Structure Based Antibody-Like Peptidomimetics


Abstract
:1. Introduction
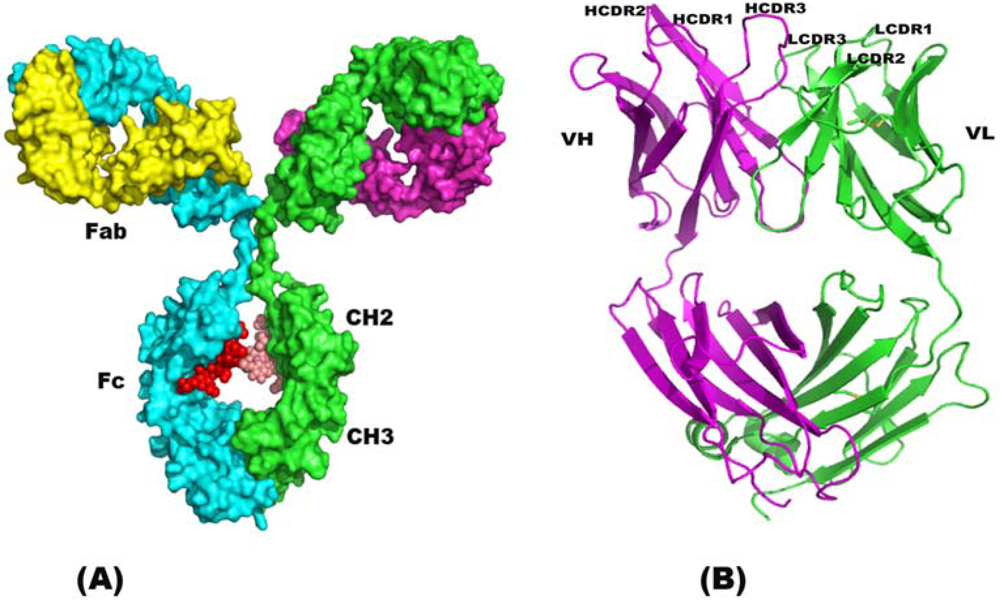
2. Structure of Immunoglobulin

2.1. Single Chain Antibody
2.2. Humanization of Monoclonal Antibody
2.3. Proteins to Peptides
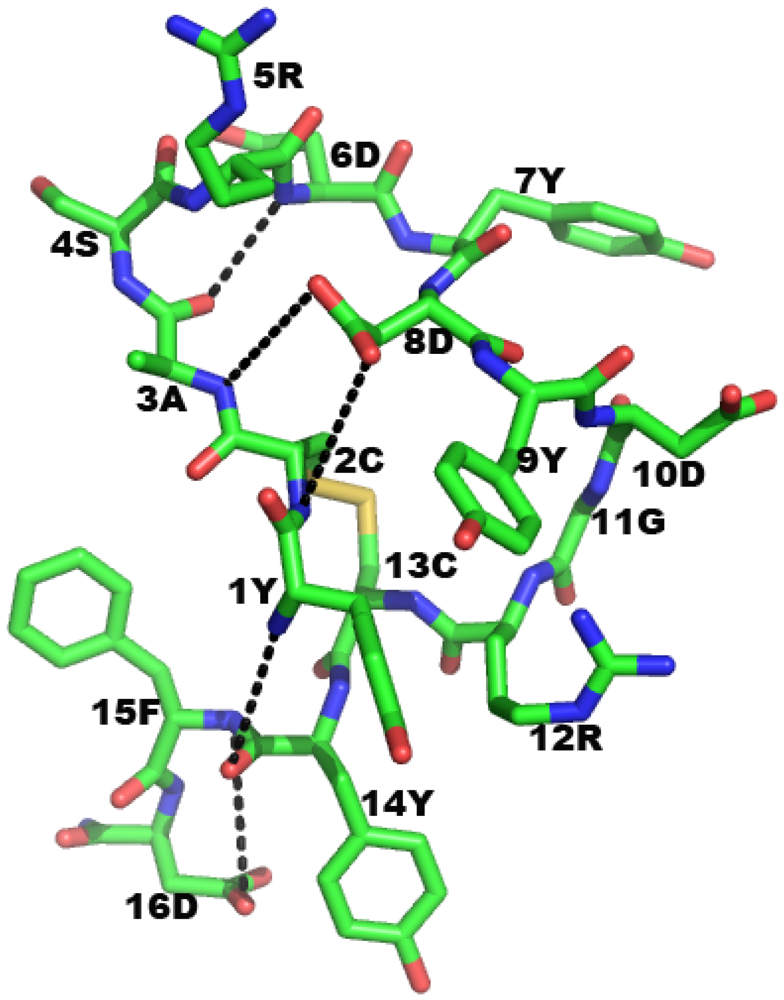
2.4. Mimicry of Antigen by Peptides
2.5. CDR Based Viral Inhibitors
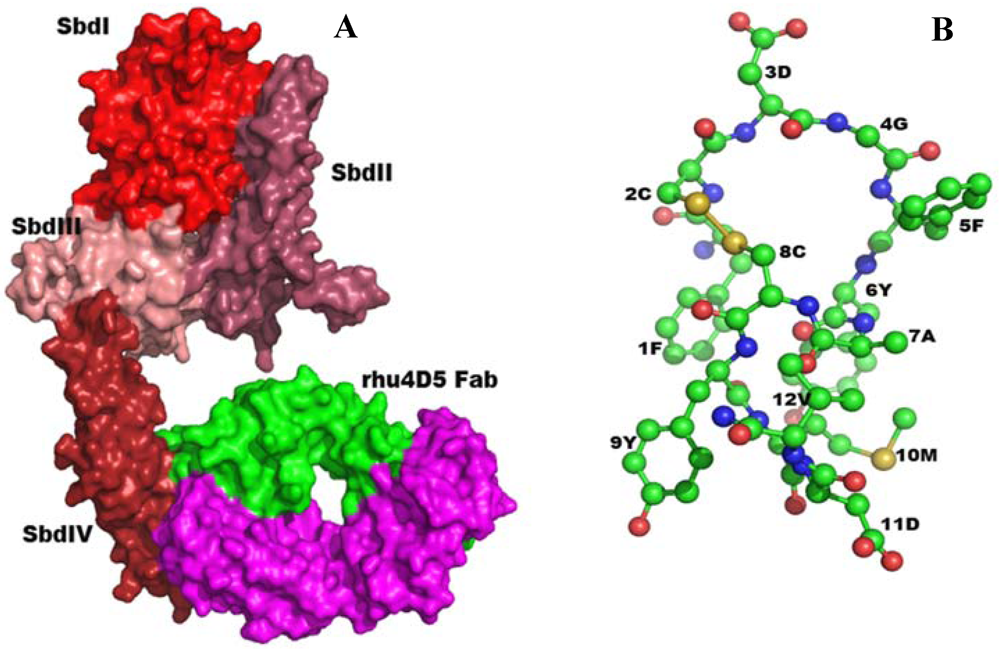
2.5.1. Anti-Reovirus Mimetics
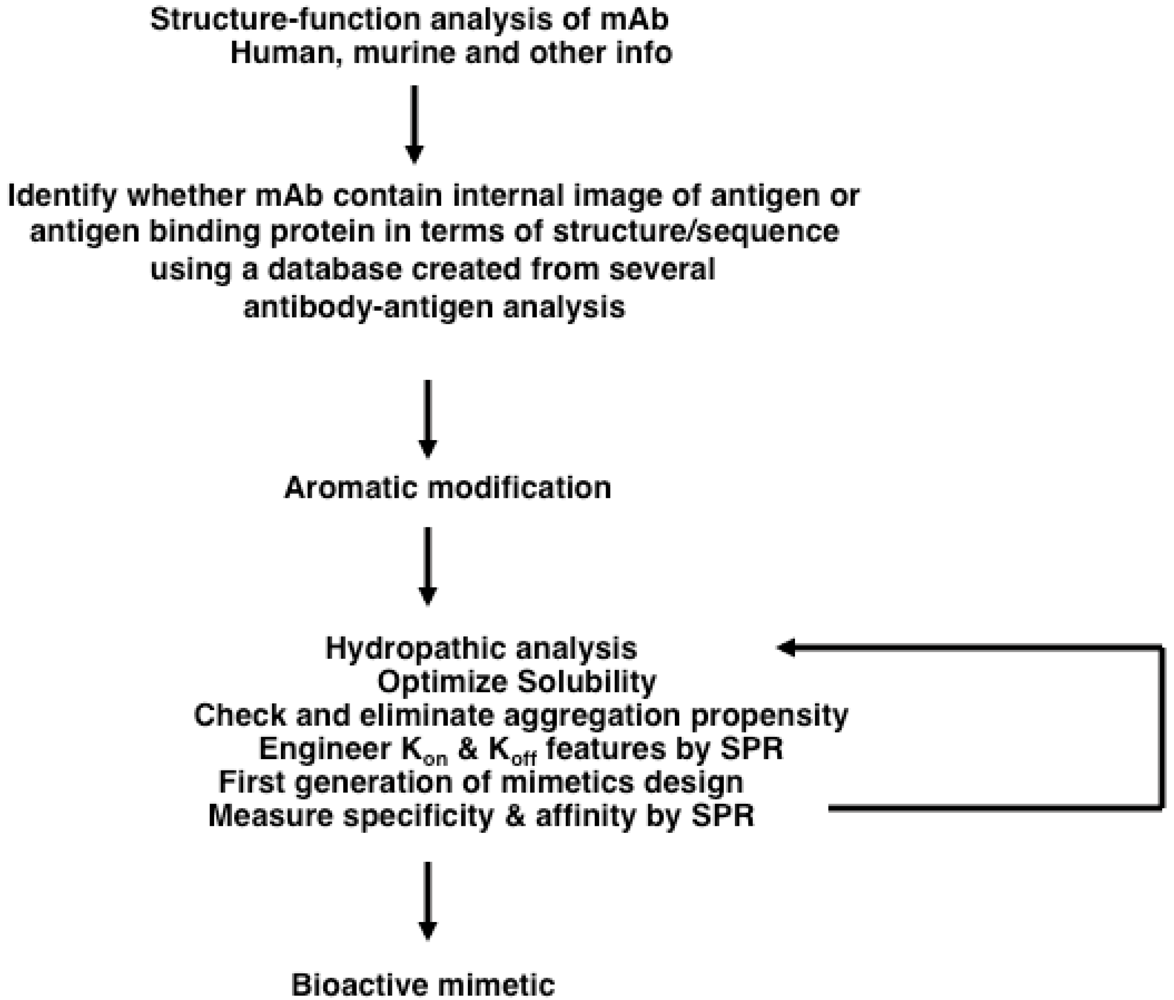
2.5.2. Structure-Based Design of Antibody-like Mimetics

2.6. CDR Based p185erbB2/neu Receptor Inhibitors
2.6.1. Comparison of Rat and Human Forms of Monoclonal Antibodies

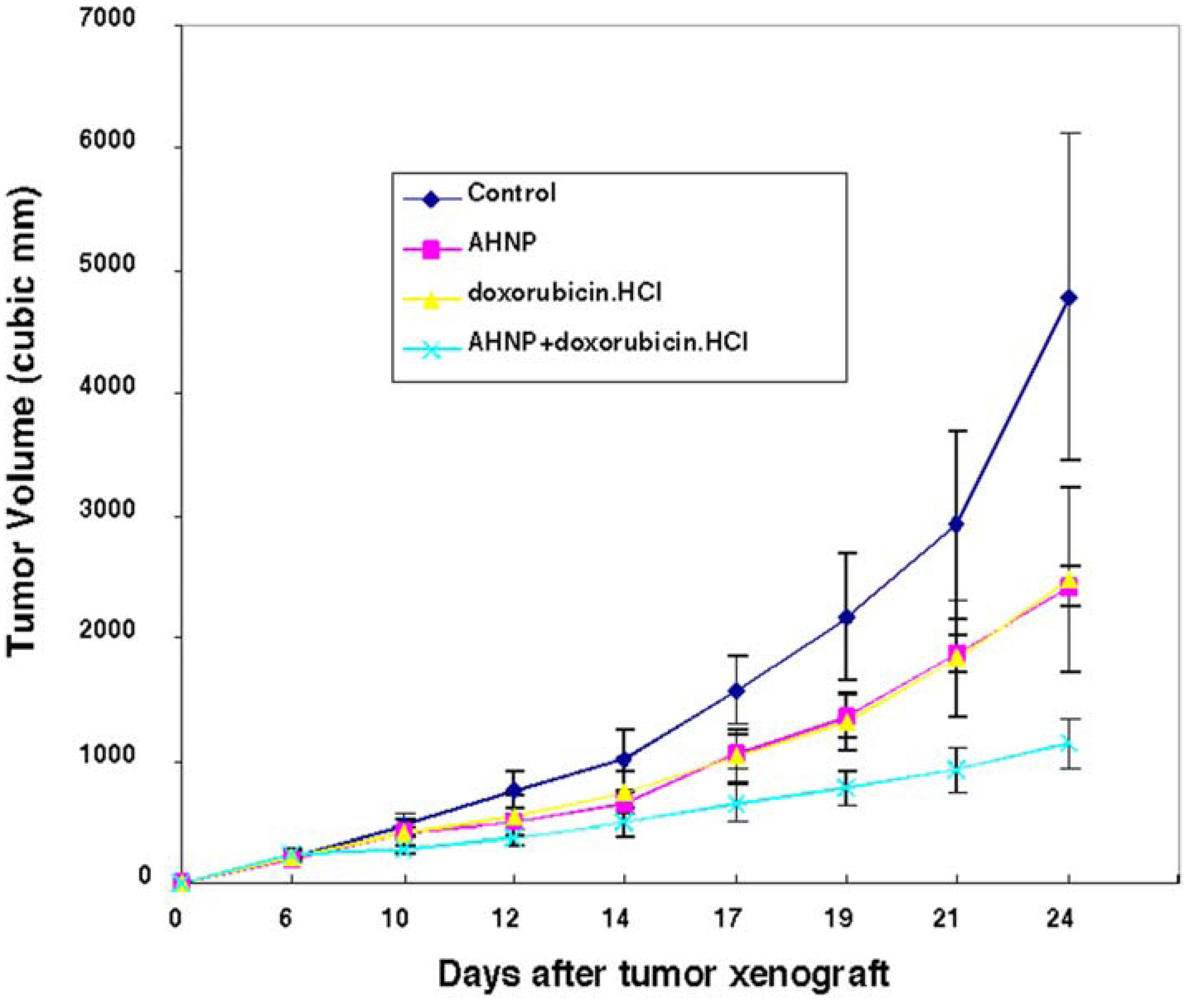
2.6.2. In Vivo Tumor Growth Inhibition by AHNP

2.7. AHNP Function Is Context Independent in Terms of Adjacent Peptidic Regions
2.8. AHNP Is a Novel Small Molecular Probe for p185erbB2/neu Biology, Diagnosis, Drug Delivery and Therapeutics
2.9. CDR Based Epidermal Growth Factor Receptor (EGFR) Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Mimetic | Concentration (μg/mL) | % Inhibition | |
|---|---|---|---|
| T6-17 (Her2++) | A431 (EGFR++) | ||
| AHNP | 10.0 | 73.92 | 19.07 |
| AERP | 10.0 | 10.08 | 15.44 |
| AERP-AHNP | 10.0 | 92.82 | 22.51 |
| h4D5 (trastuzumab) | 1.0 | 51.07 | 8.77 |
2.10. AERP as Single-Photon Emission Computed Tomography (SPECT)-Agent for Tumor Imaging
3. Future Direction for Peptides as Therapeutic Agent
4. Conclusions
Conflict of Interest
Acknowledgements
Reference
- Buckel, P. Recombinant proteins for therapy. Trends Pharmacol. Sci. 1996, 17, 450–456. [Google Scholar] [CrossRef]
- An, Z. Monoclonal antibodies—A proven and rapidly expanding therapeutic modality for human diseases. Protein Cell 2010, 1, 319–330. [Google Scholar] [CrossRef]
- Saragovi, H.U.; Fitzpatrick, D.; Raktabutr, A.; Nakanishi, H.; Kahn, M.; Greene, M.I. Design and synthesis of a mimetic from an antibody complementarity-determining region. Science 1991, 253, 792–795. [Google Scholar]
- Vita, C.; Vizzavona, J.; Drakopoulou, E.; Zinn-Justin, S.; Gilquin, B.; Menez, A. Novel miniproteins engineered by the transfer of active sites to small natural scaffolds. Biopolymers 1998, 47, 93–100. [Google Scholar] [CrossRef]
- Cooper, W.J.; Waters, M.L. Molecular recognition with designed peptides and proteins. Curr. Opin. Chem. Biol. 2005, 9, 627–631. [Google Scholar] [CrossRef]
- Craik, D.J.; Clark, R.J.; Daly, N.L. Potential therapeutic applications of the cyclotides and related cystine knot mini-proteins. Expert Opin. Investig. Drugs 2007, 16, 595–604. [Google Scholar] [CrossRef]
- Beck, A.; Reichert, J.M. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs 2011, 3, 415–416. [Google Scholar] [CrossRef]
- Huang, C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology. Curr. Opin. Biotechnol. 2009, 20, 692–699. [Google Scholar] [CrossRef]
- Lofblom, J.; Frejd, F.Y.; Stahl, S. Non-immunoglobulin based protein scaffolds. Curr. Opin. Biotechnol. 2011, 22, 843–848. [Google Scholar] [CrossRef]
- Bristow, A.F. Recombinant-DNA-derived insulin analogues as potentially useful therapeutic agents. Trends Biotechnol. 1993, 11, 301–305. [Google Scholar] [CrossRef]
- Johnson, I.S. Human insulin from recombinant DNA technology. Science 1983, 219, 632–637. [Google Scholar]
- Berkower, I. The promise and pitfalls of monoclonal antibody therapeutics. Curr. Opin. Biotechnol. 1996, 7, 622–628. [Google Scholar] [CrossRef]
- Elkins, K.L.; Ennist, D.L.; Winegar, R.K.; Weir, J.P. In vivo delivery of interleukin-4 by a recombinant vaccinia virus prevents tumor development in mice. Hum. Gene Ther. 1994, 5, 809–820. [Google Scholar] [CrossRef]
- Hudson, P.J. Recombinant antibody constructs in cancer therapy. Curr. Opin. Immunol. 1999, 11, 548–557. [Google Scholar] [CrossRef]
- Jones, D.R. Design and Synthesis of Nonpeptide Peptidomimetic Enzyme Inhibitors, and Synthesis and Characterization of C60O, the First Buckminsterfullerene Epoxide; Graduate School of Arts and Sciences, University of Pennsylvania: Philadelphia, PA, USA, 1993. [Google Scholar]
- Juweid, M.E.; Hajjar, G.; Swayne, L.C.; Sharkey, R.M.; Suleiman, S.; Herskovic, T.; Pereira, M.; Rubin, A.D.; Goldenberg, D.M. Phase I/II trial of (131)I-MN-14F(ab)2 anti-carcinoembryonic antigen monoclonal antibody in the treatment of patients with metastatic medullary thyroid carcinoma. Cancer 1999, 85, 1828–1842. [Google Scholar]
- McCafferty, J.; Glover, D.R. Engineering therapeutic proteins. Curr. Opin. Struct. Biol. 2000, 10, 417–420. [Google Scholar] [CrossRef]
- Cho, M.J.; Juliano, R. Macromolecular versus small-molecule therapeutics: Drug discovery, development and clinical considerations. Trends Biotechnol. 1996, 14, 153–158. [Google Scholar] [CrossRef]
- Dean, J.H.; Cornacoff, J.B.; Barbolt, T.A.; Gossett, K.A.; LaBrie, T. Pre-clinical toxicity of IL-4: A model for studying protein therapeutics. Int. J. Immunopharmacol. 1992, 14, 391–397. [Google Scholar] [CrossRef]
- Putney, S.D.; Burke, P.A. Improving protein therapeutics with sustained-release formulations. Nat. Biotechnol. 1998, 16, 153–157. [Google Scholar]
- Takakura, Y.; Hashida, M. Macromolecular carrier systems for targeted drug delivery: Pharmacokinetic considerations on biodistribution. Pharm. Res. 1996, 13, 820–831. [Google Scholar] [CrossRef]
- Thomas, J.A. Recent developments and perspectives of biotechnology-derived products. Toxicology 1995, 105, 7–22. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Boswell, C.A.; Brechbiel, M.W. Development of radioimmunotherapeutic and diagnostic antibodies: An inside-out view. Nucl. Med. Biol. 2007, 34, 757–778. [Google Scholar] [CrossRef]
- Stern, M.; Herrmann, R. Overview of monoclonal antibodies in cancer therapy: Present and promise. Crit. Rev. Oncol. Hematol. 2005, 54, 11–29. [Google Scholar] [CrossRef]
- Milenic, D.E.; Brechbiel, M.W. Targeting of radio-isotopes for cancer therapy. Cancer Biol. Ther. 2004, 3, 361–370. [Google Scholar]
- Gilliland, L.K.; Walsh, L.A.; Frewin, M.R.; Wise, M.P.; Tone, M.; Hale, G.; Kioussis, D.; Waldmann, H. Elimination of the immunogenicity of therapeutic antibodies. J. Immunol. 1999, 162, 3663–3671. [Google Scholar]
- Clark, M. Antibody humanization: A case of the ‘Emperor’s new clothes’? Immunol. Today 2000, 21, 397–402. [Google Scholar] [CrossRef]
- Niebecker, R.; Kloft, C. Safety of therapeutic monoclonal antibodies. Curr. Drug Saf. 2010, 5, 275–286. [Google Scholar] [CrossRef]
- Bhogal, N. Immunotoxicity and immunogenicity of biopharmaceuticals: Design concepts and safety assessment. Curr. Drug. Saf. 2010, 5, 293–307. [Google Scholar] [CrossRef]
- Tramontano, A.; Bianchi, E.; Venturini, S.; Martin, F.; Pessi, A.; Sollazzo, M. The making of the minibody: An engineered beta-protein for the display of conformationally constrained peptides. J. Mol. Recognit. 1994, 7, 9–24. [Google Scholar] [CrossRef]
- Kim, S.; Pang, H.B.; Kay, M.S. Peptide mimic of the HIV envelope gp120-gp41 interface. J. Mol. Biol. 2008, 376, 786–797. [Google Scholar] [CrossRef]
- Zhong, H.; Carlson, H.A. Computational studies and peptidomimetic design for the human p53-MDM2 complex. Proteins 2005, 58, 222–234. [Google Scholar]
- Casset, F.; Roux, F.; Mouchet, P.; Bes, C.; Chardes, T.; Granier, C.; Mani, J.C.; Pugniere, M.; Laune, D.; Pau, B.; et al. A peptide mimetic of an anti-CD4 monoclonal antibody by rational design. Biochem. Biophys. Res. Commun. 2003, 307, 198–205. [Google Scholar] [CrossRef]
- Cardo-Vila, M.; Arap, W.; Pasqualini, R. αvβ5 Integrin-dependent programmed cell death triggered by a peptide mimic of annexin V. Mol. Cell. 2003, 11, 1151–1162. [Google Scholar] [CrossRef]
- Bolin, D.R.; Swain, A.L.; Sarabu, R.; Berthel, S.J.; Gillespie, P.; Huby, N.J.S.; Makofske, R.; Orzechowski, L.; Perrotta, A.; Toth, K.; et al. Peptide and peptide mimetic inhibitors of antigen presentation by HLA-DR class II MHC molecules. Design, structure-activity relationships, and X-ray crystal structures. J. Med. Chem. 2000, 43, 2135–2148. [Google Scholar]
- Fukumoto, T.; Torigoe, N.; Kawabata, S.; Murakami, M.; Uede, T.; Nishi, T.; Ito, Y.; Sugimura, K. Peptide mimics of the CTLA4-binding domain stimulate T-cell proliferation. Nat. Biotechnol. 1998, 16, 267–270. [Google Scholar] [CrossRef]
- Kay, B.K.; Kurakin, A.V.; Hyde-DeRuyscher, R. From peptides to drugs via phage display. Drug Discov. Today 1998, 3, 370–378. [Google Scholar] [CrossRef]
- Phalipon, A.; Folgori, A.; Arondel, J.; Sgaramella, G.; Fortugno, P.; Cortese, R.; Sansonetti, P.J.; Felici, F. Peptide mimicry of carbohydrate structures. Res. Immunol. 1998, 149, 75–77. [Google Scholar] [CrossRef]
- Bruck, C.; Co, M.S.; Slaoui, M.; Gaulton, G.N.; Smith, T.; Fields, B.N.; Mullins, J.I.; Greene, M.I. Nucleic acid sequence of an internal image-bearing monoclonal anti-idiotype and its comparison to the sequence of the external antigen. Proc. Natl. Acad. Sci. USA 1986, 83, 6578–6582. [Google Scholar]
- Kieber-Emmons, T.; Murali, R.; Greene, M.I. Therapeutic peptides and peptidomimetics. Curr. Opin. Biotechnol. 1997, 8, 435–441. [Google Scholar] [CrossRef]
- Murali, R.; Greene, M.I. Structure-based design of immunologically active therapeutic peptides. Immunol. Res. 1998, 17, 163–169. [Google Scholar] [CrossRef]
- Horie, T.; Shen, Y.; Kajino, K.; Gaubin, M.; Bonomi, G.; Mani, J.C.; Berezov, A.; Piatier-Tonneau, D.; Guardiola, J.; Hillard, B.; et al. Study of disabling T-cell activation and inhibiting T-cell-mediated immunopathology reveals a possible inverse agonist activity of CD4 peptidomimetics. Exp. Mol. Pathol. 2002, 73, 93–103. [Google Scholar] [CrossRef]
- Williams, W.V.; Kieber-Emmons, T.; VonFeldt, J.; Greene, M.I.; Weiner, D.B. Design of bioactive peptides based on antibody hypervariable region structures. Development of conformationally constrained and dimeric peptides with enhanced affinity. J. Biol. Chem. 1991, 266, 5182–5190. [Google Scholar]
- Zhang, X.; Gaubin, M.; Briant, L.; Srikantan, V.; Murali, R.; Saragovi, U.; Weiner, D.; Devaux, C.; Autiero, M.; Piatier-Tonneau, D.; et al. Synthetic CD4 exocyclics inhibit binding of human immunodeficiency virus type 1 envelope to CD4 and virus replication in T lymphocytes. Nat. Biotechnol. 1997, 15, 150–154. [Google Scholar] [CrossRef]
- Zhang, X.; Piatier-Tonneau, D.; Auffray, C.; Murali, R.; Mahapatra, A.; Zhang, F.; Maier, C.C.; Saragovi, H.; Greene, M.I. Synthetic CD4 exocyclic peptides antagonize CD4 holoreceptor binding and T cell activation. Nat. Biotechnol. 1996, 14, 472–475. [Google Scholar] [CrossRef]
- Berezov, A.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling erbB receptors with rationally designed exocyclic mimetics of antibodies: Structure-function analysis. J. Med. Chem. 2001, 44, 2565–2574. [Google Scholar] [CrossRef]
- Park, B.-W.; Zhang, H.-T.; Wu, C.; Berezov, A.; Zhang, X.; Dua, R.; Wang, Q.; Kao, G.; O'Rourke, D.M.; Greene, M.I.; et al. Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo. Nat. Biotechnol. 2000, 18, 194–198. [Google Scholar]
- Ponde, D.E.; Su, Z.; Berezov, A.; Zhang, H.; Alavi, A.; Greene, M.I.; Murali, R. Development of anti-EGF receptor peptidomimetics (AERP) as tumor imaging agent. Bioorg. Med. Chem. Lett. 2011, 21, 2550–2553. [Google Scholar]
- Bork, P.; Holm, L.; Sander, C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar]
- Halaby, D.M.; Poupon, A.; Mornon, J. The immunoglobulin fold family: Sequence analysis and 3D structure comparisons. Protein Eng. 1999, 12, 563–571. [Google Scholar] [CrossRef]
- Buljan, M.; Bateman, A. The evolution of protein domain families. Biochem. Soc. Trans. 2009, 37, 751–755. [Google Scholar] [CrossRef]
- Dermody, T.S.; Kirchner, E.; Guglielmi, K.M.; Stehle, T. Immunoglobulin superfamily virus receptors and the evolution of adaptive immunity. PLoS Pathog. 2009, 5, e1000481. [Google Scholar] [CrossRef]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System, Version 1.3r1; Schrodinger LLC: San Diego, CA, USA, 2010. [Google Scholar]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.E., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- MacCallum, R.M.; Martin, A.C.; Thornton, J.M. Antibody-antigen interactions: contact analysis and binding site topography. J. Mol. Biol. 1996, 262, 732–745. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M.; Tramontano, A.; Levitt, M.; Smith-Gill, S.J.; Air, G.; Sheriff, S.; Padlan, E.A.; Davies, D.; Tulip, W.R.; et al. Conformations of immunoglobulin hypervariable regions. Nature 1989, 342, 877–883. [Google Scholar]
- Bhat, T.N.; Bentley, G.A.; Boulot, G.; Greene, M.I.; Tello, D.; Dall'Acqua, W.; Souchon, H.; Schwarz, F.P.; Mariuzza, R.A.; Poljak, R.J. Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc. Natl. Acad. Sci. USA 1994, 91, 1089–1093. [Google Scholar]
- Lescar, J.; Pellegrini, M.; Souchon, H.; Tello, D.; Poljak, R.J.; Peterson, N.; Greene, M.; Alzari, P.M. Crystal structure of a cross-reaction complex between Fab F9.13.7 and guinea fowl lysozyme. J. Biol. Chem. 1995, 270, 18067–18076. [Google Scholar]
- Rees, A.R.; Staunton, D.; Webster, D.M.; Searle, S.J.; Henry, A.H.; Pedersen, J.T. Antibody design: Beyond the natural limits. Trends Biotechnol. 1994, 12, 199–206. [Google Scholar] [CrossRef]
- Decanniere, K.; Desmyter, A.; Lauwereys, M.; Ghahroudi, M.A.; Muyldermans, S.; Wyns, L. A single-domain antibody fragment in complex with RNase A: Non-canonical loop structures and nanomolar affinity using two CDR loops. Structure 1999, 7, 361–370. [Google Scholar] [CrossRef]
- Spinelli, S.; Frenken, L.G.; Hermans, P.; Verrips, T.; Brown, K.; Tegoni, M.; Cambillau, C. Camelid heavy-chain variable domains provide efficient combining sites to haptens. Biochemistry 2000, 39, 1217–1222. [Google Scholar]
- Kettleborough, C.A.; Saldanha, J.; Heath, V.J.; Morrison, C.J.; Bendig, M.M. Humanization of a mouse monoclonal antibody by CDR-grafting: The importance of framework residues on loop conformation. Protein Eng. 1991, 4, 773–783. [Google Scholar] [CrossRef]
- Essig, N.Z.; Wood, J.F.; Howard, A.J.; Raag, R.; Whitlow, M. Crystallization of single-chain Fv proteins. J. Mol. Biol. 1993, 234, 897–901. [Google Scholar] [CrossRef]
- Arndt, K.M.; Muller, K.M.; Pluckthun, A. Factors influencing the dimer to monomer transition of an antibody single-chain Fv fragment. Biochemistry 1998, 37, 12918–12926. [Google Scholar]
- Dolezal, O.; Pearce, L.A.; Lawrence, L.J.; McCoy, A.J.; Hudson, P.J.; Kortt, A.A. ScFv multimers of the anti-neuraminidase antibody NC10: Shortening of the linker in single-chain Fv fragment assembled in V(L) to V(H) orientation drives the formation of dimers, trimers, tetramers and higher molecular mass multimers. Protein Eng. 2000, 13, 565–574. [Google Scholar] [CrossRef]
- Holliger, P.; Prospero, T.; Winter, G. Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef]
- Kortt, A.A.; Malby, R.L.; Caldwell, J.B.; Gruen, L.C.; Ivancic, N.; Lawrence, M.C.; Howlett, G.J.; Webster, R.G.; Hudson, P.J.; Colman, P.M. Recombinant anti-sialidase single-chain variable fragment antibody. Characterization, formation of dimer and higher-molecular-mass multimers and the solution of the crystal structure of the single-chain variable fragment/sialidase complex. Eur. J. Biochem. 1994, 221, 151–157. [Google Scholar]
- Freund, C.; Ross, A.; Pluckthun, A.; Holak, T.A. Structural and dynamic properties of the Fv fragment and the single-chain Fv fragment of an antibody in solution investigated by heteronuclear three-dimensional NMR spectroscopy. Biochemistry 1994, 33, 3296–3303. [Google Scholar] [CrossRef]
- Raag, R.; Whitlow, M. Single-chain Fvs. FASEB J. 1995, 9, 73–80. [Google Scholar]
- Colcher, D.; Bird, R.; Roselli, M.; Hardman, K.D.; Johnson, S.; Pope, S.; Dodd, S.W.; Pantoliano, M.W.; Milenic, D.E.; Schlom, J. In vivo tumor targeting of a recombinant single-chain antigen-binding protein. J. Natl. Cancer Inst. 1990, 82, 1191–1197. [Google Scholar] [CrossRef]
- Huston, J.S.; McCartney, J.; Tai, M.S.; Mottola-Hartshorn, C.; Jin, D.; Warren, F.; Keck, P.; Oppermann, H. Medical applications of single-chain antibodies. Int. Rev. Immunol. 1993, 10, 195–217. [Google Scholar] [CrossRef]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992, 52, 3402–3408. [Google Scholar]
- Olafsen, T.; Wu, A.M. Antibody vectors for imaging. Semin. Nucl. Med. 2010, 40, 167–181. [Google Scholar] [CrossRef]
- Rahbarizadeh, F.; Ahmadvand, D.; Sharifzadeh, Z. Nanobody; an old concept and new vehicle for immunotargeting. Immunol. Invest. 2011, 40, 299–338. [Google Scholar] [CrossRef]
- Kontermann, R.E. Alternative antibody formats. Curr. Opin. Mol. Ther. 2010, 12, 176–183. [Google Scholar]
- Mabry, R.; Snavely, M. Therapeutic bispecific antibodies: The selection of stable single-chain fragments to overcome engineering obstacles. IDrugs 2010, 13, 543–549. [Google Scholar]
- Thakur, A.; Lum, L.G. Cancer therapy with bispecific antibodies: Clinical experience. Curr. Opin. Mol. Ther. 2010, 12, 340–349. [Google Scholar]
- Nelson, A.L. Antibody fragments: Hope and hype. MAbs 2010, 2, 77–83. [Google Scholar] [CrossRef]
- Balzar, M.; Winter, M.J.; de Boer, C.J.; Litvinov, S.V. The biology of the 17-1A antigen (Ep-CAM). J. Mol. Med. 1999, 77, 699–712. [Google Scholar] [CrossRef]
- Fye, K.H. New treatments for rheumatoid arthritis. Available and upcoming slow-acting antirheumatic drugs. Postgrad. Med. 1999, 106, 82–85. [Google Scholar]
- Hudson, P.J. Recombinant antibody fragments. Curr. Opin. Biotechnol. 1998, 9, 395–402. [Google Scholar] [CrossRef]
- Waldmann, T.A. Monoclonal antibodies in diagnosis and therapy. Science 1991, 252, 1657–1662. [Google Scholar]
- Attanasio, R.; Kennedy, R.C.; Allan, J.S.; Maino, V.C.; Buck, D.; Kanda, P. Anti-idiotypic antibodies of a predefined specificity generated against CDR3VH synthetic peptides define a private anti-CD4 idiotype. Mol. Immunol. 1990, 27, 513–522. [Google Scholar] [CrossRef]
- Schroff, R.W.; Foon, K.A.; Beatty, S.M.; Oldham, R.K.; Morgan, A.C., Jr. Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer Res. 1985, 45, 879–885. [Google Scholar]
- Shawler, D.L.; Bartholomew, R.M.; Smith, L.M.; Dillman, R.O. Human immune response to multiple injections of murine monoclonal IgG. J. Immunol. 1985, 135, 1530–1535. [Google Scholar]
- Roguska, M.A.; Pedersen, J.T.; Henry, A.H.; Searle, S.M.; Roja, C.M.; Avery, B.; Hoffee, M.; Cook, S.; Lambert, J.M.; Blattler, W.A.; et al. A comparison of two murine monoclonal antibodies humanized by CDR-grafting and variable domain resurfacing. Protein Eng. 1996, 9, 895–904, Erratum: Protein Eng. 1997, 10, 181. [Google Scholar]
- Hamilton, R.G. Monoclonal antibodies in the diagnosis and treatment of human diseases. Ann. Biol. Clin. (Paris) 1989, 47, 575–581. [Google Scholar]
- Powelson, J.A.; Knowles, R.W.; Delmonico, F.L.; Kawai, T.; Mourad, G.; Preffer, F.K.; Colvin, R.B.; Cosimi, A.B. CDR-grafted OKT4A monoclonal antibody in cynomolgus renal allograft recipients. Transplantation 1994, 57, 788–793. [Google Scholar]
- Winter, G.; Milstein, C. Man-made antibodies. Nature 1991, 349, 293–299. [Google Scholar] [CrossRef]
- Roguska, M.A.; Pedersen, J.T.; Keddy, C.A.; Henry, A.H.; Searle, S.J.; Lambert, J.M.; Goldmacher, V.S.; Blattler, W.A.; Rees, A.R.; Guild, B.C. Humanization of murine monoclonal antibodies through variable domain resurfacing. Proc. Natl. Acad. Sci. USA 1994, 91, 969–973. [Google Scholar]
- Almagro, J.C.; Fransson, J. Humanization of antibodies. Front. Biosci. 2008, 13, 1619–1633. [Google Scholar]
- King, D.J.; Turner, A.; Farnsworth, A.P.; Adair, J.R.; Owens, R.J.; Pedley, R.B.; Baldock, D.; Proudfoot, K.A.; Lawson, A.D.; Beeley, N.R.; et al. Improved tumor targeting with chemically cross-linked recombinant antibody fragments. Cancer Res. 1994, 54, 6176–6185. [Google Scholar]
- Coussens, L.; Yang-Feng, T.L.; Liao, Y.C.; Chen, E.; Gray, A.; McGrath, J.; Seeburg, P.H.; Libermann, T.A.; Schlessinger, J.; Francke, U.; et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985, 230, 1132–1139. [Google Scholar]
- Nakanishi, H.; Ramurthy, S.; Raktabutr, A.; Shen, R.; Kahn, M. Peptidomimetics of the immunoglobulin supergene family—A review. Gene 1993, 137, 51–56. [Google Scholar] [CrossRef]
- Moore, G.J. Designing peptide mimetics. Trends Pharmacol. Sci. 1994, 15, 124–129. [Google Scholar] [CrossRef]
- Vicari, D.; Foy, K.C.; Liotta, E.M.; Kaumaya, P.T. Engineered conformation-dependent VEGF peptide mimics are effective in inhibiting VEGF signaling pathways. J. Biol. Chem. 2011, 286, 13612–13625. [Google Scholar]
- Eckhardt, B.; Grosse, W.; Essen, L.O.; Geyer, A. Structural characterization of a beta-turn mimic within a protein-protein interface. Proc. Natl. Acad. Sci. USA 2010, 107, 18336–18341. [Google Scholar]
- Valadon, P.; Nussbaum, G.; Oh, J.; Scharff, M.D. Aspects of antigen mimicry revealed by immunization with a peptide mimetic of Cryptococcus neoformans polysaccharide. J. Immunol. 1998, 161, 1829–1836. [Google Scholar]
- Wrighton, N.C.; Farrell, F.X.; Chang, R.; Kashyap, A.K.; Barbone, F.P.; Mulcahy, L.S.; Johnson, D.L.; Barrett, R.W.; Jolliffe, L.K.; Dower, W.J. Small peptides as potent mimetics of the protein hormone erythropoietin. Science 1996, 273, 458–464. [Google Scholar]
- Hruby, V.J. Conformational and topographical considerations in the design of biologically active peptides. Biopolymers 1993, 33, 1073–1082. [Google Scholar] [CrossRef]
- Langston, S. Peptidomimetics and small molecule design. Drug Discov. Today 1997, 2, 254–256. [Google Scholar] [CrossRef]
- Qabar, M.; Urban, J.; Sia, C.; Klein, M.; Kahn, M. Pharmaceutical applications of peptidomimetics. Lett. Pept. Sci. 1996, 3, 25–30. [Google Scholar] [CrossRef]
- Hasegawa, A.; Cheng, X.; Kajino, K.; Berezov, A.; Murata, K.; Nakayama, T.; Yagita, H.; Murali, R.; Greene, M.I. Fas-disabling small exocyclic peptide mimetics limit apoptosis by an unexpected mechanism. Proc. Natl. Acad. Sci. USA 2004, 101, 6599–6604. [Google Scholar]
- Hasegawa, A.; Takasaki, W.; Greene, M.I.; Murali, R. Modifying TNFalpha for therapeutic use: A perspective on the TNF receptor system. Mini. Rev. Med. Chem. 2001, 1, 5–16. [Google Scholar] [CrossRef]
- Takasaki, W.; Kajino, Y.; Kajino, K.; Murali, R.; Greene, M.I. Structure-based design and characterization of exocyclic peptidomimetics that inhibit TNFα binding to its receptor. Nat. Biotechnol. 1997, 15, 1266–1270. [Google Scholar] [CrossRef]
- Cheng, X.; Kinosaki, M.; Takami, M.; Choi, Y.; Zhang, H.; Murali, R. Disabling of receptor activator of nuclear factor-kappaB (RANK) receptor complex by novel osteoprotegerin-like peptidomimetics restores bone loss in vivo. J. Biol. Chem. 2004, 279, 8269–8277. [Google Scholar]
- Heath, D.J.; Vanderkerken, K.; Cheng, X.; Gallagher, O.; Prideaux, M.; Murali, R.; Croucher, P.I. An osteoprotegerin-like peptidomimetic inhibits osteoclastic bone resorption and osteolytic bone disease in myeloma. Cancer Res. 2007, 67, 202–208. [Google Scholar]
- Ta, H.M.; Nguyen, G.T.; Jin, H.M.; Choi, J.; Park, H.; Kim, N.; Hwang, H.Y.; Kim, K.K. Structure-based development of a receptor activator of nuclear factor-kappaB ligand (RANKL) inhibitor peptide and molecular basis for osteopetrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 20281–20286. [Google Scholar]
- Benkirane, N.; Guichard, G.; Briand, J.P.; Muller, S.; Brown, F.; van Regenmortel, M.H. Mimicry of viral epitopes with retro-inverso peptides of increased stability. Dev. Biol. Stand. 1996, 87, 283–291. [Google Scholar]
- de Simone, G.; Lombardi, A.; Galdiero, S.; Nastri, F.; Della Morte, R.; Staiano, N.; Pedone, C.; Bolognesi, M.; Pavone, V. Hirunorms are true hirudin mimetics. The crystal structure of human alpha-thrombin-hirunorm V complex. Protein Sci. 1998, 7, 243–253. [Google Scholar]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef]
- Kemp, D.S. Peptidomimetics and the template approach to nucleation of beta-sheets and alpha-helices in peptides. Trends Biotechnol. 1990, 8, 249–255. [Google Scholar] [CrossRef]
- Chen, P.P.; Fong, S.; Normansell, D.; Houghten, R.A.; Karras, J.G.; Vaughan, J.H.; Carson, D.A. Delineation of a cross-reactive idiotype on human autoantibodies with antibody against a synthetic peptide. J. Exp. Med. 1984, 159, 1502–1511. [Google Scholar] [CrossRef]
- McMillan, S.; Seiden, M.V.; Houghten, R.A.; Clevinger, B.; Davie, J.M.; Lerner, R.A. Synthetic idiotypes: The third hypervariable region of murine anti- dextran antibodies. Cell 1983, 35, 859–863. [Google Scholar] [CrossRef]
- Meek, K.; Takei, M.; Dang, H.; Sanz, I.; Dauphinee, M.J.; Capra, J.D.; Talal, N. Anti-peptide antibodies detect a lupus-related interspecies idiotype that maps to H chain CDR2. J. Immunol. 1990, 144, 1375–1381. [Google Scholar]
- Rini, J.M.; Schulze-Gahmen, U.; Wilson, I.A. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science 1992, 255, 959–965. [Google Scholar]
- Tormo, J.; Blaas, D.; Parry, N.R.; Rowlands, D.; Stuart, D.; Fita, I. Crystal structure of a human rhinovirus neutralizing antibody complexed with a peptide derived from viral capsid protein VP2. EMBO J. 1994, 13, 2247–2256. [Google Scholar]
- Fecondo, J.V.; Pavuk, N.C.; Silburn, K.A.; Read, D.M.; Mansell, A.S.; Boyd, A.W.; McPhee, D.A. Synthetic peptide analogs of intercellular adhesion molecule 1 (ICAM-1) inhibit HIV-1 replication in MT-2 cells. AIDS Res. Hum. Retroviruses 1993, 9, 733–740. [Google Scholar] [CrossRef]
- Tashiro, K.; Nagata, I.; Yamashita, N.; Okazaki, K.; Ogomori, K.; Tashiro, N.; Anai, M. A synthetic peptide deduced from the sequence in the cross-region of laminin A chain mediates neurite outgrowth, cell attachment and heparin binding. Biochem. J. 1994, 302, 73–79. [Google Scholar]
- Yayon, A.; Aviezer, D.; Safran, M.; Gross, J.L.; Heldman, Y.; Cabilly, S.; Givol, D.; Katchalski-Katzir, E. Isolation of peptides that inhibit binding of basic fibroblast growth factor to its receptor from a random phage-epitope library. Proc. Natl. Acad. Sci USA 1993, 90, 10643–10647. [Google Scholar]
- Balass, M.; Heldman, Y.; Cabilly, S.; Givol, D.; Katchalski-Katzir, E.; Fuchs, S. Identification of a hexapeptide that mimics a conformation-dependent binding site of acetylcholine receptor by use of a phage-epitope library. Proc. Natl. Acad. Sci USA 1993, 90, 10638–10642. [Google Scholar]
- Malby, R.L.; Tulip, W.R.; Harley, V.R.; McKimm-Breschkin, J.L.; Laver, W.G.; Webster, R.G.; Colman, P.M. The structure of a complex between the NC10 antibody and influenza virus neuraminidase and comparison with the overlapping binding site of the NC41 antibody. Structure 1994, 2, 733–746. [Google Scholar] [CrossRef]
- Kang, C.Y.; Brunck, T.K.; Kieber-Emmons, T.; Blalock, J.E.; Kohler, H. Inhibition of self-binding antibodies (autobodies) by a VH-derived peptide. Science 1988, 240, 1034–1036. [Google Scholar]
- Maier, C.C.; LeBoeuf, R.D.; Zhou, S.R.; Whitaker, J.N.; Jarpe, M.A.; Blalock, J.E. The structure of a myelin basic protein-associated idiotope. J. Neuroimmunol. 1993, 46, 235–243. [Google Scholar] [CrossRef]
- Mazza, G.; Ollier, P.; Somme, G.; Moinier, D.; Rocca-Serra, J.; van Rietschoten, J.; Theze, J.; Fougereau, M. A structural basis for the internal image in the idiotypic network: antibodies against synthetic Ab2-D regions cross-react with the original antigen. Ann. Inst. Pasteur Immunol. 1985, 136D, 259–269. [Google Scholar]
- Taub, R.; Gould, R.J.; Garsky, V.M.; Ciccarone, T.M.; Hoxie, J.; Friedman, P.A.; Shattil, S.J. A monoclonal antibody against the platelet fibrinogen receptor contains a sequence that mimics a receptor recognition domain in fibrinogen. J. Biol. Chem. 1989, 264, 259–265. [Google Scholar]
- Taub, R.; Hsu, J.C.; Garsky, V.M.; Hill, B.L.; Erlanger, B.F.; Kohn, L.D. Peptide sequences from the hypervariable regions of two monoclonal anti-idiotypic antibodies against the thyrotropin (TSH) receptor are similar to TSH and inhibit TSH-increased cAMP production in FRTL-5 thyroid cells. J. Biol. Chem. 1992, 267, 5977–5984. [Google Scholar]
- van Cleave, V.H.; Naeve, C.W.; Metzger, D.W. Do antibodies recognize amino acid side chains of protein antigens independently of the carbon backbone? J. Exp. Med. 1988, 167, 1841–1848. [Google Scholar] [CrossRef]
- Williams, W.V.; Guy, H.R.; Rubin, D.H.; Robey, F.; Myers, J.N.; Kieber-Emmons, T.; Weiner, D.B.; Greene, M.I. Sequences of the cell-attachment sites of reovirus type 3 and its anti-idiotypic/antireceptor antibody: modeling of their three-dimensional structures. Proc. Natl. Acad. Sci. USA 1988, 85, 6488–6492. [Google Scholar]
- Prammer, K.V.; Boyer, J.; Ugen, K.; Shattil, S.J.; Kieber-Emmons, T. Bioactive Arg-Gly-Asp conformations in anti-integrin GPIIb-IIIa antibodies. Receptor 1994, 4, 93–108. [Google Scholar]
- Williams, W.V.; London, S.D.; Weiner, D.B.; Wadsworth, S.; Berzofsky, J.A.; Robey, F.; Rubin, D.H.; Greene, M.I. Immune response to a molecularly defined internal image idiotope. J. Immunol. 1989, 142, 4392–4400. [Google Scholar]
- Williams, W.V.; Moss, D.A.; Kieber-Emmons, T.; Cohen, J.A.; Myers, J.N.; Weiner, D.B.; Greene, M.I. Development of biologically active peptides based on antibody structure. Proc. Natl. Acad. Sci. USA 1989, 86, 5537–5541. [Google Scholar]
- Noseworthy, J.H.; Fields, B.N.; Dichter, M.A.; Sobotka, C.; Pizer, E.; Perry, L.L.; Nepom, J.T.; Greene, M.I. Cell receptors for the mammalian reovirus. I. Syngeneic monoclonal anti- idiotypic antibody identifies a cell surface receptor for reovirus. J. Immunol. 1983, 131, 2533–2538. [Google Scholar]
- Co, M.S.; Gaulton, G.N.; Fields, B.N.; Greene, M.I. Isolation and biochemical characterization of the mammalian reovirus type 3 cell-surface receptor. Proc. Natl. Acad. Sci. USA 1985, 82, 1494–1498. [Google Scholar]
- Gaulton, G.; Co, M.S.; Greene, M.I. Anti-idiotypic antibody identifies the cellular receptor of reovirus type 3. J. Cell. Biochem. 1985, 28, 69–78. [Google Scholar] [CrossRef]
- Poljak, R.J.; Amzel, L.M.; Chen, B.L.; Phizackerley, R.P.; Saul, F. Structure and specificity of antibody molecules. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1975, 272, 43–51. [Google Scholar] [CrossRef]
- Nakajima, H.; Mizuta, N.; Sakaguchi, K.; Fujiwara, I.; Yoshimori, A.; Takahashi, S.; Takasawa, R.; Tanuma, S. Development of HER2-antagonistic peptides as novel anti-breast cancer drugs by in silico methods. Breast Cancer 2008, 15, 65–72. [Google Scholar] [CrossRef]
- Nakajima, H.; Mizuta, N.; Sakaguchi, K.; Fujiwara, I.; Yoshimori, A.; Magae, J.; Tanuma, S. Enhancement of paclitaxel-induced apoptosis in HER2-overexpressing human breast cancer cells by a pertuzumab mimetic peptide, HRAP. J. Biosci. Bioeng. 2010, 110, 250–253. [Google Scholar] [CrossRef]
- Murali, R. University of Pennsylvania, Philadelphia, PA, USA, 2004; Unpublished work.
- Akiyama, T.; Yamada, Y.; Ogawara, H.; Richert, N.; Pastan, I.; Yamamoto, T.; Kasuga, M. Site-specific antibodies to the erbB oncogene product immunoprecipitate epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 1984, 123, 797–802. [Google Scholar] [CrossRef]
- King, C.R.; Kraus, M.H.; Aaronson, S.A. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science 1985, 229, 974–976. [Google Scholar]
- Akiyama, T.; Sudo, C.; Ogawara, H.; Toyoshima, K.; Yamamoto, T. The product of the human c-erbB-2 gene: A 185-kilodalton glycoprotein with tyrosine kinase activity. Science 1986, 232, 1644–1646. [Google Scholar]
- van de Vijver, M.; van de Bersselaar, R.; Devilee, P.; Cornelisse, C.; Peterse, J.; Nusse, R. Amplification of the neu (c-erbB-2) oncogene in human mammmary tumors is relatively frequent and is often accompanied by amplification of the linked c-erbA oncogene. Mol. Cell. Biol. 1987, 7, 2019–2023. [Google Scholar]
- di Fiore, P.P.; Pierce, J.H.; Kraus, M.H.; Segatto, O.; King, C.R.; Aaronson, S.A. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 1987, 237, 178–182. [Google Scholar]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar]
- Gullick, W.J. The role of the epidermal growth factor receptor and the c-erbB-2 protein in breast cancer. Int. J. Cancer Suppl. 1990, 5, 55–61. [Google Scholar] [CrossRef]
- Maguire, H.C., Jr.; Greene, M.I. Neu (c-erbB-2), a tumor marker in carcinoma of the female breast. Pathobiology 1990, 58, 297–303. [Google Scholar] [CrossRef]
- Kern, J.A.; Schwartz, D.A.; Nordberg, J.E.; Weiner, D.B.; Greene, M.I.; Torney, L.; Robinson, R.A. p185neu expression in human lung adenocarcinomas predicts shortened survival. Cancer Res. 1990, 50, 5184–5187. [Google Scholar]
- Cohen, J.A.; Weiner, D.B.; More, K.F.; Kokai, Y.; Williams, W.V.; Maguire, H.C., Jr.; LiVolsi, V.A.; Greene, M.I. Expression pattern of the neu (NGL) gene-encoded growth factor receptor protein (p185neu) in normal and transformed epithelial tissues of the digestive tract. Oncogene 1989, 4, 81–88. [Google Scholar]
- Williams, T.M.; Weiner, D.B.; Greene, M.I.; Maguire, H.J. Expression of c-erbB-2 in human pancreatic adenocarcinomas. Pathobiology 1991, 59, 46–52. [Google Scholar] [CrossRef]
- Drebin, J.A.; Link, V.C.; Stern, D.F.; Weinberg, R.A.; Greene, M.I. Down-modulation of an oncogene protein product and reversion of the transformed phenotype by monoclonal antibodies. Cell 1985, 41, 697–706. [Google Scholar]
- Drebin, J.A.; Link, V.C.; Weinberg, R.A.; Greene, M.I. Inhibition of tumor growth by a monoclonal antibody reactive with an oncogene-encoded tumor antigen. Proc. Natl. Acad. Sci. USA 1986, 83, 9129–9133. [Google Scholar] [CrossRef]
- Drebin, J.A.; Stern, D.F.; Link, V.C.; Weinberg, R.A.; Greene, M.I. Monoclonal antibodies identify a cell-surface antigen associated with an activated cellular oncogene. Nature 1984, 312, 545–548. [Google Scholar] [CrossRef]
- di Fiore, P.P.; Segatto, O.; Aaronson, S.A. Cloning, expression, and biological effects of erbB-2/neu gene in mammalian cells. Meth. Enzymol. 1991, 198, 272–277. [Google Scholar] [CrossRef]
- Fendly, B.M.; Winget, M.; Hudziak, R.M.; Lipari, M.T.; Napier, M.A.; Ullrich, A. Characterization of murine monoclonal antibodies reactive to either the human epidermal growth factor receptor or HER2/neu gene product. Cancer Res. 1990, 50, 1550–1558. [Google Scholar]
- Baselga, J.; Norton, L.; Albanell, J.; Kim, Y.M.; Mendelsohn, J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998, 58, 2825–2831. [Google Scholar]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar]
- Hudziak, R.M.; Lewis, G.D.; Winget, M.; Fendly, B.M.; Shepard, H.M.; Ullrich, A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol. Cell. Biol. 1989, 9, 1165–1172. [Google Scholar]
- Zhang, H.; Wang, Q.; Montone, K.T.; Peavey, J.E.; Drebin, J.A.; Greene, M.I.; Murali, R. Shared antigenic epitopes and pathobiological functions of anti-p185her2/neu monoclonal antibodies. Exp. Mol. Pathol. 1999, 67, 15–25. [Google Scholar] [CrossRef]
- Zhang, X. Structural and functional mapping of immunoglobulin domains. Ph.D. Dissertation, University of Pennsylvania, Philadelphia, PA, 1997. [Google Scholar]
- Graciani, N.R.; Tsang, K.Y.; McCutchen, S.L.; Kelly, J.W. Amino acids that specify structure through hydrophobic clustering and histidine-aromatic interactions lead to biologically active peptidomimetics. Bioorg. Med. Chem. 1994, 2, 999–1006. [Google Scholar] [CrossRef]
- McDonnell, J.M.; Fushman, D.; Cahill, S.M.; Sutton, B.J.; Cowburn, D. Solution structures of Fc epsilon RI alpha-chain mimics: A beta-hairpin peptide and its retroenantiomer. J. Am. Chem. Soc. 1997, 119, 5321–5328. [Google Scholar]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef]
- Shepard, H.M.; Lewis, G.D.; Sarup, J.C.; Fendly, B.M.; Maneval, D.; Mordenti, J.; Figari, I.; Kotts, C.E.; Palladino, M.J.; Ullrich, A.; et al. Monoclonal antibody therapy of human cancer: Taking the HER2 protooncogene to the clinic. J. Clin. Immunol. 1991, 11, 117–127. [Google Scholar] [CrossRef]
- Sliwkowski, M.X.; Lofgren, J.A.; Lewis, G.D.; Hotaling, T.E.; Fendly, B.M.; Fox, J.A. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin. Oncol. 1999, 26, 60–70. [Google Scholar]
- Baselga, J.; Albanell, J.; Molina, M.A.; Arribas, J. Mechanism of action of trastuzumab and scientific update. Semin. Oncol. 2001, 28, 4–11. [Google Scholar]
- Berezov, A.; Chen, J.; Liu, Q.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling receptor ensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 2002, 277, 28330–28339. [Google Scholar]
- Masuda, K.; Richter, M.; Song, X.; Berezov, A.; Murali, R.; Greene, M.I.; Zhang, H. AHNP-streptavidin: A tetrameric bacterially produced antibody surrogate fusion protein against p185her2/neu. Oncogene 2006, 25, 7740–7746. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, X.; Richter, M.; Greene, M.I. A sensitive and high-throughput assay to detect low-abundance proteins in serum. Nat. Med. 2006, 12, 473–477. [Google Scholar] [CrossRef]
- Cohen, J.A.; Williams, W.V.; Geller, H.M.; Greene, M.I. Anti-reovirus receptor antibody accelerates expression of the optic nerve oligodendrocyte developmental program. Proc. Natl. Acad. Sci. USA 1991, 88, 1266–1270. [Google Scholar]
- Murali, R.; Liu, Q.; Cheng, X.; Berezov, A.; Richter, M.; Furuchi, K.; Greene, M.I.; Zhang, H. Antibody like peptidomimetics as large scale immunodetection probes. Cell. Mol. Biol. (Noisy-le-grand) 2003, 49, 209–216. [Google Scholar]
- Teicher, B.A. Physiologic mechanisms of therapeutic resistance. Blood flow and hypoxia. Hematol. Oncol. Clin. North Am. 1995, 9, 475–506. [Google Scholar]
- Fantin, V.R.; Berardi, M.J.; Babbe, H.; Michelman, M.V.; Manning, C.M.; Leder, P. A bifunctional targeted peptide that blocks HER-2 tyrosine kinase and disables mitochondrial function in HER-2-positive carcinoma cells. Cancer Res. 2005, 65, 6891–6900. [Google Scholar]
- Welt, S.; Ritter, G. Antibodies in the therapy of colon cancer. Semin. Oncol. 1999, 26, 683–690. [Google Scholar]
- Guillemard, V.; Nedev, H.N.; Berezov, A.; Murali, R.; Saragovi, H.U. HER2-mediated internalization of a targeted prodrug cytotoxic conjugate is dependent on the valency of the targeting ligand. DNA Cell Biol. 2005, 24, 350–358. [Google Scholar]
- Schneider, J.W.; Chang, A.Y.; Garratt, A. Trastuzumab cardiotoxicity: Speculations regarding pathophysiology and targets for further study. Semin. Oncol. 2002, 29, 22–28. [Google Scholar]
- Schneider, J.W. Personal communicatiion. VA Boston Healthcare System and Harvard Medical School: Boston, MA, USA, 2000. [Google Scholar]
- Levine, D.H.; Ghoroghchian, P.P.; Freudenberg, J.; Zhang, G.; Therien, M.J.; Greene, M.I.; Hammer, D.A.; Murali, R. Polymersomes: A new multi-functional tool for cancer diagnosis and therapy. Methods 2008, 46, 25–32. [Google Scholar] [CrossRef]
- Murali, R.; Levine, D.H.; Hammer, D.A. University of Pennsylvania: Philadelphia, PA, USA, 2008; Unpublished work.
- Afshar, S.; Asai, T.; Morrison, S.L. Humanized ADEPT comprised of an engineered human purine nucleoside phosphorylase and a tumor targeting peptide for treatment of cancer. Mol. Cancer Ther. 2009, 8, 185–193. [Google Scholar] [CrossRef]
- Tan, M.; Lan, K.H.; Yao, J.; Lu, C.H.; Sun, M.; Neal, C.L.; Lu, J.; Yu, D. Selective inhibition of ErbB2-overexpressing breast cancer in vivo by a novel TAT-based ErbB2-targeting signal transducers and activators of transcription 3-blocking peptide. Cancer Res. 2006, 66, 3764–3772. [Google Scholar]
- Murali, R.; Greene, M. University of Pennsylvania: Philadelphia, PA, USA, 1999; Unpublished work.
- Wada, T.; Myers, J.N.; Kokai, Y.; Brown, V.I.; Hamuro, J.; LeVea, C.M.; Greene, M.I. Anti-receptor antibodies reverse the phenotype of cells transformed by two interacting proto-oncogene encoded receptor proteins. Oncogene 1990, 5, 489–495. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Murali, R.; Greene, M.I. Structure Based Antibody-Like Peptidomimetics. Pharmaceuticals 2012, 5, 209-235. https://doi.org/10.3390/ph5020209
Murali R, Greene MI. Structure Based Antibody-Like Peptidomimetics. Pharmaceuticals. 2012; 5(2):209-235. https://doi.org/10.3390/ph5020209
Chicago/Turabian StyleMurali, Ramachandran, and Mark I. Greene. 2012. "Structure Based Antibody-Like Peptidomimetics" Pharmaceuticals 5, no. 2: 209-235. https://doi.org/10.3390/ph5020209
APA StyleMurali, R., & Greene, M. I. (2012). Structure Based Antibody-Like Peptidomimetics. Pharmaceuticals, 5(2), 209-235. https://doi.org/10.3390/ph5020209