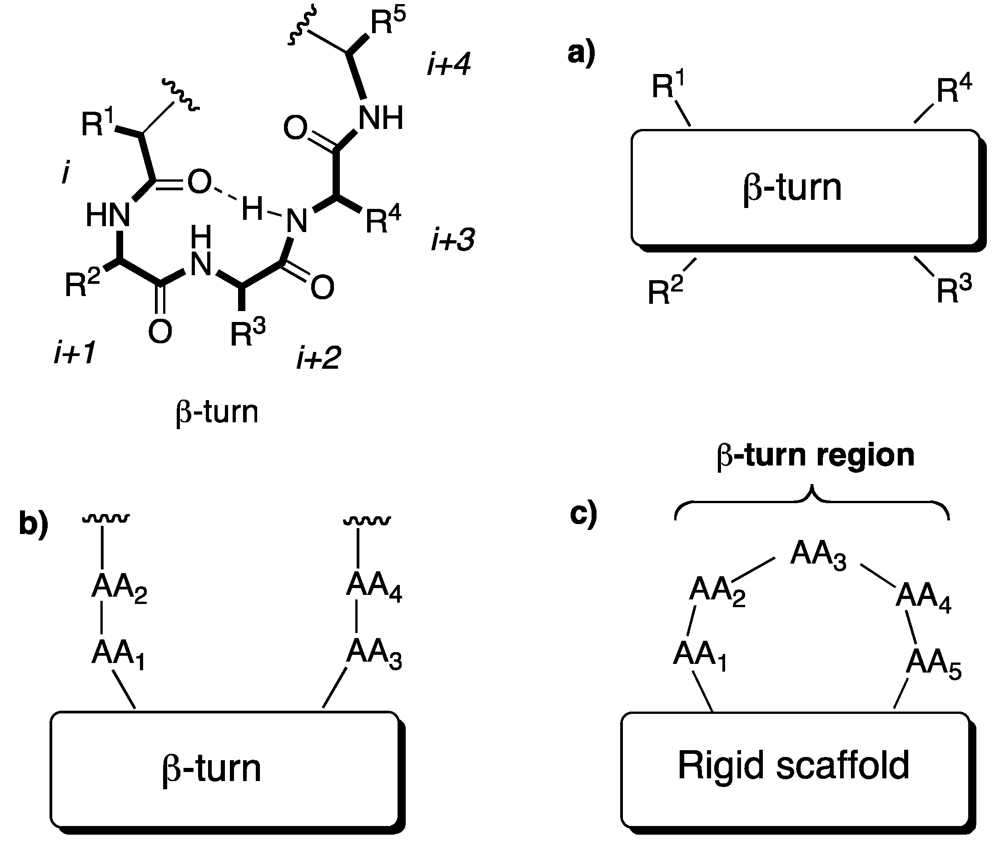
The β-turn is a common feature in biologically active peptides and is defined as any tetrapeptide sequence, with a 10-membered intramolecularly H-bonded ring, in which the C
α(i) to C
α(i+3) distance varies from 4 to 7 Angstrom (
Figure 4).
There are at least 14 types of β-turn structures, described in literature [
26]. These conformers used as models have been developed for linear and short peptides. In natural proteins, turn fragments can adopt an even larger variety of conformations, due to stabilization provided by the remaining portion of molecule. Although there has been much discussion in the literature on what constitutes a β-turn mimic and how different types of mimics have to be characterized, these can be roughly classified into three broad classes, illustrated in
Figure 4.
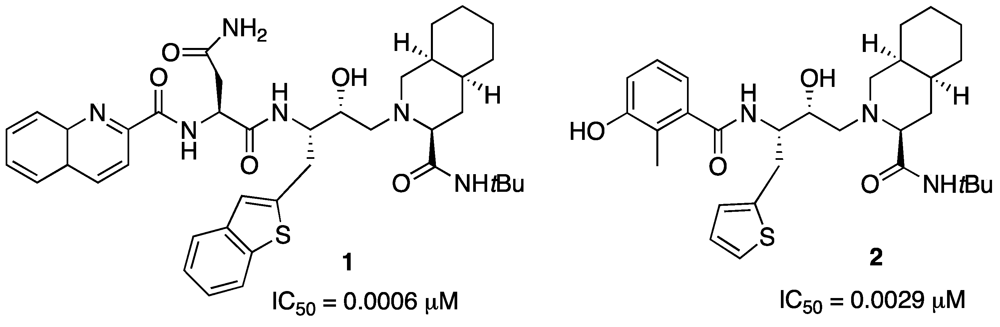
Aiming to construct a β-turn mimic with a heterocyclic scaffold we hypothesized an opportune synthetic approach, in particular directed to the preparation of potential active structure for the treatment of Alzheimer’s disease, either as β-secretase or as β-amyloid aggregation inhibitor.
Figure 4.
β-turn and induced β-turn. (a) internal β-turn mimics; (b) β-hairpin mimics (in which a rigid scaffold induces reversal of the peptide chain); (c) external β-turn inducers.
Figure 4.
β-turn and induced β-turn. (a) internal β-turn mimics; (b) β-hairpin mimics (in which a rigid scaffold induces reversal of the peptide chain); (c) external β-turn inducers.
β-Secretase (memapsin 2 or BACE1), discovered in 1999 [
27,
28], is one of the two proteases that cleave the β-amyloid precursor protein (APP) to produce a 40–42 residue amyloid β-peptide (Aβ) in the human brain. Accumulation of Aβ results in the formation of amyloid plaques and neurofibrillary tangles. The neurotoxicity of Aβ is ultimately responsible for brain inflammation, neuronal death, dementia and Alzheimer’s disease [
29]. Consequently therapeutic inhibition of memapsin 2 has emerged as one of the most active areas of today’s drug development for the intervention of Alzheimer’s disease.
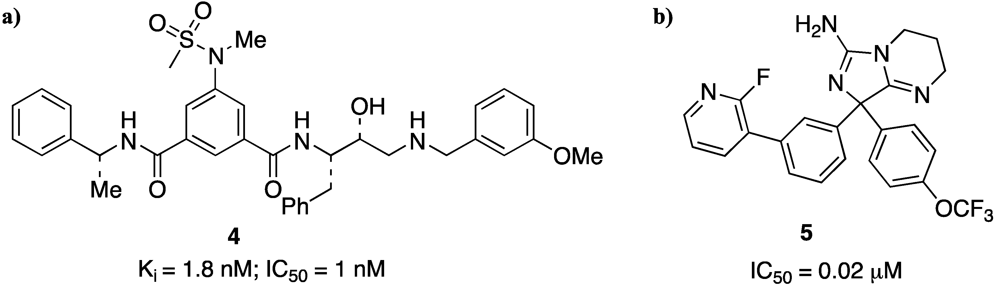
It is known that β-secretase is an aspartic protease, for which the inhibition mechanism and the design of transition state analogues through the successful development of HIV-1 protease inhibitor drugs are well previously described [
30]. So, as first approach to the design and synthesis of potential inhibitors, Ghosh and co-authors reported [
31] a series of Leu-Ala isosteres which led to the first potent transition-state inhibitor (CTS-21166) that is actually in advanced clinical trials. This compound (an optimization of the structure is reported in
Figure 5a) represents the first small peptidomimetic inhibitor modifying therapy for Alzheimer’s disease.
Figure 5.
Two potential β-secretase inhibitors. (a) GRL-8234; (b) heteroarylaminoimidazole.
Figure 5.
Two potential β-secretase inhibitors. (a) GRL-8234; (b) heteroarylaminoimidazole.
The newest generation BACE1 inhibitors are low molecular weight molecules with excellent cell permeability, have little or no peptidic character, and possess enhanced pharmacokinetic profiles [
32,
33,
34,
35]. Actually many lines of research are concerned with the synthesis and study of small-molecule-containing heterocyclic rings as β-secretase inhibitors [
36,
37] (
Figure 5b).
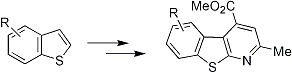
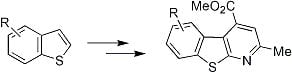
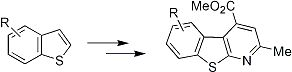
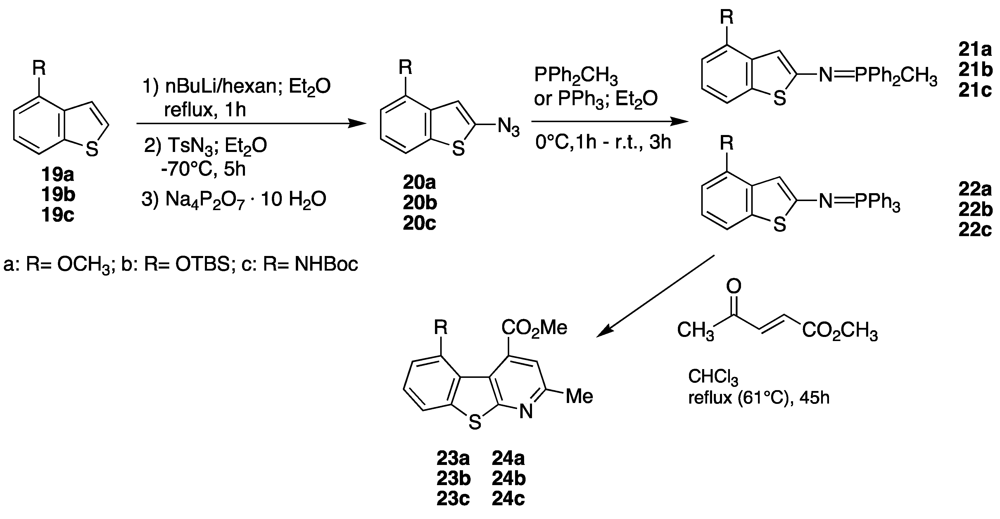
4. Scaffold for Pseudopeptides: Design and Synthesis of Heteroaromatic Tricyclic Structures
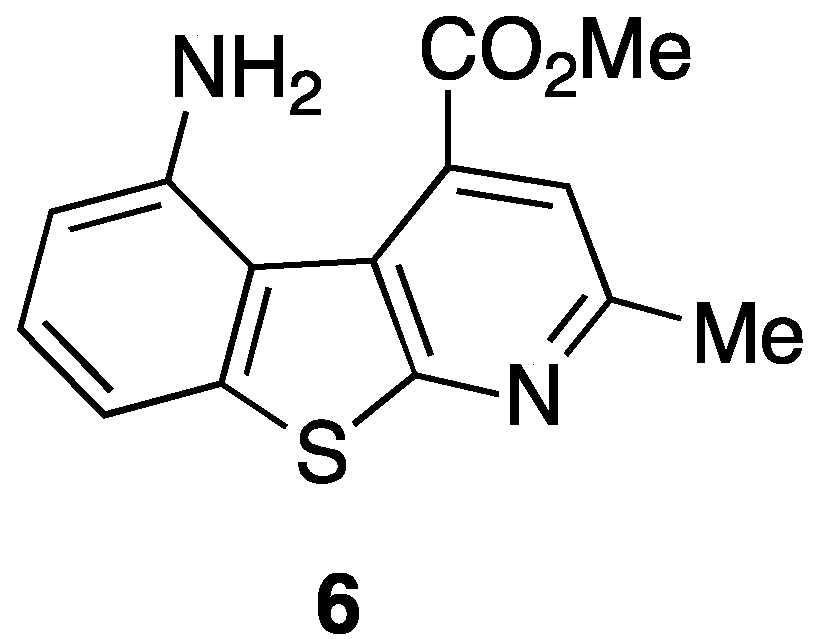
Our long interest in the preparation of heterocycles and their application in medicinal chemistry prompted us to design and synthesize new tricyclic compounds as potential scaffolds for pseudopeptides and/or as β-secretase inhibitors. Recently [
38], a particular structure that mimics a biological turn has just been synthetized in our laboratory. The molecule is shown in
Figure 6.
Figure 6.
5-Amino-4-carbomethoxy-2-methylbenzothienopyridine.
Figure 6.
5-Amino-4-carbomethoxy-2-methylbenzothienopyridine.
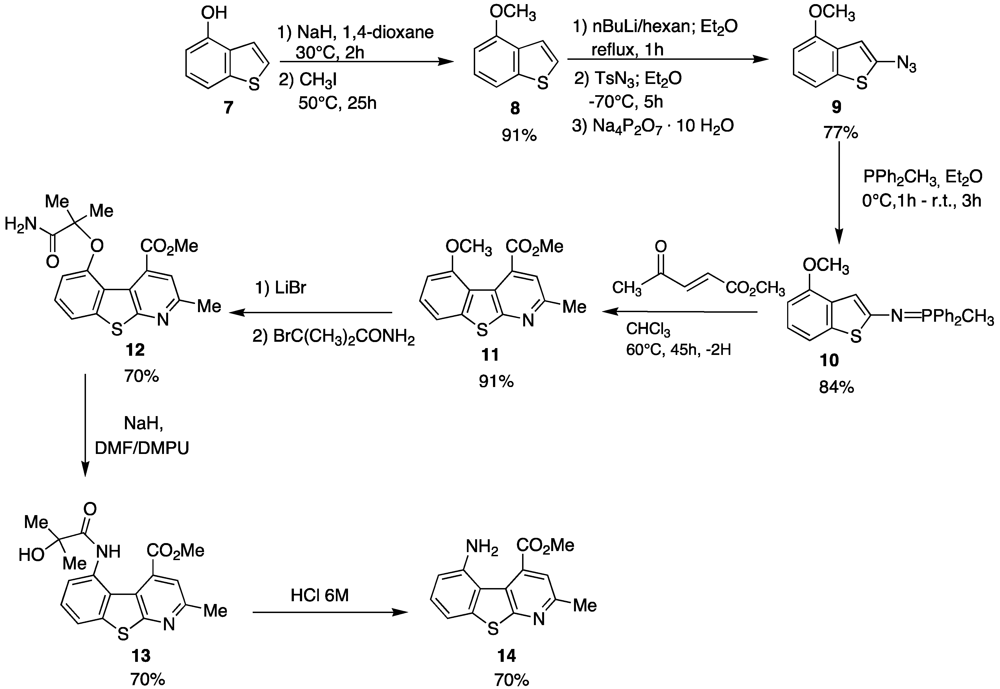
As can be seen such a benzothienopyridine bears two interesting groups in the 4- and 5-positions of the tricyclic structure, a carbomethoxy and an amine function, respectively, that immediately suggest its possible use as a pseudopeptide scaffold as well as a β-secretase inhibitor. For its preparation an innovative synthetic route was developed (
Scheme 2).
Scheme 2.
Synthetic route affording 5-amino-4-carbomethoxy-2-methylbenzothienopyridine.
Scheme 2.
Synthetic route affording 5-amino-4-carbomethoxy-2-methylbenzothienopyridine.
The depicted synthetic approach is based on four principal reactions:
- (1)
An azido-transfer reaction that furnishes the azido precursors [
39];
- (2)
A Staudinger reaction that transforms the azido group into an iminophosphorane, a powerful tool for the construction of nitrogen containing heterocycles [
40,
41,
42,
43,
44]
- (3)
A tandem aza-Wittig/electrocyclization of iminophosphoranes with suitable α,β-unsaturated carbonyl compounds;
- (4)
Finally, a Smiles rearrangement that affords the benzothienopyridine [
45,
46,
47,
48]
It is well known that benzothienopyridines are of pharmacological interest due to their isosterism with indolopyridines [
49,
50] and to their reported activity as antibacterial [
51], antiallergic [
52] and anxiolytic agents [
53], so the development of a facile preparation seems to be of timely interest.
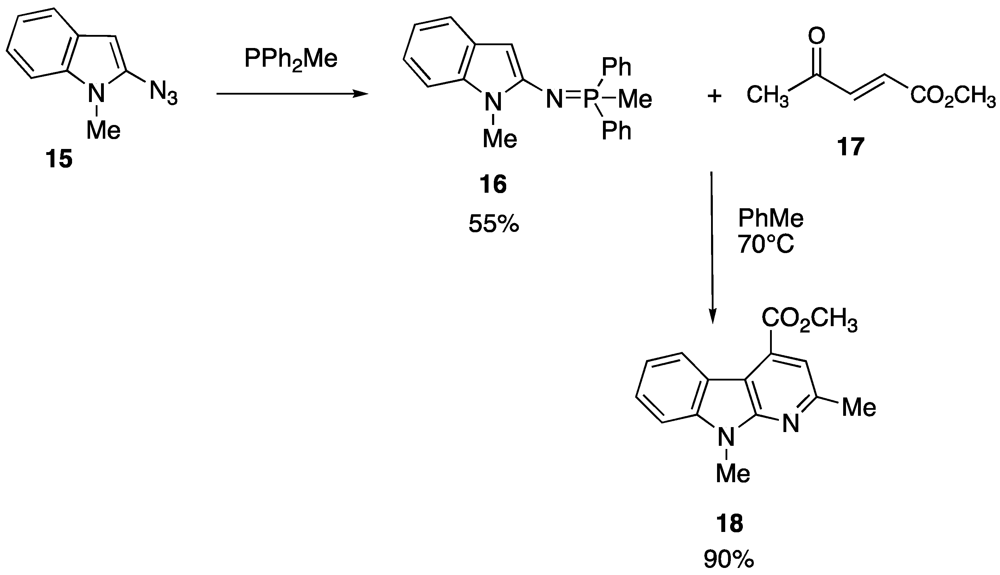
Our experience in heterocyclic chemistry suggested us also to extend the use of Staudinger reaction (and subsequent aza-Wittig/electrocyclization reaction) either to indole, benzofuran or to substituted benzothienyl ring. As an example of such a methodology for the preparation of a benzofuropyridine [
54] and α-carboline [
55] we reported the reaction scheme realized for 2-azido-
N-methylindole (
Scheme 3).
Scheme 3.
Synthesis of α-carboline.
Scheme 3.
Synthesis of α-carboline.
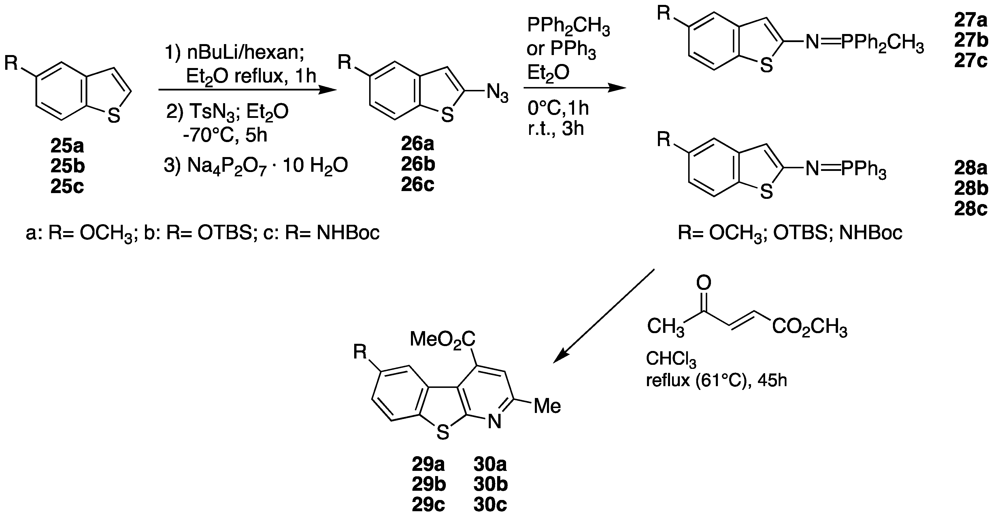
This efficient strategy was then applied on 4- and 5-substituted benzothiophenes; preliminary results are shown in the next
Scheme 4 and
Scheme 5, and
Table 1 and
Table 2, [
56,
57].
Table 1.
Synthesis of 5-amino (or hydroxy)-4-carbomethoxy-2-methylbenzothienopyridines.
Table 1.
Synthesis of 5-amino (or hydroxy)-4-carbomethoxy-2-methylbenzothienopyridines.
| Entry | Azide (%yield) | Phosphorane (%yield) | Benzothienopyridine (%yield) |
|---|
| 1 | 20a (77%) | 21a (84%) | 23a (91%) |
| 2 | 20b (80%) | 21b (40%) | 23b (14%) |
| 3 | 20c (30%) | 21c (no product) | 23c (no product) |
| 4 | 20a (77%) | 22a (33%) | 24a (40%) |
| 5 | 20b (80%) | 22b (20%) | 24b (no product) |
| 6 | 20c (30%) | 22c (no product) | 24c (no product) |
Table 2.
Synthesis of 4-carbomethoxy-6-hydroxy-(or amine)-2-methylbenzothienopyridines.
Table 2.
Synthesis of 4-carbomethoxy-6-hydroxy-(or amine)-2-methylbenzothienopyridines.
| Entry | Azide (%yield) | Phosphorane (%yield) | Benzothienopyridine (%yield) |
|---|
| 1 | 26a (55%) | 27a (58%) | 29a (35%) |
| 2 | 26b (28%) | 27b (79%) | 29b (13%) |
| 3 | 26c (62%) | 27c (49%) | 29c (no product) |
| 4 | 26a (55%) | 28a (74%) | 30a (52%) |
| 5 | 26b (28%) | 28b (81%) | 30b (58%) |
| 6 | 26c (62%) | 28c (no product) | 30c (no product) |
Scheme 4.
Synthesis of 5-amino (or hydroxy)-4-carbomethoxy-2-methylbenzothienopyridines.
Scheme 4.
Synthesis of 5-amino (or hydroxy)-4-carbomethoxy-2-methylbenzothienopyridines.
Scheme 5.
Synthesis of 4-carbomethoxy-6-hydroxy-(or amine)-2-methylbenzothienopyridines.
Scheme 5.
Synthesis of 4-carbomethoxy-6-hydroxy-(or amine)-2-methylbenzothienopyridines.
As can be seen, the presence of a t-butyldimethylsilylether functionality in the 4-position of the benzothiophene plays an important different effect: In fact the azido transfer reaction was favoured, while the Staudinger reaction was unfavoured, in particular when triphenylphosphine was used.
Probably, such an effect was due to the hindrance of the substituent either on phosphorous or on oxygen. In presence of a methoxy group at the 4-position of benzothiophene the effect on the azido formation was the same (we hypothesized a predominant electronic effect), but it had the opposite effect on iminophosporane synthesis.
The NH-Boc substituent merits particular: In this case the presence of the hydrogen on the N-function represents a drawback during lithiation, despite the hindrance of the t-butyl group; hence the chemical yields were unsatisfactory, even with excess of butyl lithium.
A different result was obtained when the same synthetic route was used on 5-substituted benzothiophene, precursor of 4-carbomethoxy-6-hydroxy-(or amine)-2-methylbenzothienopyridine (
Figure 11). As can be observed it seems that electronic effects were prevalent for all considered substrates, both in the azido transfer and in the Staudinger reaction leading to iminophosphoranes, but the final cyclization is still not an optimized procedure. Further studies on the synthesis of the described tricyclic structures and their potential biological activity on β-secretase are in progress.
5. Experimental
5.1. General
Column chromatography was carried out on Merck silica gel (0.063–0.200 mm particle size) by progressive elution with petroleum ether/ethyl acetate or petroleum ether/diethyl ether mixtures.
1H- and
13C-NMR spectra were normally recorded for CDCl
3 solutions on a Bruker AM 300 MHz or on Varian INOVA 400 and 500 MHz instruments. IR spectra were registered on a JASCO FT/IR 460 Plus. Mass spectra were obtained with a Hewlett-Packard 5971 mass-selective detector on a Hewlett-Packard GC/MS 6890-5973 system. Dichloromethane, chloroform and carbon tetrachloride were dried with anhydrous CaCl
2; diethyl ether and 1,4-dioxane were dried using sodium/benzophenone. Dry dimethylformamide was commercially available. 4-Hydroxybenzo[
b]thiophene was prepared according to the literature [
38]. 2-Bromo-2-methylpropanamide was synthesized as recently reported [
38] and used in Smiles rearrangement as reported in the same paper to obtain the suitable 2-aryloxy-2-methylpropanamide. 5-Aminobenzo[
b]thiophene was commercially available (Maybridge Trevillet, Tintagel, Cornwall, UK).
5.2. Synthesis of 4,5-Disubstituted Benzothieno[2,3-b]pyridine Precursors
4-Methoxybenzo[b]thiophene (
19a). Compound
19a was synthesized from 4-hydroxybenzo[
b]-thiophene according to a known procedure [
38]. Thick red oil (yield 91%). Found: C, 65.85; H, 4.88; S, 19.56%. C
9H
8OS requires C, 65.83; H, 4.91; S, 19.52%;
1H-NMR (300 MHz, CDCl
3) (ppm): δ
H 7.65–7.5 (m, 2H), 7.45–7.38 (m, 2H), 6.84–6.80 (m, 1H), 3.99 (s, 3H);
13C-NMR (75 MHz, CDCl
3) (ppm): δ
C 155.0, 141.2, 130.5, 125.3, 124.6, 120.5, 115.0, 104.1, 55.9; MS
m/z: 164 (M
+), 149 (100).
4-(Tert-butyldimethylsilyloxy)benzo[b]thiophene (
19b). Compound
19b was synthesized from 4-hydroxybenzothiophene according to a known procedure [
38]. Thick oil (yield 75%). Found: C, 63.59; H, 7.59; S, 12.15%. C
14H
20OSSi requires: C, 63.58; H, 7.62; S, 12.12%;
1H-NMR (300 MHz, CDCl
3) (ppm): δ
H 7.72–7.67 (m, 2H), 7.53–7.37 (m, 2H), 6.99–6.93 (m, 1H), 1.35 (s, 9H), 0.50 (s, 6H);
13C-NMR (75 MHz, CDCl
3) (ppm): δ
C 150.7, 141.5, 133.5, 125.3, 124.3, 121.0, 115.7, 113.1, 25.7, 18.5, 20.4; MS
m/z: 264 (M
+), 207 (100).
Tert-butyl benzo[b]thiophen-4-yl carbamate (
19c). 4-Hydroxybenzo[
b]thiophene (135 mg, 0.9 mmol) was converted to
N-(benzo[
b]thiophen-4-yl)-2-hydroxy-2-methylpropanamide (190 mg, 90%) via a Smiles rearrangement according to a known procedure [
38]. The
N-(benzo[
b]thiophen-4-yl)-2-hydroxy-2-methylpropanamide was dissolved in HCl 6M (10 mL) and the resulting mixture was stirred at 100 °C for 4 h. After cooling to 25 °C the mixture was slowly treated with a solution of NaOH 2M until neutrality, then extracted twice with diethyl ether and dried over sodium sulphate. Removal of the solvent gave the 4-aminebenzo[
b]thiophene as a black thick oil (107 mg, 90%).
To a solution of 4-aminobenzo[
b]thiophene (107 mg, 0.72 mmol) in dry dichloromethane (4 mL), di-
tert-butyl dicarbonate (BOC) (205 mg, 0.94 mmol) was added and the resulting mixture was stirred in an inert atmosphere for 15 h at room temperature [
56]. After solvent removal, the crude was dissolved into ethyl acetate and the organic phase was washed several times with water, once with brine and then dried over sodium sulphate and finally concentrated under vacuum. The crude product was chromatographed on silica gel, using petroleum ether/ethyl acetate 7:3 as eluent, to give
19c as a thick light pink oil (108 mg, 60%).
1H-NMR (400 MHz, CDCl
3) (ppm):δ
H 7.82–7.80 (m, 1H), 7.60–7.58 (m, 1H), 7.43–7.41 (m, 1H), 7.33–7.30 (m, 1H), 6.72 (s, 1H), 1.54 (s, 9H);
13C-NMR (125 MHz, CDCl
3) (ppm): δ
C 152.9, 140.5, 132.7, 131.1, 125.9, 124.9, 119.1, 117.8, 115.1, 80.8, 28.3; MS:
m/z 249 (M
+); 193 (100).
5.3. Synthesis of Azides 20a,b,c
2-Azido-4-methoxy-1-benzo[b]thiophene (
20a). Compound
20a was synthesized from
19a according to a known procedure [
38]. Thick yellow oil (yield 77%). Found: C, 52.65; H, 3.47; N, 20.45; S, 15.63%. C
9H
7N
3OS requires: C, 52.67; H, 3.44; N, 20.47; S, 15.62%; IR ν
max/cm
−1 2113 (N
3);
1H-NMR (300 MHz, CDCl
3) (ppm): δ
H 7.35-7.20 (m, 2H); 7.05 (s, 1H); 6.80-6.75 (m, 1H); 3.95 (s, 3H);
13C-NMR (75 MHz, CDCl
3) (ppm): δ
C 152.7, 139.5, 138.2, 125.3, 124.6, 117.1, 115.7, 110.3, 50.5.
[(2-Azidobenzo[b]thiophen-4-yl)oxy](tert-butyl)dimethylsilane (
20b). Compound
20b was synthesized from
19b according to a known procedure [
38]. Thick yellow oil (yield 80%). Found: C, 55.07; H, 6.21; N, 13.78; S, 10.44%. C
14H
19N
3OSSi requires C, 55.05; H, 6.27; N, 13.76; S, 10.50%; IR ν
max/cm
−1 2110 (N
3);
1H-NMR (300 MHz, CDCl
3) (ppm): δ
H 7.50–7.40 (m, 1H), 7.19–7.11 (m, 1H), 6.89 (s, 1H), 6.72–6.68 (m, 1H), 1.07 (s, 9H), 0.27 (s, 6H);
13C-NMR (75 MHz, CDCl
3) (ppm): δ
C 142.7, 125.3, 124.5, 121.0, 115.6, 115.0, 113.6, 108.3, 25.8, 15.9.
Tert-butyl (2-azidobenzo[b]thiophen-4-yl)carbamate 20c. Compound 19c (108 mg, 0,43 mmol) in dry diethyl ether (7 mL) was treated, under an inert atmosphere, with 1.6 M n-butyl lithium in hexane (0.65 mmol) and refluxed for 1 h. Then the reaction mixture was cooled to −70 °C and, slowly, tosylazide (119 mg, 0.60 mmol) in dry diethyl ether (3 mL) was added. After 5 h at this temperature the obtained triazene salt was filtered under vacuum, washed with dry diethyl ether and treated at 0 °C with an aqueous solution of sodium pyrophosphate decahydrate (316 mg, 0.7 mmol, in 5 mL of water). After 15 min of stirring at this temperature the suspension was filtered on a Buckner funnel and extracted twice with diethyl ether and then with ethyl acetate. After solvent removal, the crude product was purified by chromatography on Florisil (eluent petroleum ether/ethyl acetate 8:2) giving the title compound (38 mg, 30%) as a thick light yellow oil. Compound 19c is unstable, so it was only characterized by IR and 1H-NMR spectroscopy. IR νmax/cm−1 2125 (N3); 1H-NMR (400 MHz, CDCl3) (ppm): δH 7.73 (s, 1H), 7.55–7.53 (m, 1H), 7.38–7.36 (m, 1H), 7.27–7.24 (m, 1H), 6.66 (s, 1H), 1.49 (s, 9H).
5.4. Synthesis of Imminophosphoranes 21a,b,c-22a,b,c
[(4-Methoxy-1-benzothiophen-2-yl)imino]-(methyl)diphenylphosphorane 21a. This compound was prepared from the azide
20a and methyldiphenylphosphine (1eq) according to a known procedure [
38]. Chromatography with petroleum ether/ethyl acetate 7:3 as eluent gave the title compound (yield 84%) as a red oil. Found: C, 70.04; H, 5.36; N, 3.69; S, 8.50%. C
22H
20NOPS requires C, 70.01; H, 5.34; N, 3.71; S, 8.49%;
1H-NMR (300 MHz, CDCl
3) (ppm): δ
H 7.62–7.50 (m, 5H), 7.43–7.32 (m, 5H), 6.98–6.90 (m, 1H), 6.87–6.80 (m, 1H), 6.53–6.48 (m, 1H), 6.08 (s, 1H), 3.70 (s, 3H), 2.25–2.20 (d, 3H,
JPH = 12.8 Hz);
13C-NMR (75 MHz, CDCl
3) (ppm): δ
C 157.0, 152.3, 140.2, 135.3, 133.0, 132.5, 132.0, 131.7, 131.4, 130.0 129.5, 128.2, 125.8, 121.0, 114.3, 101.5, 55.5, 15.5.
4-(Tert-butyldimethylsilyl)oxy)-N-(methyldiphenylphosphoranyliene)benzo[b]thiophen-2-amine 21b. This compound was prepared from the azide
20b and methyldiphenylphosphine (1eq) according to a known procedure [
38]. Chromatography with petroleum ether/ethyl acetate 7:3 as eluent gave the title compound (88 mg, 40%) as a brown powder, mp 90–92 °C (diethyl ether). (Found: C, 67.66; H, 6.79; N, 2.99; S, 6.73%. C
27H
32NOPSSi requires C, 67.89; H, 6.75; N, 2.93; S, 6.71%);
1H-NMR (300 MHz, CDCl
3) (ppm): δ
H 7.83–7.77 (m, 5H), 7.58–7.37 (m, 5H), 7.11–7.06 (m, 1H), 6.87–6.77 (m, 1H), 6.52–6.49 (m, 1H), 6.02 (s, 1H), 2.25–2.20 (d, 3H,
JPH = 12.8 Hz); 0.95 (s, 9H), 0.1 (s, 6H);
13C-NMR (75 MHz, CDCl
3) (ppm): δ
C 168.3, 151.7, 147.5, 135.8, 134.7, 134.0, 132.2, 131.2, 130.8, 129.5, 129.0, 128.8, 120.9, 116.1, 113.2, 101.9, 25.7, 18.3, 14.5, 13.8.
Tert-butyl 2-(methyldiphenylphosphoranylidene)aminobenzo[b]thiophen-4-yl)carbamate 21c. Compound 20c (62 mg, 0.21 mmol) in dry diethyl ether (3 mL) was added dropwise to a solution of methyldiphenylphosphine (41 mg, 0.20 mmol) in dry dichloromethane (3 mL) at 0 °C, in nitrogen atmosphere. After 2 h, the reaction mixture was allowed to room temperature, finally it was concentrated under vacuum. No desired product was recovered but only degradation products of the starting materials.
[(4-Methoxy-1-benzothiophen-2-yl)imino]-triphenylphosphorane 22a. This compound was prepared from the azide
20a (100 mg, 0.49 mmol) and triphenylphosphine (128 mg, 0.49 mmol) following a reported procedure [
38]. Chromatography on silica gel with petroleum ether/ethyl acetate 7:3 as eluent gave the title compound (72 mg, 33%).
1H-NMR (500 MHz, CDCl
3) (ppm): δ
H 7.74–7.70 (m, 5H), 7.52–7.50 (m, 5H), 7.46–7.42 (m, 5H), 7.00–6.98 (d, 1H,
JPH = 10 Hz), 6.88–6.85 (m, 1H), 6.51 (d, 1H,
JPH = 5 Hz), 6.24 (s, 1H), 3.73 (s, 3H);
13C-NMR (125 MHz, CDCl
3) (ppm): δ
C 161.2, 140.9, 132.6, 131.2, 128.7, 125.4, 123.3 121.4, 115.2, 108.6, 98.9, 56.2.
[(4-[tert-Butyl(dimethyl)silyl]oxy-1-benzothiophen-2-yl)imino]-triphenylphosphorane 22b. This compound was prepared from the azide
20b (66 mg, 0.22 mmol) and triphenylphosphine (59 mg, 0.22 mmol) following a reported procedure [
38]. Chromatography on silica gel with petroleum ether/ethyl acetate 7:3 as eluent gave the title compound (24 mg, 20%) as a brown/green oil.
1H-NMR (500 MHz, CDCl
3) (ppm): δ
H 7.74–7.70 (m, 5H), 7.50–7.49 (m, 5H), 7.42–7.41 (m, 5H), 7.01 (d, 1H,
JPH = 11 Hz), 6.80–6.76 (m, 1H), 6.46 (d, 1H,
JPH = 7 Hz), 6.03 (s, 1H), 0.95 (s, 9H), 0.1 (s, 6H);
13C-NMR (125 MHz, CDCl
3) (ppm): δ
C 185.8, 166.9, 147.2, 135.5, 134.5, 132.9, 132.8, 132.7, 128.8, 128.7, 120.8, 114.7, 112.9, 80.3, 25.8, 23.8, 18.1, 1.0, −4.4.
Tert-butyl (2((triphenylphosphoranylidene)amino)benzo[b]thiophen-4-yl)carbamate 22c. Compound 20c (105 mg, 0.4 mmol) in dry dichloromethane (5 mL) was added dropwise to a solution of triphenylphosphine (117 mg, 0.44 mmol) in dry dichloromethane (5 mL) at 0 °C, in nitrogen atmosphere. After 2 h, the reaction mixture was allowed to reach room temperature and stirred over night; finally it was concentrated under vacuum. No desired product was recovered but only degradation products of the starting materials.
5.5. General Procedure for Synthesis of (1)Benzothieno[2,3-b]pyridines 23a,b, 24a
A solution of the appropriate iminophosphorane
22a,b (0.12 mmol) in dry chloroform (3 mL) was treated with methyl
trans-4-oxo-2-pentenoate (0.12 mmol) and then stirred in an inert atmosphere for 34 h at 45 °C. After removal of the solvent, the crude product was chromatographed on silica gel, using petroleum ether/ethyl acetate 7:3 as eluent. For yields see
Table 1.
Methyl 5-methoxy-2-methyl(1)benzothieno[2,3-b]pyridine-4-carboxylate 23a–24a. This compound was obtained as a thick oil. (Found: C, 62.72; H, 4.58; N, 4.85; S, 11.13%. C15H13NO3S requires C, 62.70; H, 4.56; N, 4.87; S, 11.16%); 1H-NMR (300 MHz, CDCl3) (ppm): δH 7.75–7.70 (m, 1H), 7.57–7.50 (m, 1H), 7.47–7.40 (m, 1H), 6.95–6.90 (m, 1H), 3.98 (s, 6H), 2.72 (s, 3H); 13C-NMR (75 MHz, CDCl3) (ppm): δC 169.4, 167.8, 156.8, 139.4, 138.2, 132.5, 131.2, 129.1, 128.8, 118.2, 115.6, 106.6, 55.9, 30.3; MS: m/z = 287 (M+).
Methyl 5-[tert-butyl(dimethyl)silyl]oxy-2-methyl(1)benzothieno[2,3-b]pyridine-4-carboxylate 23b. This compound was obtained as a thick oil. (Found: C, 62.00; H, 6.53; N, 3.58; S, 8.24%. C20H25NO3SSi requires: C, 61.98; H, 6.50; N, 3.61; S, 8.27%); 1H-NMR (300 MHz, CDCl3) (ppm): δH 7.50–7.25 (m, 3H), 7.00–6.95 (m, 1H), 3.90 (s, 3H), 2.72 (s, 3H), 0.81 (s, 9H), 0.15 (s, 6H); 13C-NMR (75 MHz, CDCl3) (ppm): δC 169.5, 154.2, 151.9, 150.5, 149.5, 141.5, 131.7, 128.4, 117.7, 117.0, 116.2, 53.9, 29.8, 26.2, 25.7, 19.8, 18.5, −0.4; MS: m/z: 387 (M+).
5.6. Synthesis of 4,6-Disubstituted Benzothieno[2,3-b]pyridine Precursors
5-Methoxybenzo[b]thiophene 25a. Compound
25a was prepared from commercial 4-methoxybenzenethiol (2 g, 14 mmol) according to a known procedure [
57]. Chromatography on silica gel, using petroleum ether/ethyl acetate 9:1 as eluent, furnished the title compound
25a as a thick fragrant yellow oil (218 mg, 31%).
1H-NMR (500 MHz, CDCl
3) (ppm): δ
H 7.67 (d, 1H,
JPH = 10 Hz), 7.37 (d, 1H,
JPH = 5 Hz), 7.22–7.20 (m, 2H), 6.94–6.93 (d, 1H,
JPH = 5 Hz), 3.81 (s, 3H);
13C-NMR (125 MHz, CDCl
3) (ppm): δ
C 159.3, 141.0, 132.9, 127.9, 123.8, 123.6, 121.5, 106.7, 55.9; MS: m/z = 164 (M
+).
5-Hydroxybenzo[b]thiophene. At 0 °C, to the substrate 25a dissolved in chlorobenzene was added dropwise a solution of BBr3·S(CH3)2 1 M in CH2Cl2. After about 30 min at 0 °C the solution is brought to reflux for 22 h. The reaction was quenched by adding about 50 mL of water and extracting the organic product three times with CH2Cl2 (25 mL). The combined organic phases were washed first with water, then with brine. The reaction product is purified by chromatography on silica gel (petroleum ether/diethyl ether 8:2 as eluent) to give 395 mg (81%) of 5-hydroxybenzo[b]thiophene as a light pink solid. Mp 89–92 °C. 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.73 (d, 1H, JPH = 10 Hz ), 7.46 (d, 1H, JPH = 5 Hz), 7.27 (d, 1H, JPH = 5 Hz), 7.22 (d, 1H, JPH = 5 Hz); 6.94 (d, 1H, JPH = 5 Hz); 5.04 (s;1H). 13C-NMR (125 MHz, CDCl3) (ppm): δC 153.4, 141.1, 133.6, 128.1, 123.5, 116.2, 114.6, 108.8; MS: m/z 150 (M+).
(Benzo[b]thiophen-5-yloxy)(tert-butyl)dimethylsilane 25b. 5-Hydroxybenzo[b]thiophene (320 mg, 2.13 mmol) was dissolved in 8 mL of dry dichloromethane and to this solution were added tert-butyldimethylsilylchloride (3.48 mmol), imidazole (6.68 mmol) and a catalytic amount of 4-dimethylaminopyridine. The resulting mixture was stirred in inert atmosphere for 3 h at room temperature, then was filtered and washed with saturated ammonium chloride solution and twice with water. After solvent removal the crude product was chromatographed on silica gel, using petroleum ether as eluent, to give 25b as a thick light brown oil (434 mg, 83%). 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.47 (d, 1H, JPH = 15 Hz), 7.19 (d, 1H, JPH = 15 Hz), 7.04 (d, 1H, JPH = 15 Hz), 7.02–6.99 (m, 1H), 6.70–6.68 (m, 1H), 0.79 (s, 9H), 0.30–0.00 (m, 6H); 13C-NMR (125 MHz, CDCl3) (ppm): δC 153.0, 140.9, 132.8, 127.3, 123.4, 122.9, 118.7, 113.5, 29.7, 25.7. MS: m/z 264 (M+); 207 (100).
Tert-butyl benzo[b]thiophen-5-yl-carbamate 25c. To a solution of benzo[b]thiophen-5-amine (78 mg, 0.52 mmol) in dry dichloromethane (2 mL) was added di-tert-butyl dicarbonate (BOC) (148 mg, 0.68 mmol). The resulting mixture was stirred in an inert atmosphere for 15 h at room temperature. Then, after solvent removal, the crude was dissolved in ethyl acetate and the resulting organic phase was washed several times with water, once with brine and then dried over sodium sulphate. After solvent removal, the crude product was chromatographed on silica gel, using petroleum ether/ethyl acetate 7:3 as eluent, to give 25c as a thick colourless oil (103 mg, 80%). 1H-NMR (400 MHz, CDCl3) (ppm): δH 8.01 (s, 1H), 7.76 (d, 1H, JPH = 8 Hz), 7.43 (d, 1H, JPH = 4 Hz); 7.26 (d, 1H, JPH = 4 Hz); 7.22 (d, 1H, JPH = 8 Hz), 6.64 (s, 1H), 1.56 (s, 9H); 13C-NMR (125 MHz, CDCl3) (ppm): δC 153.0, 140.3, 135.2, 134.4, 127.3, 123.8, 122.6, 116.8, 113.0, 80.5, 29.3. MS: m/z 249 (M+); 193 (100).
5.7. Synthesis of Azides 26a, 26b, 26c
2-Azido-5-methoxy-1-benzothiophene 26a. Compound 25a (300 mg, 1.8 mmol) in dry diethyl ether (4 mL) was treated, in an inert atmosphere, with 1.6M n-butyllithium in hexane (2.7 mmol) and refluxed for 1 h. Then the reaction mixture was cooled to −70 °C and tosyl azide (380 mg, 1.9 mmol) in dry diethyl ether (4 mL) was added dropwise. After 4 h at this temperature, the obtained triazene salt was filtered under vacuum, washed with dry diethyl ether and then treated at 0 °C with an aqueous solution of sodium pyrophosphate decahydrate (868 mg, 1.9 mmol, in 9 mL of water). After 15 min of stirring at this temperature the suspension was filtered on a Buckner filter and extracted twice with diethyl ether and then with ethyl acetate until the organic phase appeared colourless. Then, after solvent removal, the crude product was purified by chromatography on Florisil (eluent petroleum ether/ethyl acetate 8:2) giving the title compound (206 mg, 55%) as a thick orange oil. Azide 26a is unstable; it was characterized by IR and 1H-NMR spectroscopy. ΙR νmax/cm−1 2117 (N3); 1H-NMR (400 MHz, CDCl3) (ppm): δH 7.35–7.20 (m, 1H), 7.07 (s, 1H), 6.95 (d, 1H, JPH = 24 Hz), 6.75 (s, 1H), 3.84 (s, 3H).
((2-Azidobenzo[b]thiophen-5-yl)oxy)(tert-butyl)dimethylsilane 26b. Compound 25b (434 mg, 1.64 mmol) in dry diethyl ether (9 mL) was treated, in an inert atmosphere, with 1.6 M n-butyllithium in hexane (1.64 mmol) and refluxed for 1 h. Then the reaction mixture was cooled to −78 °C and, slowly, tosylazide (378 mg, 1.92 mmol) in dry diethyl ether (9 mL) was added. After 5 h at this temperature the obtained triazene salt was filtered on a Buckner filter, washed with dry diethyl ether and then treated at 0 °C with an aqueous solution of sodium pyrophosphate decahydrate (0.9 g, 2.02 mmol) in water (9 mL). After 15 min of stirring at this temperature the suspension was filtered on a Buckner filter and extracted twice with diethyl ether and then with ethyl acetate until the organic phase appeared colourless. Then, after solvent removal, the crude product was purified by chromatography on Florisil (eluent petroleum ether) giving the title compound (141 mg, 28%) as a thick yellow oil. Azide 26b is unstable; it was characterized by IR and 1H-NMR spectroscopy. IR νmax/ cm−1 2116 (N3). 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.72–7.70 (d, 1H, JPH = 10 Hz), 7.43 (d, 1H, JPH = 10 Hz ), 7.27 (s, 1H), 6.73 (s, 1H), 1.03 (s, 9H), 0.23 (m, 6H).
Tert-butyl (2-azidobenzo[b]thiophen-5-yl)carbamate 26c. Compound 24c (141 mg, 0.56 mmol) in dry diethyl ether (3 mL) was treated, under a nitrogen atmosphere, with 1.6 M n-butyllithium in hexane (500 μL, 0.70 mmol) and refluxed for 1 h. Then the reaction mixture was cooled to −78 °C and tosylazide (135 mg, 0.68 mmol) in dry diethyl ether (3 mL) was added dropwise. After 5 h at this temperature, the reaction mixture was treated at 0 °C with an aqueous solution of sodium pyrophosphate decahydrate (412 mg; 0.92 mmol, in 3 mL of water). After 15 min of stirring at this temperature the suspension was extracted several times with diethyl ether and then with ethyl acetate until the organic phase appeared colourless and dried over sodium sulphate. Then, after solvent removal, the crude product was purified by chromatography on Florisil (eluant petroleum ether/diethyl ether 8:2) giving the title compound (150 mg, 62%) as a thick yellow oil. IR νmax/cm−1 2126 (N3). 1H-NMR (400 MHz, CDCl3) (ppm): δH 8.01 (s, 1H), 7.86 (d, 1H, JPH = 8 Hz), 7.41 (s, 1H), 7.21 (d, 1H, JPH = 4 Hz), 6.55 (s, 1H), 1.55 (s, 9H). 13C-NMR (100 MHz, CDCl3) (ppm): δC 152.5, 140.3, 135.2, 134.2, 123.8, 122.7, 116.8, 113.0, 98.8, 80.6, 28.4.
5.8. Synthesis of Imminophosphoranes 27a,b,c-28a,b
[(5-Methoxy-1-benzothiophen-2-yl)imino]-(methyl)diphenylphosphorane 27a. Compound 26a (46 mg, 0.22 mmol) in dry diethyl ether (2 mL) was added dropwise to a solution of methyldiphenylphosphine (44 mg, 0.22 mmol) in dry diethyl ether (2 mL) at 0 °C, in nitrogen atmosphere. After 2 h, the reaction mixture was allowed to room temperature for another hour and then was concentrated under vacuum. The residual material was purified by chromatography on silica gel (eluent petroleum ether/ethyl acetate 7:3). The title compound (83 mg, 58%) was obtained as a brown solid. Mp 135–136°C. 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.71–7.66 (m, 5H), 7.47–7.45 (m, 5H), 7.41–7.40 (m, 1H), 7.18 (d, 1H, JPH = 15 Hz), 6.72 (s, 1H), 6.52 (d, 1H, JPH = 10 Hz), 5.88 (s, 1H), 3.70 (s, 3H), 2.08 (d, 3H, JPH = 15 Hz); 13C-NMR (100 MHz, CDCl3) (ppm): δC 159.6, 157.5, 142.6, 132.5, 131.8, 131.7, 130.8, 129.8, 129.3, 129.1 126.4, 122.1, 109.4, 105.3, 105.2, 105.1, 103.6, 55.7, 15.1.
5-((Tert-butyldimethylsilyl)oxy)-N-(methyldiphenylphosphoranylidene)benzo[b]thiophen-2-amine 27b. Compound 26b (62 mg, 0.21 mmol) in dry dichloromethane (2 mL) was added dropwise to a solution of methyldiphenylphosphine (42 mg, 0.21 mmol) in dry dichloromethane (2 mL) at 0 °C, in nitrogen atmosphere. After 2 h, the reaction mixture was taken to room temperature for another hour and then was concentrated under vacuum. The residual material was crystallized from diethyl ether and characterized. The title compound (79 mg, 79%) was obtained as a green thick oil. 1H NMR (500 MHz, CDCl3) (ppm): δH 7.58–7.56 (m, 4H), 7.39–7.37 (m, 4H), 7.21–7.19 (m, 2H), 6.82 (d, 1H, JPH = 5 Hz), 6.73 (s, 1H), 6.44 (d, 1H, JPH = 10 Hz), 5.90 (s, 1H), 2.23 (d, 3H, JPH = 10 Hz), 0.83 (s, 9H), 0.19 (d, 6H); 13C NMR (125 MHz, CDCl3) (ppm): δC 155.0, 140.8, 132.8, 131.3, 130.1, 128.70, 127.5, 123.3, 114.6, 98.9, 30.2, 25.9, 25.3.
Tert-butyl(2-((methyldiphenylphosphoranylidene)amino)benzo[b]thiophen-5-yl)carbamate 27c. Compound 26c (100 mg, 0.34 mmol) in dry dichloromethane (3 mL) was added dropwise to a solution of methyldiphenylphosphine (85 mg, 0.42 mmol) in dry dichloromethane (3 mL) at 0 °C, in nitrogen atmosphere. After 2 h, the reaction mixture was taken to room temperature for another hour and then was concentrated under vacuum. The residual material was crystallized from diethyl ether and characterized. The title compound (77 mg, 49%) was obtained as white needles. Mp 112–114 °C. 1H-NMR (500 MHz, CDCl3) (ppm): δH 8.58 (s, 1H), 7.81 (d, 1H, JPH = 10 Hz), 7.73–7.62 (m, 5H) 7.54–7.39 (m, 5H), 7.23 (s, 1H), 6.98–6.97 (d, 1H, JPH = 5 Hz), 5.18 (s, 1H), 2.38 (s, 3H), 2.27 (s, 9H). 13C-NMR (125 MHz, CDCl3) (ppm): δC 152.5, 143.0, 140.0, 137.0, 135.5, 129.2, 128.2, 126.3, 123.3, 123.0, 116.4, 115.9, 98.8, 79.5, 38.7, 28.4, 25.2.
[(5-Methoxy-1-benzothiophen-2-yl)imino]-triphenylphosphorane 28a. Compound 26a (125 mg, 0.61 mmol) in dry diethyl ether (5 mL) was added dropwise to a solution of triphenylphosphine (162 mg, 0.62 mmol) in dry diethyl ether (5 mL) at 0 °C, in nitrogen atmosphere. After 2 h, the reaction mixture was allowed to room temperature, then was concentrated under vacuum. The residual material (a very dark and thick oil) was purified by chromatography on silica gel (eluent petroleum ether/ethyl acetate 8:2). The title compound (200 mg, 74%) was obtained as a brown oil. 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.81–7.77 (m, 5H), 7.61–7.57 (m, 5H), 7.52–7.47 (m, 5H), 7.3 (s, 1H), 6.80 (d, 1H, JPH = 5 Hz), 6.63–6.60 (m, 1H), 6.07 (s, 1H), 3.78 (s, 3H); 13C-NMR (100 MHz, CDCl3) (ppm): δC 162.2, 160.5, 144.2, 136.3, 134.8, 132.7, 131.0, 129.7, 129.5, 129.2, 126.8, 122.7, 109.1, 105.7, 105.4, 105.1, 103.1.
5-((Tert-butyldimethylsilyl)oxy)-N-(triphenylphosphoranylidene)benzo[b]thiophen-2-amine 28b. Compound 26b (66 mg, 0.22 mmol) in dry dichloromethane (2 mL) was added dropwise to a solution of triphenylphosphine (60 mg, 0.23 mmol) in dry dichloromethane (2 mL) at 0 °C, in nitrogen atmosphere. After 2 h, the reaction mixture was taken to room temperature for another hour and then was concentrated under vacuum. The crude was purified on silica gel using petroleum ether/dichloromethane 1:1 as eluent to give the title compound (97 mg, 81%) as a yellow solid. Mp 163–165 °C. 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.82–7.78 (m, 5H), 7.60–7.50 (m, 10H), 7.23 (d, 1H, JPH = 10 Hz), 7.01 (s, 1H), 6.75 (s, 1H), 6.53 (d, 1H, JPH = 10 Hz), 0.99 (s, 9H), 0.19 (m, 6H); 13C-NMR (125 MHz, CDCl3) (ppm): δC 152.6, 132.9, 132.9, 132.6, 130.8, 128.8, 125.3, 121.6, 114.5, 112.5, 98.8, 29.7, 25.8.
5.9. General Procedure for the Synthesis of (1)-Benzothieno[2,3-b]pyridines 29a,b, 30a,b
A solution of the appropriate iminophosphorane
27a,b, 28a,b in dry chloroform was treated with methyl
trans 4-oxo-2-pentenoate (1.1 eq) and then stirred in inert atmosphere at 61 °C. After removal of the solvent, the crude product was chromatographed on silica gel, using petroleum ether/ethyl acetate 9:1 as eluent. For yields see
Table 2.
Methyl 6-methoxy-2-methyl(1)benzothieno[2,3-b]pyridine-4-carboxylate 29a-30a. This compound was obtained as a thick oil. 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.95 (s, 1H), 7.75 (d, 1H, JPH = 12 Hz), 7.47 (s, 1H), 7.09–7.07 (d, 1H, JPH = 12 Hz), 4.10 (s, 3H), 3.91 (s, 3H), 2.74 (s, 3H); 13C-NMR (125 MHz, CDCl3) (ppm): δC 168.2, 160.0, 160.3, 142.4, 136.6, 133.5, 132.2, 131.2, 129.5, 126.4, 120.9, 115.6, 106.5, 55.8, 27.5; MS: m/z 287 (M+).
Methyl 6-((Tert-butyldimethylsilyl)oxy)-2-methylbenzo[4,5]thieno[2,3-b]pyridine-4-carboxylate (29b-30b). Thick red oil. 1H-NMR (500 MHz, CDCl3) (ppm): δH 7.84 (s, 1H); 7.72–7.71 (m, 1H), 7.43 (s, 1H), 7.07 (d, 1H, JPH = 5 Hz), 4.09 (s, 3H), 2.74 (s, 3H), 1.05 (s, 9H), 0.35 (s, 6H); 13C-NMR (125 MHz, CDCl3) (ppm): δC 165.9, 159.0, 151.1, 142.8, 133.6, 132.9, 127.5, 126.5, 124.9, 120.9, 114.6, 112.6, 51.6, 30.2, 25.9, 23.5, −0.9; MS: m/z 387 (M+).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

